






Abstract
FimH-mediated bacterial invasion and polymorphonuclear neutrophil (PMN) transmigration across human brain microvascular endothelial cells (HBMECs) are required for the pathogenesis of Escherichia coli meningitis. However, the underlying mechanism remains unclear. This study demonstrated that the TnphoA mutant (22A33) and FimH-knockout mutant (ΔFimH) of E coli strain E44, which resulted in inactivation of FimH, were less invasive and less effective in promoting PMN transmigration than their wild-type strain. FimH protein induced PMN transmigration, whereas calmodulin inhibitor significantly blocked this effect. Moreover, immunofluorescence and co-immunoprecipitation analysis indicated that colocalized CD48 and α7 nAChR formed a complex on the surface of HBMECs that is associated with increased cofilin dephosphorylation, which could be remarkably enhanced by FimH+ E44. Our study concluded that FimH-induced E coli K1 invasion and PMN migration across HBMECs may be mediated by the CD48-α7nAChR complex in lipid rafts of HBMEC via Ca2+ signaling and cofilin dephosphorylation.
Neonatal bacterial meningitis (NBM) remains a devastating disease associated with high mortality and morbidity despite advanced antibiotic treatments [1, 2]. Bacterial penetration through the blood-brain barrier (BBB) and polymorphonuclear neutrophil (PMN) recruitment to the central nervous system (CNS) often lead to serious neurological sequelae [3–8]. A better understanding of molecular and cellular mechanisms has important implications for preventing NBM and treating Escherichia coli K1. Escherichia coli K1 is the most common Gram-negative bacterial pathogen that causes sepsis and meningitis in neonates [9, 10]. FimH, the tip of the fimbrial shaft, is responsible for the mannose-sensitive binding ability of E coli type 1 fimbriae [11–13]. It plays an important role in bacterial adhesion and invasion, but its mechanism is not entirely clear [5, 14–16]. In response to bacterial invasion, numerous PMNs migrate through human brain microvascular endothelial cells (HBMECs) to the inflammatory site. Although PMN transmigration across HBMECs is involved in host defense against meningitic bacterial pathogens, it also leads to significant damage to the CNS. IbeA, vimentin (the primary receptor of IbeA), and PSF (the coreceptor) contribute to E coli K1 penetration and PMN transmigration across HBMECs, as both in vivo and in vitro findings show [3].
Kim et al [17] and McArdel et al [18] revealed that FimH-induced E coli K1 adhesion and invasion involve the binding of FimH and its receptor, CD48, on the membrane of HBMECs. Alpha 7 nicotinic acetylcholine receptor (α7 nAChR), an essential regulator of inflammation, plays a detrimental role in various inflammatory processes, including E coli K1 penetration and PMN recruitment in HBMECs [19, 20]. Previous research has confirmed the coexistence of CD48 and α7 nAChR in the lipid raft [18, 21]. It is likely that their coexistence serves as a common signal platform and the 2 might form a protein complex for signal transduction. Furthermore, the CD48 and α7 nAChR complex in the lipid raft of HBMECs might contribute to FimH-mediated biological effects.
The question of how this complex regulates bacterial invasion and PMN migration across HBMECs needs to be addressed. Changes in free intracellular Ca2+ can stimulate cytoskeleton reorganization, a critical step in bacterial invasion [22, 23]. This reorganization may be attributed to the plasticity of actin dynamics, which is associated with cytoskeletal proteins that bind to and are regulated by Ca2+. Cofilin, one of the most important actin depolymerization factors, mediates host-pathogen interactions in response to pathogen stimulation. Dephosphorylation of cofilin (activated state) is important for E coli-induced cytoskeleton rearrangement, which promotes PMN transmigration across the endothelial cell barrier. These phenomena prompt further examination of whether calcium signaling and the dephosphorylation status of cofilin are involved in the FimH-induced E coli invasion of HBMECs and PMN transmigration across HBMECs.
This study first explored the role of FimH in E coli K1 invasion and PMN transmigration across HBMECs. The abilities of 3 bacterial strains (FimH+E coli [E44], FimH-mutant strain [22A33], and FimH-knockout strain [ΔFimH]) to invade HBMECs and induce PMN transmigration across HBMECs were compared. Furthermore, a series of experiments were performed to identify the roles of the CD48/α7nAChR interaction in the lipid raft of HBMECs, calcium signaling, and the phosphorylation status of cofilin in FimH-mediated E coli K1 invasion and PMN transmigration across HBMECs.
MATERIALS AND METHODS
Human Brain Microvascular Endothelial Cell Culture
Human brain microvascular endothelial cells were cultured as described in the previous studies [20, 21, 24]. In brief, HBMECs were routinely cultured in Roswell Park Memorial Institute 1640 medium, and E coli invasion assay was performed in experimental medium (more details are provided in the Supplementary Material).
