






Abstract
Seasonal influenza virus infection causes a range of disease severity, including lower respiratory tract infection with respiratory failure. We evaluated the association of common variants in interferon (IFN) regulatory genes with susceptibility to critical influenza infection in children.
We performed targeted sequencing of 69 influenza-associated candidate genes in 348 children from 24 US centers admitted to the intensive care unit with influenza infection and lacking risk factors for severe influenza infection (PICFlu cohort, 59.4% male). As controls, whole genome sequencing from 675 children with asthma (CAMP cohort, 62.5% male) was compared. We assessed functional relevance using PICFlu whole blood gene expression levels for the gene and calculated IFN gene signature score.
Common variants in DDX58, encoding the retinoic acid–inducible gene I (RIG-I) receptor, demonstrated association above or around the Bonferroni-corrected threshold (synonymous variant rs3205166; intronic variant rs4487862). The intronic single-nucleotide polymorphism rs4487862 minor allele was associated with decreased DDX58 expression and IFN signature (P < .05 and P = .0009, respectively) which provided evidence supporting the genetic variants’ impact on RIG-I and IFN immunity.
We provide evidence associating common gene variants in DDX58 with susceptibility to severe influenza infection in children. RIG-I may be essential for preventing life-threatening influenza-associated disease.
Seasonal influenza infection causes high morbidity with an estimated 140 000–710 000 hospitalizations annually between 2010 and 2020 [1]. Although influenza infection most often causes self-limited and uncomplicated disease, individuals with underlying chronic disorders are at higher risk for developing complications, including death. However, healthy children and adolescents can also develop critical illness requiring life support, particularly during pandemics when disease is prevalent in young healthy individuals [2]. In any given individual, host genetics and innate and adaptive immunity influence susceptibility to and outcome of influenza virus infection. Among these factors, type I interferon (IFN) and downstream IFN-regulated responses are important for early antiviral host defense by suppressing virus replication. Rare variants in genes mediating the type I IFN response (including TLR3, IRF7, and IRF9) have been reported in a small number of individuals critically ill with influenza virus infection [3–5].
Common variants in IFN-associated genes have also been reported to be associated with influenza disease susceptibility in children and adults [6], with the strongest support for interferon-inducible transmembrane protein 3 (IFITM3) [7]. Moreover, different IFN activity levels in early viral infection have been associated with different ancestries and correlate with cis-expression quantitative trait loci (eQTL), which explains ancestry-associated differences [8]. Therefore, it is important to identify common variants for eQTL as well as rare ones for loss-of-function mutations in genes linked to type I IFN signaling and clinical outcome.
From an immunologic standpoint, children differ developmentally from adults, for example having more plasmacytoid dendritic cells that are a major producer of type I IFN responses [9]. Therefore, the impact of genetic risk factors for susceptibility to influenza virus infection may differ depending upon age. Because of this, we evaluated for potential association of common variants in IFN regulatory genes in a pediatric cohort admitted to the intensive care unit (ICU) with severe disease. Children enrolled in a longitudinal asthma study served as the comparison cohort, using covariate adjustment for age, sex, and ethnicity.
METHODS
Study Populations
The Pediatric Intensive Care Influenza (PICFlu) investigators were from 24 North American sites (Supplementary Appendix) from the larger Pediatric Acute Lung Injury and Sepsis Investigators (PALISI) research network. Children (<18 years of age) with confirmed influenza infection admitted to the pediatric intensive care units (PICUs) at 24 sites were enrolled (2008–2016). Influenza virus infection was confirmed from clinical testing of respiratory samples [10] and/or by research testing on nasopharyngeal samples using multiplex polymerase chain reaction–based testing. Children with immunosuppressive or severe underlying respiratory, metabolic cardiac, neurologic, or other condition(s) predisposing them to more severe influenza infection were ineligible.
