






Abstract
Additional therapeutic strategies could benefit efforts to reverse the recent increase in malaria cases in sub-Saharan Africa, which mostly affects young children. A primary candidate is dihydroartemisinin + piperaquine (DHA + PPQ), which is effective for uncomplicated malaria treatment, seasonal malaria chemoprevention, and intermittent preventive treatment. In Southeast Asia, Plasmodium falciparum parasites acquired PPQ resistance, mediated primarily by mutations in the P falciparum chloroquine resistance transporter PfCRT. The recent emergence in Africa of DHA-resistant parasites creates an imperative to assess whether PPQ resistance could emerge in African parasites with distinct PfCRT isoforms.
We edited 2 PfCRT mutations known to mediate high-grade PPQ resistance in Southeast Asia into GB4 parasites from Gabon. Gene-edited clones were profiled in antimalarial concentration-response and fitness assays.
The PfCRT F145I mutation mediated moderate PPQ resistance in GB4 parasites but with a substantial fitness cost. No resistance was observed with the PfCRT G353V mutant. Both edited clones became significantly more susceptible to amodiaquine, chloroquine, and quinine.
A single PfCRT mutation can mediate PPQ resistance in GB4 parasites, but with a growth defect that may preclude its spread without further genetic adaptations. Our findings support regional use of drug combinations that exert opposing selective pressures on PfCRT.
Antimalarial drug resistance presents a major barrier to the elimination of Plasmodium falciparum parasites, the causative agent of the most lethal form of human malaria [1]. Malaria control efforts have been further exacerbated by the coronavirus disease 2019 pandemic, and morbidity and mortality in 2020 rose worldwide to an estimated 241 million cases and 670 000 deaths, predominantly affecting young African children [2].
Artemisinin-based combination therapies (ACTs) became the first-line treatment for P falciparum in Africa in the 2000s, decades after resistance to the former first-line drugs chloroquine (CQ) and sulfadoxine-pyrimethamine (SP) had swept through the region [3]. Artemisinin-based combination therapies consist of a fast-acting artemisinin derivative, which rapidly kills drug-sensitive asexual blood stage parasites yet has a very short plasma half-life (typically <1–2 hours), combined with a longer-acting partner drug that clears the residual infection [4]. In Africa, the most frequently used ACTs are artemether + lumefantrine (LMF) and artesunate + amodiaquine. A third ACT, dihydroartemisinin + piperaquine (DHA + PPQ), has shown 99% efficacy in curing uncomplicated malaria in African settings, as per a recent meta-analysis [5, 6]. Dihydroartemisinin + PPQ is also proving to be effective for seasonal malaria chemoprevention (defined by the World Health Organization as the intermittent administration of full treatment courses of an antimalarial medicine to children during the malaria season in areas of highly seasonal transmission) as well as intermittent preventative treatment of pregnant women and infants (involving the administration of full therapeutic courses of an antimalarial medicine to pregnant women and infants to prevent adverse consequences of malaria infection), with DHA + PPQ having an advantage over combinations containing SP that can encounter SP-resistant parasites. Dihydroartemisinin + PPQ offers long preventive activity, allowing excellent efficacy when used monthly for chemoprevention. Piperaquine has the pharmacokinetic advantage of an extended terminal half-life (∼20–30 days, compared to ∼3 days for LMF), thereby providing posttreatment prophylactic activity [7]. The recent emergence and spread of PPQ resistance in Southeast (SE) Asia [8, 9], however, raises questions about whether PPQ might succumb to resistance in African parasites.