Bacterial Strains and Media
Escherichia coli RS218 (O18:K1:H7) is isolated from the cerebrospinal fluid of a neonate with E coli meningitis [20, 21]. E44 is a rifampin-resistant mutant derivative of E coli K1 strain RS218. 22A33 is a TnphoA insertion mutant of FimB of E44, which resulted in inactivation of FimH. The TnphoA mutants were made as described previously [25]. ΔFimH is FimH gene knockout strain, and the details for preparing ΔFimH are provided in the Supplementary Material. All strains were grown overnight at 37°C in Luria-Bertani broth supplemented with rifampin (100 μg/mL).
Bacterial Invasion
The HBMECs were cultured as described previously [20, 24]. The confluent monolayers of HBMECs were incubated with 1 × 107 colony-forming units (CFU) of E coli E44 or its mutant 22A33 or ΔFimH for 1.5 hours at 37°C to allow invasion to occur. Results were expressed as percentage relative invasion. For inhibitor treatment, the cells were pretreated with the calmodulin (CaM) antagonist trifluoperazine ([TFP]; 5 μM) for 1 hour before exposure to E44, 22A33, and ΔFimH.
Intracellular Ca2+ Concentration Measurements
The intracellular Ca2+ concentration ([Ca2+]i) of HBMECs was examined as previously described [20]. In brief, after baseline, [Ca2+]i was measured for 120 seconds, E44 or 22A33 (1 × 108 CFU) was added to stimulate the cells, and the fluorescence intensity ratio at 340 and 380 nm excitation wavelengths (F340/F380) was calculated to determine the change in [Ca2+]i. All measurements were performed in at least 3 separate experiments. More details are provided in the Supplementary Material.
Immunofluorescence and Confocal Microscopic Assays
To observe the expression of intercellular adhesion molecule (ICAM)-1, CD48, and α7 nAChR on HBMECs, HBMECs were seeded onto 12-well plates, with sterile round coverslip in each well, and incubated with E44 or 22A33 or ΔFimH (1 × 106 CFU) at 37°C for 2 hours. After immunofluorescent staining, the cells were examined with a confocal microscope. Green and red fluorescent signals (excitation wavelengths at 488 nm and 555 nm, respectively) represented the distribution of CD48 protein and α7 nAChR protein, respectively; a yellow signal indicated the superposition of red and green fluorescence. The average fluorescence intensity was calculated by dividing the total fluorescence intensity with the total fluorescence area of the cells. The antibodies and reagents used are described in the Supplementary Material.
Polymorphonuclear Neutrophil Transmigration Assay In Vitro
The PMN transmigration assay was performed using the transwell cell migration model described by Che et al [3] and Chi et al [20] with slight modifications. In brief, different doses of E coli strains (E44, 22A33, and ΔFimH; 105–107 CFU) or purified FimH protein (0.01–1 µg/mL) were added to the lower chambers for 2 hours. The PMN were then added to the upper chamber of migration model at a concentration of 1 × 106 cells per well at 37°C, 5% CO2 for 3 hours (E44, 22A33, and ΔFimH groups) or 6 hours (purified FimH protein) to migrate over HBMEC monolayers. In the time course experiment, HBMECs were incubated with the E coli strain (E44, 22A33, and ΔFimH; 5 × 106 CFU) or FimH protein (0.5 µg/mL) in the lower chambers for 2 hours, and 1 × 106 PMN were added to the upper chamber and incubated for either 1–4 hours or 2–24 hours. At the end of the incubation, the migrated PMN cells were collected from the lower chamber and counted in a blinded fashion using a hemacytometer. The final results of PMN transmigration were expressed as the percentage of PMN cells across the HBMEC monolayer. For inhibitor treatment, the CaM inhibitor TFP (5 μM) was added to the transwell cell culture chamber 1 hour before E coli stimulation. Each cell group was tested in 3 separate experiments. The method for FimH protein expression and purification is described in the Supplementary Material.
Preparation of Lipid Rafts From Human Brain Microvascular Endothelial Cells
The preparation of lipid rafts was performed according to the method described previously [21]. In brief, HBMECs were seeded and grown in 100-mm dishes; after they grew to a 70%–80% confluence, the cells were incubated in either experimental media (control) or medium supplied with E44 (5 × 107/plate) for 2 hours. After incubation, the preparation of lipid rafts from HBMECs was performed on ice following the procedures described in our previous report [21]. Target proteins in the gradient fractions were detected by WEstern blotting.