For the comparison, the Childhood Asthma Management Program (CAMP) longitudinal cohort was used (enrollment 1993–1995). All participants in CAMP were aged 5–12 years at enrollment and had mild to moderate persistent asthma for at least 6 months in the previous year. The mild to moderate severity classification was confirmed using a self-reported diary score and peak expiratory flow readings over a 28-day period using criteria as previously described [11]. Children with severe asthma or other chronic medical conditions were excluded.
Subject recruitment and study procedures for both PICFlu and CAMP have been previously described in detail [12, 13]. Written informed consent from parents and/or the subject’s assent were obtained for each childhood study protocol. Study protocols were approved by the institutional review boards (IRBs) of Boston Children’s Hospital (PICFlu) and Brigham and Women’s Hospital (CAMP) in Boston, Massachusetts (IRB numbers X08-11-0534 and IRB-P00037375), and by local IRBs at each recruitment site for each study.
Genotyping Data
Genomic DNA from PICFlu patients were extracted from peripheral blood using the Gentra Puregene Blood kit (Qiagen). Targeted resequencing using Illumina TruSeq Custom Amplicon kit (TSCA; Illumina, San Diego, California) was performed to generate genotype information for 69 IFN pathway and related genes (Supplementary Table 1). In short, oligonucleotide probes were designed to generate 425 bp amplicons using Illumina DesignStudio tool to cover exonic regions and some potential regulatory intronic regions of the targeted genes. Sequencing libraries were prepared following the TSCA library preparation guide using 200 ng of genomic DNA. Up to 96 samples were barcoded using Illumina TruSeq index primers and the multiplexed libraries were pooled for sequencing. Sequencing of amplicons was performed using MiSeq V3 Sequencing Kit (Illumina) and run on a MiSeq Illumina sequencer at a 2 × 300 bp read-length configuration with dual indexing.
The whole genome sequencing data for CAMP were generated as part of the National Heart, Lung, and Blood Institute Trans-Omics for Precision Medicine (TOPMed) Program [14]. Details regarding the laboratory methods, data processing, and quality control are described (TOPMed accession released on dbGaP, accession number phs001726).
After combining genotyping data in both cohorts, we included only single-nucleotide polymorphisms (SNPs) with <1% SNP missing rate, allele frequency >0.1%, and Hardy-Weinberg proportions (P > 10−5) for variant quality control. This yielded 1268 variants tested for association with severe influenza virus infection in children.
Gene Expression Measurements
PAXgene RNA vacutainers (PreAnalytix) with 1–2.5 mL whole blood, collected within the first 72 hours of ICU admission, were incubated at room temperature for 18–24 hours (minimum of 2 hours) and frozen at –80°C. Total RNA was extracted using the PAXgene Blood RNA kit (Qiagen) and QIAcube instrument as recommended by the manufacturer, then quality checked using the RNA 6000 Nano kit and Agilent 2100 Bioanalyzer (Agilent Technologies). Gene expression of various messenger RNA targets was measured using a NanoString custom-designed probe panel according to the manufacturer with the modification that an estimated 85 ng RNA was run for each sample (NanoString Technologies, Seattle, Washington) [15]. Data were quality checked and normalized to 6 housekeeping genes (B2M, DECR1, GUSB, HPRT1, PPIB, POLG).
The expression levels of 10 type I IFN signature genes (IFI27, IFI44L, IFIT1, ISG15, RSAD2, STAT1, STAT2, IRF7, MX1, OAS3) were used for the IFN score calculation. The IFN scores were calculated by the geometric mean of the NanoString counts of selected IFN signature genes for each sample as described by Kim et al [16]. Gene expression levels for log-transformed DDX58 (retinoic acid–inducible gene I [RIG-I]) counts and the IFN scores were compared in different subject groups using 1-way analysis of variance test.