High-grade PPQ resistance can be mediated by mutations in the P falciparum CQ resistance transporter PfCRT, which spans the digestive vacuole membrane [10]. In SE Asia, these mutations arose in strains harboring multicopy plasmepsins 2 and 3 (pm 2/3), which is thought to augment PPQ resistance, as well as mutations in K13 that drive resistance to artemisinin derivatives [11–14]. The spread of PPQ- and DHA-resistant parasites has led to DHA + PPQ treatment failure rates exceeding 50% across several sites in SE Asia [9]. An association between mutant PfCRT and PPQ resistance in vitro, and an increased risk of parasite recurrence post-DHA + PPQ treatment, was first described in Cambodia with the PfCRT F145I mutation [15]. The resistance threshold has been defined in vitro as >10% of ring-stage parasites surviving a 48-hour exposure to 200 nM PPQ, using the PPQ survival assay (PSA) [11, 12]. In vitro resistance is also evidenced by incomplete parasite killing at elevated PPQ concentrations, causing a substantial increase in the IC90 (the drug concentration that inhibits parasite growth by 90%) [14, 16]. In Cambodian parasites, this F145I mutation arose in the CQ-resistant PfCRT Dd2 isoform. The Dd2 isoform already carried 8 point mutations relative to the wild-type (3D7) allele (Table 1). F145I, or another prevalent mutation G353V, conferred PPQ resistance in vitro when edited into the pfcrt locus in Dd2 parasites (a strain that carries a single copy of pm 2/3) [16]. Both of these PfCRT mutations substantially reduced growth rates (ie, caused a fitness cost) in edited Dd2 parasites.
PfCRT Haplotypes for Parasite Lines Used in This Study
| Parasite Linea | Edited | PfCRT Haplotype | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 74 | 75 | 76 | 145 | 220 | 271 | 326 | 353 | 356 | 371 | ||
| Dd23D7 (WT PfCRT) | Yes | M | N | K | F | A | Q | N | G | I | R |
| Dd2 (parent) | No | I | E | T | F | S | E | S | G | T | I |
| Dd2Dd2 + F145I | Yes | I | E | T | I | S | E | S | G | T | I |
| GB4 (parent) | No | I | E | T | F | S | E | N | G | I | I |
| GB4GB4 (editing control) | Yes | I | E | T | F | S | E | N | G | I | I |
| GB4GB4 + F145I | Yes | I | E | T | I | S | E | N | G | I | I |
| GB4GB4 + G353V | Yes | I | E | T | F | S | E | N | V | I | I |
| Parasite Linea | Edited | PfCRT Haplotype | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 74 | 75 | 76 | 145 | 220 | 271 | 326 | 353 | 356 | 371 | ||
| Dd23D7 (WT PfCRT) | Yes | M | N | K | F | A | Q | N | G | I | R |
| Dd2 (parent) | No | I | E | T | F | S | E | S | G | T | I |
| Dd2Dd2 + F145I | Yes | I | E | T | I | S | E | S | G | T | I |
| GB4 (parent) | No | I | E | T | F | S | E | N | G | I | I |
| GB4GB4 (editing control) | Yes | I | E | T | F | S | E | N | G | I | I |
| GB4GB4 + F145I | Yes | I | E | T | I | S | E | N | G | I | I |
| GB4GB4 + G353V | Yes | I | E | T | F | S | E | N | V | I | I |
Abbreviations: PfCRT, Plasmodium falciparum chloroquine resistance transporter; WT, wild type.
Dd2Dd2 (the pfcrt editing control) is not listed, because this study only used Dd23D7 and Dd2Dd2 + F145I for statistical comparisons.
PfCRT Haplotypes for Parasite Lines Used in This Study
| Parasite Linea | Edited | PfCRT Haplotype | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 74 | 75 | 76 | 145 | 220 | 271 | 326 | 353 | 356 | 371 | ||
| Dd23D7 (WT PfCRT) | Yes | M | N | K | F | A | Q | N | G | I | R |
| Dd2 (parent) | No | I | E | T | F | S | E | S | G | T | I |
| Dd2Dd2 + F145I | Yes | I | E | T | I | S | E | S | G | T | I |
| GB4 (parent) | No | I | E | T | F | S | E | N | G | I | I |
| GB4GB4 (editing control) | Yes | I | E | T | F | S | E | N | G | I | I |
| GB4GB4 + F145I | Yes | I | E | T | I | S | E | N | G | I | I |
| GB4GB4 + G353V | Yes | I | E | T | F | S | E | N | V | I | I |
| Parasite Linea | Edited | PfCRT Haplotype | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 74 | 75 | 76 | 145 | 220 | 271 | 326 | 353 | 356 | 371 | ||
| Dd23D7 (WT PfCRT) | Yes | M | N | K | F | A | Q | N | G | I | R |
| Dd2 (parent) | No | I | E | T | F | S | E | S | G | T | I |
| Dd2Dd2 + F145I | Yes | I | E | T | I | S | E | S | G | T | I |
| GB4 (parent) | No | I | E | T | F | S | E | N | G | I | I |
| GB4GB4 (editing control) | Yes | I | E | T | F | S | E | N | G | I | I |
| GB4GB4 + F145I | Yes | I | E | T | I | S | E | N | G | I | I |
| GB4GB4 + G353V | Yes | I | E | T | F | S | E | N | V | I | I |
Abbreviations: PfCRT, Plasmodium falciparum chloroquine resistance transporter; WT, wild type.