Co-Immunoprecipitation Assay and Western Blot
Exogenous and endogenous co-immunoprecipitation (Co-IP) assays were used to detect the interaction between CD48 and α7 nAChR. For the exogenous protein interaction, the pcDNA3.1-α7 nAChR-Flag and pcDNA3.1-CD48-Myc were transfected into 293T cells. PcDNA3.1-Flag and pcDNA3.1-Myc were used as controls. The cells were harvested after 24 hours of transfection for Co-IP. For the endogenous protein interaction, the cells were seeded onto a 10-cm dish and collected for detection when they grew to 80% confluence. The Co-IP assay was performed using a protein G-agarose kit according to the instructions provided by the manufacturer with slight modifications. The detailed procedures for Co-IP and Western blotting are described in the Supplementary Material.
Statistical Analysis
The results are presented as the mean ± standard. Statistical significance was determined using the unpaired Student’s t test or analysis of variance followed by Fisher’s post hoc test for comparisons between 2 groups or multiple comparisons, respectively. SPSS version 20.0 was used for analysis, and P < .05 was considered statistically significant.
RESULTS
Role of Virulence Factor FimH in Escherichia coli K1 Penetration and Polymorphonuclear Neutrophil Transmigration Across Human Brain Microvascular Endothelial Cells
A bacterial invasion assay was performed to investigate the role of FimH with type 1 fimbriae in E coli K1 invasion of HBMECs by comparing the abilities of wild-type E coli strains E44 (FimH+), 22A33 (FimH mutant strain), and FimH-knockout strain (ΔFimH) to invade HBMECs in vitro. As Figure 1 shows, the invasion ability of 22A33 and ΔFimH across HBMECs declined approximately 50% compared with that of the wild-type strain, E44 (P < .001). This significant decline suggested that FimH might play a crucial role in the process of E coli K1 invasion across HBMECs (Figure 1A).
![Effect of FimH on Escherichia coli K1 penetration and polymorphonuclear neutrophil (PMN) transmigration across human brain microvascular endothelial cells (HBMECs). (A) Wild strain E44 (FimH+) had a stronger ability to invade HBMECs compared with FimH mutant strain 22A33 and FimH knockout strain ΔFimH. Results are presented as relative invasion. (B) Dose- and time-dependent induction of PMN transmigration across HBMECs by E44, 22A33, and ΔFimH stimulation. (Left) E44, 22A33, and ΔFimH stimulation at different doses for 3 hours; (Right) E44, 22A33, and ΔFimH (5 × 106 colony-forming units [CFU]) stimulation for 1–4 hours. (C) Dose- and time-dependent induction of PMN transmigration in response to purified FimH protein. (Left) FimH stimulation at different doses for 6 hours; (Right) stimulation with 0.5 µg/mL purified FimH for 0–24 hours. Values are means ± standard deviation of 3 independent experiments conducted in triplicate. *, P < .05, **, P < .01, and ***, P < .001 compared with the nontreated control group. E44, wild-type E coli K1 strain; 22A33, FimH mutant E coli K1 strain; ΔFimH, FimH knockout E coli K1 strain.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/219/3/10.1093_infdis_jiy531/1/m_jiy53101.jpeg?Expires=1750226275&Signature=Ns3OdGgCra84uRxRl4llbyr5n7gNEpK~464cG-ElA4oix7020APJPwZXSPR0a9QmsZZnawbeiOl-K0bNhdgVGJzTF7KkOMP8Q0I11pITrdwzSfd6FNjUiDpHrveiDDUbTZp6v-nlCVi4Ab7VbPZvIY7Hmssbvluxi29J70IoO082F82ManVbXS9mGm6VHqRGo912I2kkDtMzvMxRuF1azWed2~dhNyBbpebKnbhRQjRdWdB7KQBc59dZZk-mReXZtQjlF~z2QfNZQpx5DJusfdIFhYV~ygLO9cnWrY5wocwDNfA3-7HxfLNwEhh3aIWUqkcDWQsl8nieZ-j0-UzCwA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of FimH on Escherichia coli K1 penetration and polymorphonuclear neutrophil (PMN) transmigration across human brain microvascular endothelial cells (HBMECs). (A) Wild strain E44 (FimH+) had a stronger ability to invade HBMECs compared with FimH mutant strain 22A33 and FimH knockout strain ΔFimH. Results are presented as relative invasion. (B) Dose- and time-dependent induction of PMN transmigration across HBMECs by E44, 22A33, and ΔFimH stimulation. (Left) E44, 22A33, and ΔFimH stimulation at different doses for 3 hours; (Right) E44, 22A33, and ΔFimH (5 × 106 colony-forming units [CFU]) stimulation for 1–4 hours. (C) Dose- and time-dependent induction of PMN transmigration in response to purified FimH protein. (Left) FimH stimulation at different doses for 6 hours; (Right) stimulation with 0.5 µg/mL purified FimH for 0–24 hours. Values are means ± standard deviation of 3 independent experiments conducted in triplicate. *, P < .05, **, P < .01, and ***, P < .001 compared with the nontreated control group. E44, wild-type E coli K1 strain; 22A33, FimH mutant E coli K1 strain; ΔFimH, FimH knockout E coli K1 strain.