Statistical Analysis
Demographic characteristics and clinical features between groups were compared using t tests or χ2 tests, as appropriate, using the R statistical software 4.03. We performed generalized logistic regression of 2 cohorts adjusting for age, sex, and 10 principal components for genetic ancestry under the additive model. Association analysis was done by using PLINK 1.9 (downloaded on 24 January 2020). P values were considered significant at the Bonferroni-corrected threshold for testing 1268 variants (0.05/1268 = 3.94 × 10−5). Linkage disequilibrium (LD) information was estimated using the 1000 Genomes Project datasets [17, 18]. Two-tailed t tests were used in the expression level/IFN-stimulated genes (ISG) score comparison between individual groups with different genotypes.
RESULTS
The demographic characteristics of the PICFlu (n = 348) and CAMP (n = 675) cohorts for patients included in the genomics analyses are presented in Table 1. Both cohorts had approximately 60% males, but PICFlu patients were younger and had a higher proportion of Hispanic/Latino and a lower proportion of non-Hispanic White patients than CAMP. All patients in CAMP had a diagnosis of mild to moderate persistent asthma with the exclusion of severe asthma or other chronic medical conditions. By contrast, most PICFlu patients were previously healthy with no underlying diagnoses (n = 186 [53.5%]) and reported taking no prescription medications. Their most common underlying medical condition was mild intermittent asthma or reactive airway disease, reported in 24% of patients. Other conditions predisposing to severe influenza infection were excluded from enrollment in PICFlu.
Demographic Characteristics, Disease Severity, and Clinical Outcomes of the Pediatric Intensive Care Influenza and Childhood Asthma Management Program Cohorts
| Characteristic | PICFlu Cohort | CAMP Cohort | P Value |
|---|---|---|---|
| No. of participants | 348 | 675 | |
| Male sex | 207 (59.5) | 422 (62.5) | .38 |
| Age, y, median (IQR) | 6.5 (2.5–11.2) | 8.98 (7.2–10.8) | <.001 |
| Ethnic background (self-reported) | <.001 | ||
| White non-Hispanic | 188 (54) | 467 (69.2) | |
| Hispanic/Latino | 91 (26.2) | 68 (10) | |
| Black/African-American non-Hispanic | 42 (12.1) | 84 (12.4) | |
| Other | 27 (7.8) | 56 (8.3) | |
| Influenza Aa | 285 (81.9) | … | |
| Influenza Ba | 67 (19.3) | … | |
| Invasive mechanical ventilation | 254 (73) | … | |
| Hospital stay, d, median (IQR) | 10.9 (5.8–21.8) | … | |
| Hospital mortality | 23 (6.6) | … |
| Characteristic | PICFlu Cohort | CAMP Cohort | P Value |
|---|---|---|---|
| No. of participants | 348 | 675 | |
| Male sex | 207 (59.5) | 422 (62.5) | .38 |
| Age, y, median (IQR) | 6.5 (2.5–11.2) | 8.98 (7.2–10.8) | <.001 |
| Ethnic background (self-reported) | <.001 | ||
| White non-Hispanic | 188 (54) | 467 (69.2) | |
| Hispanic/Latino | 91 (26.2) | 68 (10) | |
| Black/African-American non-Hispanic | 42 (12.1) | 84 (12.4) | |
| Other | 27 (7.8) | 56 (8.3) | |
| Influenza Aa | 285 (81.9) | … | |
| Influenza Ba | 67 (19.3) | … | |
| Invasive mechanical ventilation | 254 (73) | … | |
| Hospital stay, d, median (IQR) | 10.9 (5.8–21.8) | … | |
| Hospital mortality | 23 (6.6) | … |
Data are presented as No. (%) unless otherwise indicated.
Abbreviations: CAMP, Childhood Asthma Management Program; IQR, interquartile range; PICFlu, Pediatric Intensive Care Influenza.
Four patients were positive for influenza A and B (for influenza A: 38.8% H1N1pdm2009, 18.7% H3N1, 4.6% H1N1 seasonal; 18.7% no subtype).