Dd2Dd2 (the pfcrt editing control) is not listed, because this study only used Dd23D7 and Dd2Dd2 + F145I for statistical comparisons.
In this study, we have examined whether PPQ resistance might arise in African parasites via the F145I or G353V mutations in PfCRT. This question is of particular importance given the recent detection in eastern Africa of parasites that have locally acquired K13 mutations that can cause decreased artemisinin susceptibility [17–20], which in turn could place additional selective pressure on PPQ as a partner drug [21]. This study used a laboratory-adapted African strain, GB4, whose PfCRT isoform is closely related to the SE Asian Dd2 isoform (the former has only 6 mutations and represents the most common mutant PfCRT sequence in Africa) (Table 1) [22]. Our findings identify F145I as a causal determinant of moderate PPQ resistance, yet with a fitness cost that we suspect would hinder its rapid spread in high-transmission African settings. Our cross-resistance data also suggest a path to mitigate potential resistance through local use of artesunate-amodiaquine, whose potency is enhanced against our PPQ-resistant parasites, to treat uncomplicated falciparum malaria.
METHODS
Plasmid Construction
Mutations in pfcrt (PF3D7_0709000) were edited using customized zinc-finger nucleases, as previously described [23]. The GB4 donor template was constructed by site-directed mutagenesis of the plasmid pfcrtDd2-hdhfr to remove the Dd2-specific N326S and I356T mutations (Supplementary Figure 1A). Additional mutagenesis reactions used primers p1 to p4 to introduce the F145I or G353V mutations (Supplementary Table 1), yielding the plasmids pfcrtGB4 + F145I-hdhfr or pfcrtGB4 + G353V-hdhfr.
Parasite Culturing and Transfections
GB4 asexual blood stage parasites were cultured in human red blood cells and transfected as described in [23, 24] and detailed in the Supplementary Methods.
In Vitro IC50 Drug Susceptibility Assays
Drug IC50 values were determined for the pfcrt-edited lines relative to control lines GB4 (parent strain), Dd23D7, and Dd2Dd2 + F145I. Concentration-response curves were determined for PPQ, CQ, monodesethyl (md)-CQ, md-amodiaquine (md-ADQ), quinine, mefloquine (MFQ), LMF, DHA, pyronaridine (PND), and ferroquine (FQ). Parasites were incubated at 37°C, with 0.2% starting parasitemias for the lines GB4, GB4GB4, GB4GB4 + G353V, and Dd23D7, and 0.4% starting parasitemias for GB4GB4 + F145I and Dd2Dd2 + F145I, at 1% hematocrit, across a range of drug concentrations diluted 2-fold in 96-well plates. Parasite growth in each well was assessed after 72 hours using flow cytometry. In brief, parasites were stained at 37°C for 20 minutes with 100 nM MitoTracker Deep Red and 1×SYBR green and quantified on an Accuri C6 flow cytometer [25]. For all antimalarials, in vitro IC50 values were determined by nonlinear regression analysis of concentration-response data with GraphPad Prism 8 software.
Piperaquine Survival Assay
Survival ratios were determined using a modified version of the PSA [11]. In brief, 0- to 6-hour postinvasion ring-stage parasites, which had been doubly synchronized with sorbitol (5% final volume, performed ∼18 hours apart in the prior cycle), were incubated at 1.0% starting parasitemia and 1% hematocrit with varying concentrations of PPQ (tetraphosphate tetrahydrate salt; Alfa Aesar) at 37°C for 72 hour in 96-well plates. The PPQ was dissolved in 0.5% lactic acid in water. The assay was carried out over the range of 1.6 nM–1600 nM PPQ. Parasitemias were determined via flow cytometry. Assays were repeated independently 6 times in duplicate or triplicate (Figure 1A, Supplementary Table 2). Statistical comparisons used Mann-Whitney U tests. The PPQ survival was defined as the percentage of parasites remaining relative to the no-drug control (100% survival).