The effects of E44, 22A33, and ΔFimH on PMN migration across HBMECs were compared by incubating PMNs with E coli at different concentrations for 3 hours. Although E44, 22A33, and ΔFimH promoted PMN migration across HBMECs in a dose-dependent manner, E44 showed a significantly stronger effect than 22A33 and ΔFimH (Figure 1B, left). In addition, the migration of PMN increased over time when they were incubated with E44 or 22A33 or ΔFimH (5 × 106 CFU) (Figure 1B, right). The results indicated that FimH is involved in the bacteria-induced PMN migration through HBMECs.
To further examine whether FimH contributes to the acceleration of PMN transmigration, the purified recombinant FimH protein, which was treated with polymyxin B resin to remove lipopolysaccharide (LPS) and confirmed for LPS removal (the level of residual LPS less than 1 endotoxin unit/mg protein) according to the commercial kit manufacturer recommendations, was used in the in vitro PMN migration experiment. The result showed that the number of migrated PMNs remarkably increased with the increase in FimH concentration (0 to 1 μg/mL). In addition, the increase of PMN transmigration was time-dependent between posttreatment 2 and 24 hours compared with the control group without FimH treatment (Figure 1C). These results suggested that virulence factor FimH alone can enhance PMN transmigration, thus contributing to the E coli-induced PMN behavior.
Involvement of Ca2+ Signaling in FimH-Mediated Escherichia coli K1 Penetration and Polymorphonuclear Neutrophil Transmigration Across Human Brain Microvascular Endothelial Cells
Ca2+, as an important second messenger molecule in the cell, plays an important role in cytoskeleton rearrangement, thereby regulating the interaction between bacteria and host cells. To examine whether FimH stimulation can lead to the change in the [Ca2+]i of HBMECs, a [Ca2+]i assay was performed in the presence of FimH+ E44 or FimH− 22A33 stimulation. The treatment with E44 elicited an increase in the [Ca2+]i of HBMECs, whereas [Ca2+]i was not significant affected by stimulation with 22A33 (Figure 2A).
![Involvement of Ca2+ signaling in FimH-mediated Escherichia coli K1 penetration and polymorphonuclear neutrophil (PMN) transmigration across human brain microvascular endothelial cells (HBMECs). (A) Effects of 22A33 (top panel) and E44 (bottom panel) on the intracellular Ca2+ concentration ([Ca2+]i) of HBMECs. The monolayer was monitored for intracellular calcium flux for 10 minutes with 4-second intervals under an automated fluorescent microscope. The monolayer cells were stimulated with E44 (108 colony-forming units) at the 120-second time point. The intensities of fluorescence at 340 nm and 380 nm were measured, and their ratio (340 nm/380 nm) was calculated for each time interval and plotted into a continuous line. (B) Inhibition of the calmodulin antagonist trifluoperazine (TFP) on bacterial invasion induced by E44 and 22A33. (C) Inhibition of the calmodulin antagonist TFP on PMN migration across HBMECs induced by E44 and 22A33. The HBMECs were preincubated with or without TFP (0–25 μM) for 1 hour before E44 or 22A33 stimulation. Data shown are means ± standard deviation of 3 independent experiments conducted in triplicate; *, P < .05, **, P < .01, and ***, P < .001 compared with the nontreated control group. E44, wild-type E coli K1 strain; 22A33, FimH mutant E coli K1 strain; TFP, calmodulin inhibitor trifluoperazine.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/219/3/10.1093_infdis_jiy531/1/m_jiy53102.jpeg?Expires=1750226275&Signature=1eYT~ANuma7OD0sKT5QEwTlZcvIgs7QWZSJrztbyQ5-5tVeOuPdyrJFBCkYI5C4hycFHMDub9IMXTvY5YlunopqECIDV0iPGqUTI9Z0r5XTWmlY98QC5r0~7H4mo2Zq3Wiham4KdobC44xH4HNQjq2~2zDwciQpTJhSyiBlLWtiYzBeRcPREJbVJbBun0dX04dt1HOXqfVgPWhxA3zkuSSwmkZUToJDqybBWX7s~bv47afD2e~IIp15l4Ub0ufaNg6kbpxbIS--GN~waJKwCiXY5Yl9Zy1PxNGhNXOKoRfXzwxkFBh83llRs97FIKhd0aCFR~ONPaprYTgL3moYU8A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Involvement of Ca2+ signaling in FimH-mediated Escherichia coli K1 penetration and polymorphonuclear neutrophil (PMN) transmigration across human brain microvascular endothelial cells (HBMECs). (A) Effects of 22A33 (top panel) and E44 (bottom panel) on the intracellular Ca2+ concentration ([Ca2+]i) of HBMECs. The monolayer was monitored for intracellular calcium flux for 10 minutes with 4-second intervals under an automated fluorescent microscope. The monolayer cells were stimulated with E44 (108 colony-forming units) at the 120-second time point. The intensities of fluorescence at 340 nm and 380 nm were measured, and their ratio (340 nm/380 nm) was calculated for each time interval and plotted into a continuous line. (B) Inhibition of the calmodulin antagonist trifluoperazine (TFP) on bacterial invasion induced by E44 and 22A33. (C) Inhibition of the calmodulin antagonist TFP on PMN migration across HBMECs induced by E44 and 22A33. The HBMECs were preincubated with or without TFP (0–25 μM) for 1 hour before E44 or 22A33 stimulation. Data shown are means ± standard deviation of 3 independent experiments conducted in triplicate; *, P < .05, **, P < .01, and ***, P < .001 compared with the nontreated control group. E44, wild-type E coli K1 strain; 22A33, FimH mutant E coli K1 strain; TFP, calmodulin inhibitor trifluoperazine.