Demographic Characteristics, Disease Severity, and Clinical Outcomes of the Pediatric Intensive Care Influenza and Childhood Asthma Management Program Cohorts
| Characteristic | PICFlu Cohort | CAMP Cohort | P Value |
|---|---|---|---|
| No. of participants | 348 | 675 | |
| Male sex | 207 (59.5) | 422 (62.5) | .38 |
| Age, y, median (IQR) | 6.5 (2.5–11.2) | 8.98 (7.2–10.8) | <.001 |
| Ethnic background (self-reported) | <.001 | ||
| White non-Hispanic | 188 (54) | 467 (69.2) | |
| Hispanic/Latino | 91 (26.2) | 68 (10) | |
| Black/African-American non-Hispanic | 42 (12.1) | 84 (12.4) | |
| Other | 27 (7.8) | 56 (8.3) | |
| Influenza Aa | 285 (81.9) | … | |
| Influenza Ba | 67 (19.3) | … | |
| Invasive mechanical ventilation | 254 (73) | … | |
| Hospital stay, d, median (IQR) | 10.9 (5.8–21.8) | … | |
| Hospital mortality | 23 (6.6) | … |
| Characteristic | PICFlu Cohort | CAMP Cohort | P Value |
|---|---|---|---|
| No. of participants | 348 | 675 | |
| Male sex | 207 (59.5) | 422 (62.5) | .38 |
| Age, y, median (IQR) | 6.5 (2.5–11.2) | 8.98 (7.2–10.8) | <.001 |
| Ethnic background (self-reported) | <.001 | ||
| White non-Hispanic | 188 (54) | 467 (69.2) | |
| Hispanic/Latino | 91 (26.2) | 68 (10) | |
| Black/African-American non-Hispanic | 42 (12.1) | 84 (12.4) | |
| Other | 27 (7.8) | 56 (8.3) | |
| Influenza Aa | 285 (81.9) | … | |
| Influenza Ba | 67 (19.3) | … | |
| Invasive mechanical ventilation | 254 (73) | … | |
| Hospital stay, d, median (IQR) | 10.9 (5.8–21.8) | … | |
| Hospital mortality | 23 (6.6) | … |
Data are presented as No. (%) unless otherwise indicated.
Abbreviations: CAMP, Childhood Asthma Management Program; IQR, interquartile range; PICFlu, Pediatric Intensive Care Influenza.
Four patients were positive for influenza A and B (for influenza A: 38.8% H1N1pdm2009, 18.7% H3N1, 4.6% H1N1 seasonal; 18.7% no subtype).
In PICFlu (Table 1), 82% of patients had influenza A and 19% had influenza B infection (1% both), with bacterial coinfection diagnosed within the first 72 hours of hospitalization in 112 (32%) patients, most commonly with Staphylococcus aureus (n = 61 [17.5%]). The majority of patients in the PICFlu cohort had life-threatening illness, with 119 (34%) diagnosed with acute respiratory distress syndrome. Two hundred fifty-four (73%) were intubated and received invasive mechanical ventilator support, 33 (9.5%) received extracorporeal membrane oxygenation support, and 23 (6.6%) died during hospitalization.
Genomic Association and Functional Analyses
Testing the hypothesis that the common variants were enriched in the PICFlu cohort compared to the CAMP cohort, the targeted sequencing of a panel of type I IFN–related response genes in the PICFlu cohort compared to corresponding genetic data from the CAMP cohort showed that adjustment for race and ethnicity using principal components analysis had excellent overlap (Figure 1A), and the quantile-quantile plot showed that the 2 datasets had the same distributions (Figure 1B). Therefore, genotyping bias in the genomic analysis from population structure effects was minimal. In the Manhattan plot (Figure 1C) evaluating overall pediatric susceptibility to severe influenza virus infection, only DDX58 exceeded the Bonferroni-corrected threshold in the absence of population structure effects (SNP rs3205166, P = 3.84 × 10−5).