![GB4 + F145I PfCRT confers high-grade piperaquine (PPQ) resistance. (A) The PPQ survival assay data for parental and pfcrt-edited parasite lines displayed as concentration-response curves. Dd2 and GB4 lines harboring the F145I mutation showed a survival rate of 21.3% and 7.8%, respectively, at the reference concentration of 200 nM, with the former exceeding the 10% cutoff earlier proposed to be indicative of in vitro resistance [12]. Assays were conducted on 6 separate occasions with 2–3 technical replicates. Significance between pfcrt-edited clones and their parental controls (GB4 or Dd2) was determined using Mann-Whitney U tests; **, P < .01; ***, P < .001 for GB4 and Dd2 edited lines compared to their isogenic control lines. (B) Cell morphology of parental and pfcrt-edited parasite lines. Images reveal enlarged, translucent digestive vacuoles to various degrees in parasites expressing novel PfCRT variants with F145I and G353V mutations.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/226/11/10.1093_infdis_jiac365/1/m_jiac365f1.jpeg?Expires=1749858946&Signature=QJlWKDjfEjEi8eaNLuxeko4qDjdu1eOu2m4mQEqJrIXfPP-Vp2QUQbZJF9Hb2U1pHff~6fnzGNcl47tb5CeJRRCfhgnJ4-Se5NvGFa~19SovxroRgb9G7rLFtEu3cMNwdEN8aT1xAFt6qJH2m-JeqCm-ILuPwF3BbtAfVSqMb4TNqyFj2LNj0dmdw4HEhAualsQjWoIiqA7AwXfXkfordMpG66Gjoy9VBtFMPQoa6ph~JuhyUOguzqBNEMTj5k80dFYCsi2XItS7S1ruIkCOnUZ5IHSGPpiG1GFAvU~cE9CynQG4-fMzx-itXS~ZuopwS5SXVIvV4usungcVlB1efw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
GB4 + F145I PfCRT confers high-grade piperaquine (PPQ) resistance. (A) The PPQ survival assay data for parental and pfcrt-edited parasite lines displayed as concentration-response curves. Dd2 and GB4 lines harboring the F145I mutation showed a survival rate of 21.3% and 7.8%, respectively, at the reference concentration of 200 nM, with the former exceeding the 10% cutoff earlier proposed to be indicative of in vitro resistance [12]. Assays were conducted on 6 separate occasions with 2–3 technical replicates. Significance between pfcrt-edited clones and their parental controls (GB4 or Dd2) was determined using Mann-Whitney U tests; **, P < .01; ***, P < .001 for GB4 and Dd2 edited lines compared to their isogenic control lines. (B) Cell morphology of parental and pfcrt-edited parasite lines. Images reveal enlarged, translucent digestive vacuoles to various degrees in parasites expressing novel PfCRT variants with F145I and G353V mutations.
Competitive Fitness and TaqMan Allelic Discrimination Assays
Fitness assays were performed by coculturing synchronized trophozoites of isogenic wild-type (GB4GB4) and mutant edited parasites in 1:1 ratios. Cultures were maintained in 6-well plates and sampled every 4 days over a period of 32 days. Parasite samples were freed from their red blood cells using 0.1% saponin, and genomic deoxyribonucleic acid (DNA) was extracted using the QIAmp DNA Blood Mini Kit. The percentage of the wild-type or mutant allele in each sample was determined using TaqMan allelic discrimination real-time polymerase chain reaction (PCR) assays as described in [25, 26] and detailed in the Supplementary Methods.
Structural Modeling
The single-particle cryo-EM structure of the PfCRT 7G8 isoform (PDB:6UKJ [27]) was imported into the Schrödinger molecular modeling suite (Maestro, version 2019-1). The PfCRT protein structure was analyzed using the Protein Preparation Wizard. Force field atom types and bond orders were assigned, missing atoms including hydrogens were added, tautomer/ionization states were assigned, the hydrogen bond network was optimized, and a constrained energy minimization was performed. F145I and G353V were introduced into the PfCRT 7G8 structure using the Residue and Loop Mutation tool, and then refined by a local minimization step. The final structure was visualized in Schrödinger PyMOL to display the residues of interest in specific colors.