Next, the role of the Ca2+ signal in E coli invasion of HBMECs was examined by pretreating HBMECs with a CaM inhibitor, TFP, for 60 minutes before the bacterial invasion assay. As shown in Figure 2B, TFP significantly reduced the invasion of FimH+ E44 and FimH− 22A33 to HBMECs by 48% and 13%, respectively, showing a significant difference between the 2 strains. The results suggested that Ca2+ signaling in HBMECs is required for E coli K1 invasion, especially E44 invasion.
Likewise, TFP also inhibited purified FimH-induced PMN transmigration as well as E coli-induced PMN transmigration across HBMECs (Figure 2C). After TFP treatment, the number of transmigrated PMNs declined in a dose-dependent manner. The change was more significant for the cells treated with E coli strain E44 compared with those treated with 22A33 (Figure 2C), indicating that calcium signaling mediates FimH-enhanced PMN transmigration.
Expression of Intercellular Adhesion Molecule-1 in Human Brain Microvascular Endothelial Cells Induced by FimH+Escherichia coli Strain E44 and FimH−E coli Strain ΔFimH
The endothelial adhesion molecule, ICAM-1, expressed on cell membrane, is the main receptor responsible for leukocyte adhesion on the vascular endothelium. The immunofluorescence study showed that E44 and ΔFimH infection at 37°C for 2 hours upregulated the surface expression of ICAM-1 on HBMECs, with E44 having a significantly stronger effect (Figure 3A and B). This finding was confirmed by immunoblotting analysis (Figure 3C and D). These results indicated that FimH can significantly upregulate the surface expression of ICAM-1 on HBMECs.
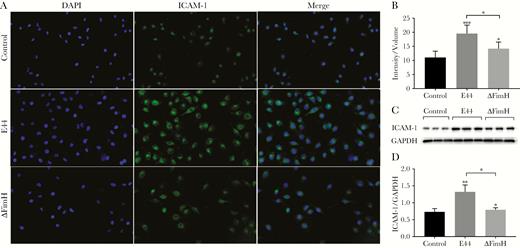
Surface expression of intercellular adhesion molecule (ICAM)-1 induced by E44 and ΔFimH. Immunofluorescence microscopy (×200; A and B) and Western blotting assay (C and D) revealed significantly increased ICAM-1 expression in human brain microvascular endothelial cells after infection with E44 or ΔFimH at 37°C for 2 hours; E44 treatment produced a more prominent increase. *, P < .05, **, P < .01 and ***, P < .001. DAPI, 4’,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; E44, wild-type Escherichia coli K1 strain; ΔFimH, FimH knockout E coli K1 strain.
Colocalization and Interaction Between CD48 and α7 Nicotinic Acetylcholine Receptor on the Lipid Rafts of Human Brain Microvascular Endothelial Cells
As shown in our previous study, a redistribution of α7 nAChR into lipid rafts was induced by either nicotine or E44 [21], α7 nAChR was mainly distributed in fractions 6–9 of the lipid raft in the control group, but it was partly redistributed to fraction 2 after E44 treatment. In this study, we found that nicotine and E44 can induce a redistribution of CD48 into the lipid raft. Lipid raft fractionation analysis demonstrated that CD48 on the HBMEC membrane was distributed in fractions 5–9 of the lipid raft in the control group but partly redistributed to fractions 1–4 of the lipid raft, especially fraction 2, in response to E44 stimulation. Both α7 nAChR and CD48 were present in fraction 2 after E44 stimulation, suggesting a possible CD48 and α7 nAChR interaction on the lipid raft (Figure 4 and our previous report [21]). We deduced that CD48 may interact with α7 nAChR to form a protein complex that serves as a signaling platform to mediate E coli invasion and PMN adhesion transmigration.