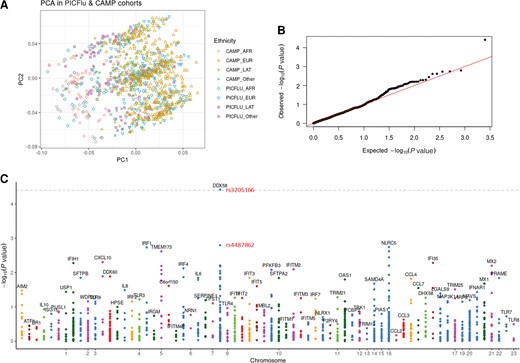
Principal components analysis plot showing adjustment for race (A), quantile–quantile plot (B), and Manhattan plot (C) for generalized logistic regression of 2 cohorts (PICFlu vs CAMP) adjusting for age, sex, and 10 principal components of genetic ancestry under the additive model. The grey dotted line in the Manhattan plot shows a Bonferroni-corrected threshold of .05 (P = 3.90 × 10−5), which only synonymous variant DDX58 rs3205166 exceeds, with intronic DDX58 rs4487862 the dot below it (P = .0016). Abbreviations: AFR, African; CAMP, Childhood Asthma Management Program; EUR, European; LAT, Latino; PC, principal component; PCA, principal components analysis; PICFlu, Pediatric Intensive Care Influenza.
DDX58 encodes RIG-I, a cytosolic viral RNA sensor that recognizes the 5′-diphosphate/triphosphate ends of double-stranded viral RNA intermediates to initiate type I IFN signaling. Among all common variants in the region of DDX58 (Supplementary Table 2), we evaluated the 2 most significant SNPs in DDX58 including rs3205166 and rs4487862 (intronic variant) for their potential functional impact on downstream immune signaling. SNP rs3205166 is a synonymous variant with a XXX (CADD) score of 0.91 whereas rs4487862 is an intronic variant with a CADD score of 3.68; both variants were not predicted to be associated with any potential splicing events, thus having no direct impact on the DDX58 gene itself.
For the PICFlu patients, the expression profiles were generated from patients with samples available that were collected early during their PICU admission. There was a mild trend that individuals with homozygous G/G had slightly higher DDX58 expression, but there was no statistical significance in DDX58 expression for patients with different genotypes of the synonymous variant rs3205166 (homozygous G/G, heterozygous G/T, and homozygous T/T). For the other intronic variant rs4487862, individuals with the homozygous G/G and heterozygous A/G genotypes (average expression log value of 2.366 and 2.389) had significantly lower DDX58 expression levels compared to those with the homozygous A/A genotype (average expression log value of 2.619; Figure 2A and Supplementary Figure 1A). Since DDX58 itself is an IFN-stimulated gene [19, 20], its expression level reflects feedback amplification of initial type I IFN responses. Therefore, we also examined expression of other IFN-stimulated genes (IFI27, IFI44L, IFIT1, ISG15, RSAD2, STAT1, STAT2, IRF7, MX1, OAS3) to assess overall type I IFN signaling associated with these DDX58 genotypes. A similar pattern was seen in the association with cumulative IFN gene signature scores and the 10 constituent IFN-stimulated genes, where rs4487862 was significantly associated with its homozygous minor allele (A/A) having the lowest score (Figure 2B, Supplementary Figures 1B and 2). These results suggested the potential decrement on the DDX58 gene expression itself and on its downstream IFN signaling pathway.