PfCRT Haplotype Analysis
This publication uses data from the MalariaGEN Plasmodium falciparum Community Project as described in [28].
RESULTS
PfCRT F145I Mediates Piperaquine Resistance in GB4 (Gabon) Parasites
To evaluate the effect of the PPQ resistance-conferring PfCRT mutations (F145I or G353V) on an African P falciparum strain, we used zinc-finger nucleases to genetically edit these mutations into the pfcrt locus in the CQ-resistant GB4 line, yielding GB4GB4 + F145I and GB4GB4 + G353V (Table 1, Supplementary Figure 1). We also generated a control line (GB4GB4) edited to express the parental isoform.
To test for PPQ resistance, we used the PSA whereby survival rates are calculated after exposing 0- to 6-hour ring-stage parasites to PPQ [11]. As a control, we tested in parallel the pfcrt-edited PPQ-resistant Dd2Dd2 + F145I line, which is an Asian parasite expressing the Dd2 + F145I isoform. For GB4GB4 + F145I, 8% survival was observed at 200 nM PPQ, and >10% survival was identified at higher concentrations (400 to 1600 nM). These survival rates were lower than Dd2Dd2 + F145I (21%–24% survival at 200 to 1600 nM PPQ) but were significantly higher than GB4 or GB4GB4 (showing a background of 1% survival at these high concentrations). In contrast, GB4GB4 + G353V showed no increased survival at high PPQ concentrations, with a significant increase only evident at 50 nM or lower (Figure 1A, Supplementary Table 2).
Morphologically, we also observed enlarged, translucent digestive vacuoles lined by hemozoin crystals in both the trophozoite and schizont stages of GB4GB4 + F145I parasites expressing the GB4 + F145I isoform. A similar phenotype was observed with PPQ-resistant Dd2Dd2 + F145I parasites (Figure 1B). Edited GB4GB4 + G353V showed only slightly bloated digestive vacuoles compared with GB4 and GB4GB4, in contrast to the distended digestive vacuoles previously observed in the PPQ-resistant Dd2Dd2 + G353V line [16].
We also measured PPQ susceptibility using 72-hour concentration-response assays with asynchronous parasites. Results showed sustained parasite growth at elevated PPQ concentrations in the GB4GB4 + F145I line, although this was less pronounced than with Dd2Dd2 + F145I. No such phenotype was observed with GB4GB4 + G353V, confirming its lack of resistance (Supplementary Figure 2, Supplementary Table 2). It is important to note the essentiality of the PSA for determining and quantifying PPQ resistance, since both GB4GB4 + F145I and GB4GB4 + G353V had lower PPQ IC50 values in this standard concentration-response assay when compared with the isogenic control line GB4GB4. These data are consistent with earlier observations that PPQ resistance is only apparent at high drug concentrations, a phenomenon that we suspect is related to concentration-dependent engagement of drug efflux via mutant PfCRT [16, 23, 29].
Piperaquine-Resistant GB4 Parasites Manifest Hypersensitivity to 4-Aminoquinoline Drugs
Edited pfcrt variants were next profiled against a panel of licensed antimalarial drugs in 72-hour concentration-dependent growth inhibition assays (Supplementary Table 3). Results showed that when compared with the isogenic control line GB4GB4, the mutant GB4GB4 + F145I line showed increased susceptibility to md-ADQ, the active metabolite of this African ACT partner drug, manifesting as a 2-fold lower mean IC50 value (Figure 2A). A similar increase in md-ADQ sensitivity was observed in the GB4GB4 + G353V line, despite this line not acquiring PPQ resistance. Both PfCRT variants also resulted in GB4 parasites becoming significantly more sensitive to quinine, used as a treatment for severe malaria (Figure 2B). We also observed that both mutations reversed GB4 parasite resistance to CQ and its metabolite md-CQ. This sensitization was most pronounced with the GB4GB4 + F145I line, which became as sensitive to CQ and md-CQ compounds as the comparator Dd23D7, a Dd2 line expressing the 3D7 wild-type PfCRT isoform (Figure 2Cand 2D). There was no significant difference in mean IC50 values for GB4GB4 + F145I and GB4GB4 + G353V, compared to the GB4 controls, with the other ACT drugs MFQ, LMF, DHA, or PND. Likewise, there was no IC50 shift against FQ, presently in human clinical trials as a novel antimalarial [30] (Supplementary Figure 3A–E).