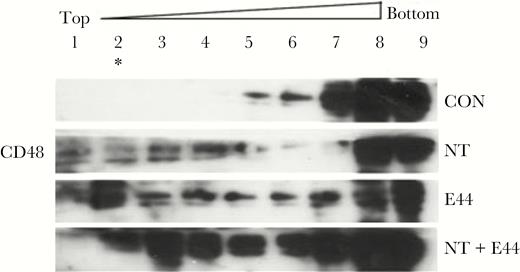
Fractionation analysis of the redistribution of CD48 in lipid rafts. Redistribution of CD48 in lipid rafts induced by nicotine (NT) and E44. Nine fractions were prepared from lysates of human brain microvascular endothelial cells treated with or without NT and E44. Compared with the control group, CD48 was partly redistributed to fractions 1–4, especially fraction 2 (*) of the lipid rafts after E44 treatment. CON, control without treatment; E44, wild-type Escherichia coli K1 strain.
Next, the colocalization of CD48 and α7 nAChR proteins on the membrane of E coli-treated HBMECs and its possible relationship with FimH adhesin were investigated by confocal microscopy using FimH+ and FimH−E coli. The findings showed that E44 significantly increased the surface expression and recruitment of CD48 and α7 nAChR, which were largely colocalized in the HBMECs when compared with the 22A33 strain (Figure 5). Furthermore, the result was verified by using the purified FimH protein. Compared with the control group and bovine serum albumin (BSA) group, FimH increased the surface expression and recruitment of CD48 and α7 nAChR on the HBMECs (Supplementary Figure S2). Our findings suggested that the colocalization and interaction of CD48 and α7 nAChR might be associated with FimH-induced HBMEC responses.
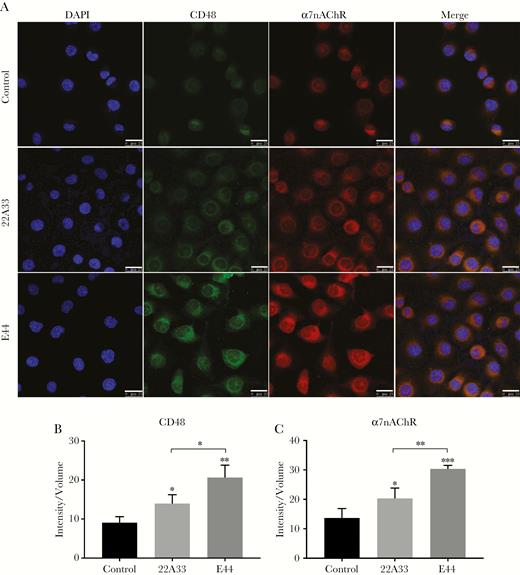
Surface expression and colocalization of CD48 and α7 nicotinic acetylcholine receptor (nAChR) on human brain microvascular endothelial cells (HBMECs) induced by E44 and 22A33 stimulation. (A) Compared with the untreated control group, both CD48 and α7 nAChR membrane expression and colocalization on the membrane of HBMECs significantly increased in the groups treated with Escherichia coli strains, particularly E44. Scale bar, 25 μM. (B and C) The average fluorescence intensities of CD48 and α7 nAChR remarkably increased in the groups treated with E coli strains, particularly E44. *, P < .05, **, P < .01 and ***, P < .001. CD48, FimH receptor; DAPI, 4’,6-diamidino-2-phenylindole; E44, wild-type E coli K1 strain; 22A33, FimH mutant E coli strain.
Furthermore, a Co-IP assay was used to verify the interaction between CD48 and α7 nAChR under both exogenous and endogenous conditions. For exogenous protein interactions, the pcDNA3.1-α7 nAChR-Flag, pcDNA3.1-CD48-Myc, and empty vector (pcDNA3.1-Flag and pcDNA3.1-Myc) were transfected into 293T cells in a 6-well plate. The cells were harvested after 24 hours of incubation and then used for protein extraction for Co-IP assay. The results showed that Flag-α7 nAChR protein could be pulled down by the protein A/G-coupled agarose beads bound with the anti-Myc antibodies, whereas Myc-CD48 protein could be pulled down by the protein A/G-coupled agarose beads bound with the anti-Flag antibodies (Figure 6A). In the endogenous IP experiment using IgG with the same origin as the control to remove nonspecific binding, the antibody against CD48 precipitated the α7 nAChR protein, whereas the antibody against α7 nAChR precipitated the CD48 protein (Figure 6B). The results proved that the CD48 and α7 nAChR proteins can interact under both exogenous and endogenous conditions.