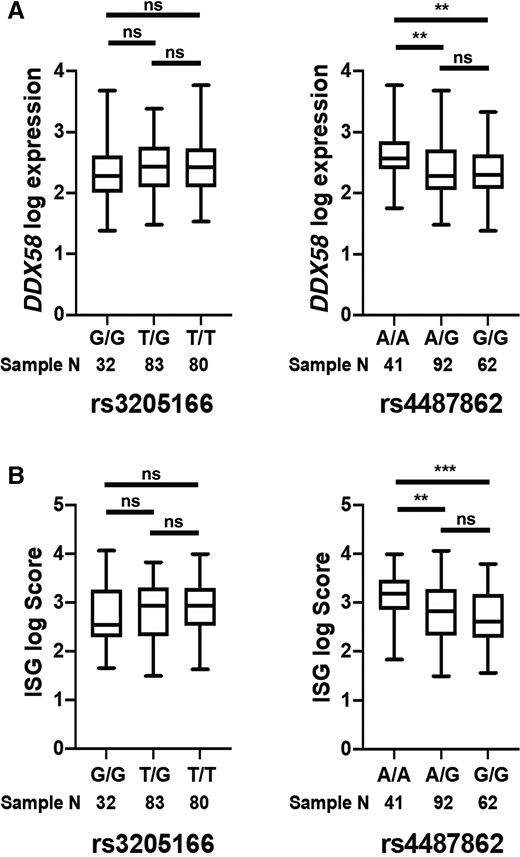
DDX58 (retinoic acid–inducible gene I) expression level (A) and interferon gene signature (ISG) score (B) according to genotyping of the DDX58 common variants rs3205166 and rs4487862. **P < .01, ***P < .001; ns, not significant.
Linkage Disequilibrium Between SNP rs4487862 and the DDX58 Promoter Region
Because we found evidence suggesting a link between SNP rs4487862 and the DDX58 expression for the biological interpretation, we examined other variants in LD with rs4487862. Strong LD between rs4487862 and rs3739674 was observed in individuals with European ancestry from CAMP or 1000 Genome projects (D′ = 1.0, R2 = 0.7490 or R2 = 0.8099, respectively; Supplementary Figures 3 and 4 and Supplementary Table 3). For the SNP rs3739674, cis-regulatory elements are present in this region as open chromatin in 95 ENCODE overlaid DNase I datasets, and epigenetic modifications also showed marks of active enhancers (histone H3 lysine 4 mono-methylation [H3K4me1] and histone H3 lysine 27 acetylation [H3K27ac] in Supplementary Figure 5). DDX58 rs3739674 was also previously reported to be associated with DDX58 expression in lymphoblastoid cell lines (P = 2.80 × 10−10) [21]. These data suggest that rs3739674 as eQTL of DDX58 should be examined more closely in future studies of influenza virus susceptibility. Unfortunately, rs3739674 is an SNP located in the promoter region of DDX58, which was not genotyped in the PICFlu cohort. Additional follow-up genetic study by whole exome sequencing or whole genome sequencing should be performed to confirm the association of rs3739674 or any other common variants or genes that were missed in this study due to technical limitations.
DISCUSSION
The type I IFN cytokine family plays a critical role in the human immune response against influenza infection, activating a complex regulatory system of innate and adaptive immune responses. In the PICFlu cohort, we used targeted sequencing to evaluate candidate genes chosen based on evidence that they could selectively impair innate immunity to influenza virus; these genes included those involved in virus nucleic acid sensing, IFN signal transduction or amplification, transcriptional regulation by type I IFN, and virus replication restriction [22, 23]. We also evaluated an additional 9 genes involved in inflammation. Among these candidate genes, we identified that common variants in DDX58 (coding for immune signaling components of the RIG-I receptor) constituted a significant locus for pediatric susceptibility to severe influenza virus infection. In addition, SNP rs4487862, which was enriched in the PICFlu cohort, was linked to lower DDX58 expression and IFN gene signature at this locus. Taken together, these results provide evidence that the signaling cascade for RIG-I–induced IFN production may contribute to influencing pediatric susceptibility to severe influenza virus infection.
In the PICFlu cohort, we previously showed that suppression of the RIG-I pathway (as assessed by stimulating whole blood ex vivo with the viral ligand polyinosinic-polycytidylic acid-low-molecular-weight/LyoVecPoly [Poly I:C]) was associated with worse clinical outcomes including death and prolonged multiple organ dysfunction [24]. Although we did not evaluate the role of rare gene variants in this study, Jorgensen and colleagues [25] reported a case of a patient with rare variants in DDX58 that could explain susceptibility to severe influenza infection during the 2009 influenza A pandemic. The majority of patients in the PICFlu cohort had influenza A infection, which underlies most of the RIG-I literature, but 19% were infected with influenza B. Investigators have shown that RIG-I is also essential for mounting an effective innate immune response to influenza B in mice [26]. Recently, SNP rs12551294 was suggested as lead cis eQTL for DDX58 [8] where upon viral RNA recognition, RIG-I leads to a signaling cascade inducing antiviral interferon-stimulated genes such as type I IFN [19, 20]. However, this could not be examined in our cohorts given the low representation of subjects with African ancestry in which this allele is common. Additional genetic efforts for similar influenza cohorts with a broad geographic region coverage will be helpful to identify genotype-phenotype association traits.