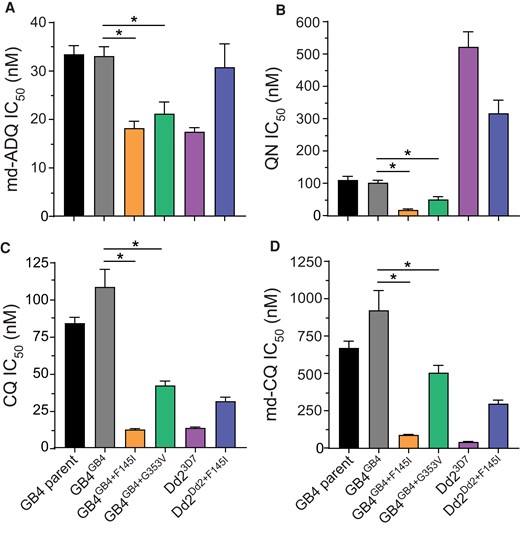
Concentration-response data for antimalarial drugs profiled against parental and pfcrt-edited parasite lines. Results show complete or partial reversal of resistance and increased sensitivity of GB4 parasites harboring the PfCRT mutations F145I and G353V, relative to the control lines and the Dd2 parasite expressing wild-type 3D7 against the drugs (A) monodesethyl-amodiaquine (md-ADQ); (B) quinine (QN); (C) chloroquine (CQ); and (D) the CQ active metabolite monodesethyl-CQ (md-CQ). Comparisons are shown between pfcrt-edited parasites and their isogenic GB4 or Dd2 controls. Significance was determined using Mann-Whitney U tests with N, n = 4.2; *, P < .05.
Piperaquine-Resistant GB4 + F145I Parasites Display a Substantial Growth Defect
To determine the relative asexual blood stage fitness conferred by the F145I and G353V mutations on the African GB4 background, we cocultured edited mutant parasites with the wild-type GB4 editing control in pairwise competitive assays (Figure 3A). The relative expansion of each line was assessed by TaqMan-based allelic discrimination quantitative PCR (qPCR) (Supplementary Tables 1 and 4). Assays were initiated with tightly synchronized trophozoites mixed in a 1:1 ratio of wild-type to mutant isogenic parasites, and cultures were maintained over a period of 32 days (∼16 generations of asexual blood stage growth). GB4GB4 + F145I parasites were fully outcompeted by GB4GB4 within 12 days (∼6 generations), illustrating a substantial fitness cost of the F145I mutation on edited GB4 parasites (Figure 3A). From these data, we calculated the fitness cost, which represents the percentage reduction in growth rate per 48-hour generation of a test line compared to its wild-type isogenic comparator. For GB4GB4 + F145I, the mean fitness cost was measured at 39.2% per generation (Figure 3B). However, the G353V mutation had no effect on the fitness of GB4 parasites (with only a 0.2% fitness cost). These data revealed that the high PSA survival rate of GB4GB4 + F145I parasites was offset by a high fitness cost (Figure 3B).
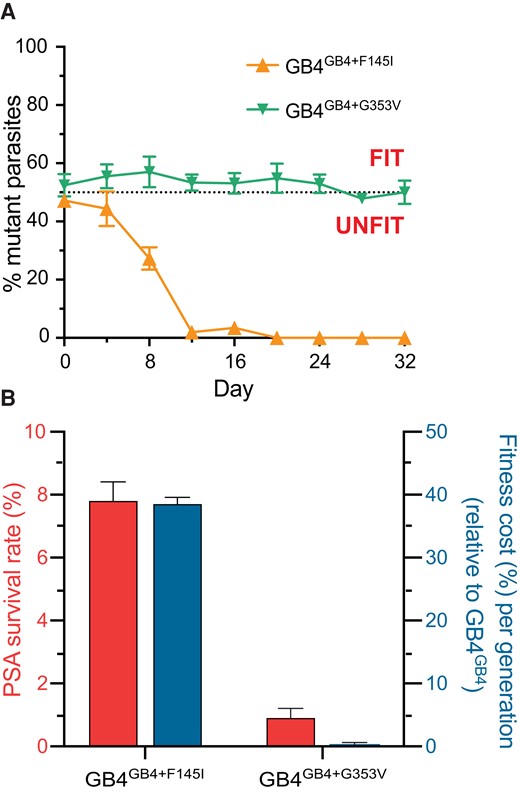
GB4GB4 + F145I exhibits a significant fitness cost. (A) Parasite fitness was determined by pairwise competition of either mutant PfCRT-edited line, F145I and G353V, with an isogenic wild-type control over 32 days. Percentages of mutant alleles relative to the wild-type allele overtime were determined using custom TaqMan allelic discrimination quantitative polymerase chain reaction assays. Results, shown as mean ± SEM, were obtained from 3 independent experiments, each performed in duplicate. (B) Piperaquine (PPQ) survival assay (PSA) survival rates at 200 nM PPQ (left Y axis, in red) and mean relative fitness costs per generation (right Y axis, in blue) are shown for GB4GB4 + F145I and GB4GB4 + G353V relative to parental GB4GB4.