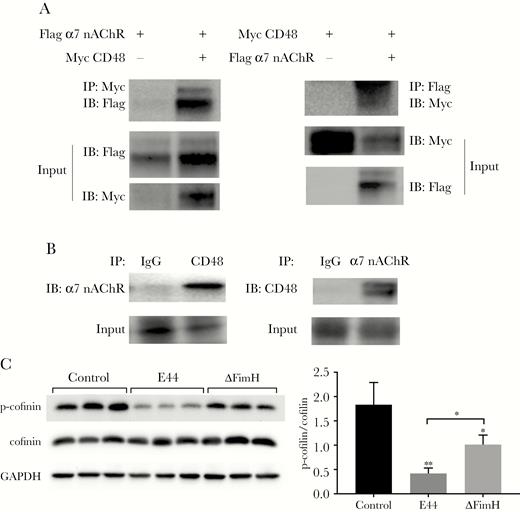
Co-immunoprecipitation (Co-IP) to verify the interaction of CD48 and α7 nicotinic acetylcholine receptor (nAChR) and cofilin dephosphorylation in E44- and ΔFimH-stimulated human brain microvascular endothelial cells (HBMECs). (A) Exogenous condition Co-IP. The 293T cells were divided into 4 groups: transfection with pcDNA3.1-α7 nAChR-Flag and pcDNA3.1-Myc (Flag-α7 nAChR+, Myc-CD48−); transfection with pcDNA3.1-α7 nAChR-Flag and pcDNA3.1-CD48-Myc (Flag-α7 nAChR+, Myc-CD48+); transfection with pcDNA3.1-CD48-Myc and pcDNA3.1-Flag (Myc-CD48+, Flag-α7nAChR−); and transfection with pcDNA3.1-CD48-Myc and pcDNA3.1-α7nAChR-Flag (Myc-CD48+, Flag-α7nAChR+). Cells harvested after 24 hours of transfection were lysed in IP lysis buffer and used for the Co-IP experiments. (B) Endogenous condition Co-IP. The HBMECs were seeded into four 10-cm dishes. After they grew to 80% confluence, the cells were harvested and lysed in IP lysis buffer for the Co-IP experiments. (C) Western blotting assay of phosphorylated cofilin and total cofilin in HBMECs infected with E44 or ΔFimH for 2 hours (left). The result demonstrated a significant decline in the phosphorylated cofilin level and no change in the total cofilin level. The phosphocofilin antibody specifically recognizes the inactivated form of cofilin (p-cofilin), and the cofilin antibody was used to detect total cofilin. (Right) Quantitative analysis of the ratio of p-cofilin/cofilin). Data shown are the means ± standard deviation of 3 independent experiments conducted in triplicate: *, P < .05 and **, P < .01; Flag α7 nAChR+, transfection with pcDNA3.1-α7 nAChR-Flag; Flag α7 nAChR−, transfection with pcDNA3.1-Flag; Myc CD48+, transfection with pcDNA3.1-CD48-Myc; Myc CD48−, transfection with pcDNA3.1-Myc. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IB, immunoblotting, Western blotting to display target protein; IgG, immunoglobulin G; Input, lysis solution was used before IP for IB to exclude the false-positive interference.
As mentioned above, the FimH of E coli K1 elicited an increase in [Ca2+]i in HBMECs (Figure 2A), which can stimulate cytoskeleton rearrangement. Hence, the effect of FimH on the phosphorylation of cofilin, an important small actin-depolymerizing protein associated with cytoskeleton rearrangement, was investigated using Western blotting. The change in cofilin phosphorylation was presented as the ratio of phosphocofilin to total-cofilin. The HBMECs treated with E44 or ΔFimH showed remarkably lower levels of phosphorylated cofilin (P < .01 and P < .05, respectively) (Figure 6C), whereas the total cofilin protein level in the cells remained unchanged. In addition, E44 stimulation decreased the level of phosphorylated cofilin more significantly than ΔFimH stimulation (P < .05) (Figure 6C). These data suggest that the virulence factor FimH of E coli K1 can induce cofilin dephosphorylation and thus mediate the E coli K1 invasion of HBMECs and PMN transmigration across HBMECs by affecting the actin cytoskeleton rearrangement.
DISCUSSION
Escherichia coli K1 is the most common cause of bacterial meningitis, and its fimbrial component FimH adhesin is considered a major bacterial virulence factor. In this study, we confirmed the role of FimH in regulating E coli K1 invasion and PMN behavior using FimH mutant E coli strain 22A33, FimH knockout strain ΔFimH, and purified FimH protein. Furthermore, the underlying mechanisms of FimH-induced responses were elucidated.