The strengths of this analysis include the well-phenotyped pediatric cohorts in ethnically diverse North American populations. The ascertainment of both cohorts on the basis of influenza or asthma was stringent, so that misclassification of severe influenza infection was highly unlikely. Our study has several limitations. First, we restricted the genomic analysis to the specified gene panel with very high biologic plausibility for the PICFlu cohort and the control CAMP cohort. Second, our sample size in the PICFlu cohort is relatively small, thus limiting statistical power. We cannot rule out the possibility that some individuals in the CAMP cohort were hospitalized with severe influenza infection, but this would likely bias toward the null for common variant analysis. Despite these constraints, we were able to detect an enrichment of a common variant in DDX58 above the Bonferroni-corrected threshold which was associated with differential expression of DDX58 and other downstream IFN-regulated genes. Lung function and atopy-related genes may also underlie severe influenza infection, and use of the pediatric asthma control group may have attenuated these associations as well as associations with IFN-related genes underlying asthma susceptibility. In addition, we did not have a wide range of influenza disease severity in the PICFlu cohort, as the majority of patients had life-threatening disease and all patients were admitted to an ICU or high-acuity unit and we were not able to evaluate the association across a range of disease severity. Previously healthy children infrequently develop life-threatening influenza disease, and we could not identify a sizeable independent validation cohort with this clinical phenotype.
In conclusion, RIG-I is important for antiviral host response to influenza infection, and we show that gene variants in DDX58 may influence this response and susceptibility to severe disease in children. Understanding RIG-I’s physiologic role in humans may lead to more effective vaccines and novel therapeutic techniques targeting this pathway. To better understand how genetics affects the susceptibility to severe pediatric influenza infection, more research in large clinically defined cohorts with diverse ethnicities and ages is required.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the authors.
Notes
Author contributions. A. G. R. designed and oversaw all aspects of the PICFlu study. S. T. W. designed and oversaw the CAMP study. Y. Z. and H. C. S. performed genotyping on PICFlu samples. S. L., Y. Z., and C. L. performed the analyses. M. N. oversaw data management and T.N. oversaw sample management. PICFlu site lead authors collected data and samples. All authors contributed to the relevant sections and approved the final manuscript.
Acknowledgments. We thank Michael Ciancanelli and Jean-Laurent Casanova for helpful discussions.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH) (AI154470 to A. R., P. T., and C. L.; AI084011 to A. R.; Division of Intramural Research to Y. Z. and H. C. S.; and 2U01HG008685 and P01HL132825 to S. T. W.). Molecular data for the TOPMed program were supported by the National Heart, Lung, and Blood Institute (NHLBI), NIH. Genome sequencing for “NHLBI TOPMed: Genetic Epidemiology of Asthma in Costa Rica” (phs000988.v4.p1) and “NHLBI TOPMed: Childhood Asthma Management Program (CAMP)” (phs001726.v1.p1) was performed at the Northwest Genomics Center (HHSN268201600032I, 3R37HL066289-13S1). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering, were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract number HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity quality control, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract number HHSN268201800001I).
References
Author notes
S. L. and Y. Z. contributed equally to this work.
H. C. S. and A. G. R. contributed equally to this work.
The PALISI PICFlu Investigator contributors are listed in the Supplementary Data.
Potential conflicts of interest. A. G. R. reports receiving reagents to her institution from Illumina Inc for work outside of this project.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}