DISCUSSION
Piperaquine presents considerable advantages as an additional partner drug for antimalarial combination therapy and chemoprevention in Africa due to its potency and long half-life. However, the emergence of resistance in SE Asia calls for investigations into the risk of resistance arising in genetically distinct African parasites. In this study, we provide evidence that moderate PPQ resistance can be acquired via a single-point mutation, F145I, introduced into the widely prevalent mutant GB4 isoform of the CQ resistance transporter, PfCRT. This gain of resistance was accompanied by a very substantial loss of parasite fitness, which we posit could preclude this mutation from being competitive in high-transmission African settings with typically mixed infections. We also find that PPQ resistance results in increased sensitization to md-ADQ, quinine, and CQ, creating a rationale for implementing regimens with different drug combinations that exert opposing selective pressures on parasite populations and thereby hinder the spread of multidrug-resistant P falciparum malaria [23].
Recent reports of mutant k13 alleles in local parasite populations in Rwanda and Uganda provided a major impetus for our study. These include clinical evidence of delayed parasite clearance after treatment with an ACT and in vitro evidence of ART resistance [17–20]. Recent data suggest that ART resistance mediated by K13 mutations does not extend to all African strains, implying an important contribution of genetic background [31]. Nonetheless, ART resistance in Africa is likely to spread, thereby imposing greater selective pressure on ACT partner drugs. Lumefantrine has not yet encountered resistance, and ADQ still remains widely effective, with these drugs exerting selective pressure in favor of wild-type and mutant pfcrt alleles, respectively [32]. Because PPQ is increasingly adopted as a curative and chemopreventive drug in Africa, the lessons learned from SE Asia can help guide its use in ways that mitigate the risk of resistance.
High-grade PPQ resistance in SE Asia is driven primarily by point mutations in PfCRT, with F145I conferring the greatest degree of resistance. In the Asian Dd2 strain, this mutation was earlier found to confer a substantial fitness cost, estimated at ∼17% per 48-hour generation of asexual blood stage growth, and in the field other mutations with reduced fitness costs have gained in prevalence (such as T93S) [29, 33]. These PfCRT mutations arose in SE Asian strains that express multiple copies of pm 2/3. These hemoglobinases are thought to augment PPQ resistance [14] and potentially alleviate some of the mutant PfCRT-mediated fitness costs. Of note, parasites harboring multicopy pm 2/3 have yet to be detected in African studies [19, 34, 35]. In SE Asia, PPQ-resistant parasites also typically harbor mutant K13 [36]. The pathway unifying these resistance determinants is hemoglobin catabolism, used by the parasites to generate peptides for protein synthesis, but also an Achilles heel because it generates toxic heme byproducts to which PPQ, CQ, and ADQ bind to prevent detoxification [37]. This process occurs in the parasite's acidic digestive vacuole, where these drugs accumulate and where pm 2/3 help proteolytically cleave hemoglobin [10]. PfCRT is known to efflux both globin-derived peptides as well as drugs depending on the structural conformation of the isoform. Impaired peptide flux is a feature common to mutant PfCRT isoforms [38–41]. Morphologically, this manifests as distended, translucent digestive vacuoles [16], as seen herein with GB4 and Dd2 parasites expressing the GB4 + F145I and Dd2 + F145I mutant PfCRT isoforms, respectively. K13 is also connected because it regulates the import of host hemoglobin into newly formed ring-stage parasites, and mutations conferring ART resistance act in part to restrict this endocytic process [42, 43].
In African GB4 parasites expressing the GB4 + F145I variant, we estimate a fitness cost of ∼39% reduced growth rate per generation compared with the isogenic parasites expressing the parental GB4. By comparison, parental GB4 itself also produced a fitness cost of 8% slower growth per generation compared with the wild-type isoform in isogenic parasites [22]. Earlier studies in Malawi, later confirmed across Africa, showed that the fitness costs observed with CQ-resistant PfCRT isoforms was sufficient for wild-type PfCRT parasites to overtake mutant isolates once CQ was no longer used clinically [44]. In The Gambia, studies showed that this attrition of mutant PfCRT could occur within a single season of malaria transmission [45]. The impact of these reduced growth rates on parasite viability at the population level is thought to be exacerbated in high-transmission African settings with frequently mixed infections, robust host immunity, and reduced per-patient use of therapeutics in the adult population, compared to SE Asian settings [46]. These observations lead us to surmise that the GB4 + F145I variant alone would not be competitive in African epidemiological settings.
Strikingly, no change in PPQ susceptibility was observed upon introducing the PfCRT G353V mutation into GB4 parasites, despite its demonstrated ability to mediate PPQ resistance in Asian strains [22]. Of note, the GB4 and Dd2 PfCRT isoforms differ at the nearby position 356, which highlights the potential influence of PfCRT amino acids on nearby residues and overall protein function (Supplementary Figure 4). These isoforms also differ at residue 326, which in its wild-type state as found in GB4 increases md-ADQ susceptibility when compared with the Dd2 N326S mutation [47]. Of note, molecular dynamics simulations based on the recently elucidated PfCRT structure provided evidence that the F145I mutation may change the conformation of helices surrounding the PfCRT binding cavity, thereby influencing drug and peptide efflux properties [27].
Our findings of increased sensitization to md-ADQ in our PPQ-resistant GB4GB4 + F145I line evokes consideration of the potential benefits of combining these 2 drugs into a triple ACT. A similar approach, combining artemether + LMF with ADQ, recently showed excellent efficacy in Cambodia and is being considered to prolong the therapeutic lifetime of artemether + LMF [48]. An alternative would be to allow multiple first-line therapies to be used concurrently in local settings, such as DHA + PPQ and artesunate-ADQ that would exert opposing selective pressures on PfCRT variants [49].
CONCLUSIONS
Our study demonstrates that PPQ resistance can arise on an African background via the acquisition of a single point mutation in PfCRT added to the CQ-resistant GB4 mutant isoform, albeit with a severe fitness cost. This GB4 isoform was earlier estimated to have a 19% prevalence in a collection of 783 sequenced African isolates present in the Pf3K data set [22, 50]. The next most common mutant PfCRT isoform, termed Cam783, had a 12% prevalence, with the CQ-sensitive wild-type (3D7) allele present in 66% of samples. Our analysis of PfCRT haplotypes in the Pf6 data set, comprising 3885 African samples, shows that mutant PfCRT (as per the available partial haplotype data) remained quite common across Western, Central, and Eastern Africa, unlike Southern Africa where almost all parasites expressed wild-type PfCRT (Supplementary Figure 5). Further studies will be important to test additional PfCRT mutations and background haplotypes [22] to fully assess the possibility of PPQ resistance arising in Africa.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Financial support. Partial funding for this work was provided by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (Grant Numbers R01 AI050234, R01 AI147628, R01 AI124678, R21 AI127581 [to D. A. F.]; R01 AI143521 [to K. J. W.]; F31 AI157410 [to L. M. H.]; and K08 AI163497 [to J. L. S.-S.]). J. L. S.-S. gratefully acknowledges support from a Doris Duke Physician Scientist Award and a Louis V. Gerstner, Jr. Scholars Award. L. M. H. was funded in part by the Columbia University Graduate Training Program in Microbiology and Immunology (Grant T32 AI106711; Program Director D. Fidock).
References
Author notes
J. L. S.-S. and L. M. H. contributed equally to this work.
Presented in part: Keystone Symposium on Malaria, April 2022, Breckenridge, CO.
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.


{kind=link}
{kind=link}
{kind=link}