The development of NBM is a complex process characterized by pathogen penetration and PMN transmigration across the BBB [26–28]. FimH plays an important role in bacterial adhesion and invasion of host cells. Currently, the mechanisms responsible for FimH adhesin-modulated E coli K1 invasion of the BBB are incompletely understood, and whether FimH adhesin contributes to the process of PMN transmigration across the BBB remains to be addressed.
Khan et al [29] reported that CD48 is a putative HBMEC receptor for FimH, and the interaction of FimH with CD48 induces E coli K1 invasion of the host cell. CD48 is anchored to the cell membrane by a GPI linkage, which allows CD48 to associate with cholesterol-rich lipid rafts and relevant signaling molecules [30, 31]. We explored whether CD48 can bind with other proteins to form a protein complex in the lipid raft and serve as a signaling platform.
α7 nicotinic acetylcholine receptor is a key inflammatory regulatory factor in sterile inflammation, and it plays an important role in the host immune response to microbial infection [20, 32, 33]. In a previous study, we found that α7 nAChR recruitment to caveolin-1-enriched lipid rafts is required for nicotine-enhanced E coli K1 invasion of HBMECs [21]. α7 nicotinic acetylcholine receptor can modulate pathogen invasion, PMN recruitment, calcium signaling, and neuronal inflammation in the host defense against meningitic infection [19, 20]. In the present study, the data from lipid raft fractionation analysis and confocal microscopy and IP assay consistently proved the colocalization and interaction of α7 nAChR and CD48 on the lipid rafts, suggesting that they may form a protein complex to synergistically regulate various signal transduction pathways. Moreover, when the wild-type E coli strain E44, 22A33, and ΔFimH were used to treat HBMECs, the results revealed that the colocalization and interaction of the 2 proteins might be an important step in mediating FimH-induced HBMEC behaviors in response to E coli infection.
Calcium signaling is involved in a variety of processes in host cells after infection with bacterial pathogens, including the expression of proinflammatory mediators and cytoskeletal reorganization [22, 23]. In this study, HBMECs treated with either E44 or 22A33 in vitro displayed increases in the intracellular calcium flow, which might contribute to pathogen penetration and PMN transmigration across the BBB. Compared with the wild-type E coli E44, 22A33 triggered a smaller change in the intracellular calcium level, implying an involvement of calcium signaling in the FimH-induced responses of HBMECs. This was verified by using the CaM antagonist, TFP. Trifluoperazine significantly reduced the bacterial invasion of HBMECs and PMN transmigration across HBMECs after E coli treatment, particularly treatment with E44. Similar results were obtained when purified FimH protein was used to treat HBMECs. Taken together, these findings suggest that the Ca2+/CaM signal pathways are involved in FimH-induced responses, including E coli K1 invasion and PMN transmigration.
Cytoskeletal rearrangements are required for bacterial entry into host cells. Actin cytoskeletal reorganization is essential for E coli K1 invasion of HBMECs, which subsequently causes infection. Cofilin, a key player in actin filament dynamics, is inactivated by phosphorylation and reactivated by dephosphorylation to mediate host-pathogen interactions. This study’s findings suggest that FimH-induced enhancement of cofilin dephosphorylation in HBMECs might further facilitate the entry of E coli into cells.
Studies have shown that the increased expression of ICAM-1 in brain microvascular endothelial cells is associated with lymphocyte adhesion and transmigration across the BBB, and an elevated ICAM-1 level is thought to reflect the destruction of the BBB [34]. This study confirmed that ICAM-1 expression on the HBMEC membrane can be elevated by bacterial infection, and this elevation can be mostly attributed to the effect of FimH, as evidenced by the fact that the wild-type FimH+ strain had a much stronger effect than the FimH− strain. Therefore, FimH may enhance the ability of PMN to pass through HBMECs by up regulating the surface expression of ICAM-1 in HBMECs.
CONCLUSIONS
In conclusion, FimH increases the formation of the CD48 and α7 nAChR complex in HBMECs after E coli infection, triggering a series of downstream responses. The increases in the intracellular Ca2+ concentration and cofilin dephosphorylation, which may promote host cell actin cytoskeletal rearrangements, enhance the E coli K1 invasion of HBMECs. In addition, the surface expression of ICAM-1 in HBMECs increases after bacterial infection, prompting PMN migration across HBMECs.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Acknowledgments. We thank Yanxiao Ji, Lingping Zhao, and Dong Xia (Wuhan University, China) for technical assistance. We also greatly appreciate the help of Dr. Hui Nie (Durham, North Carolina) for the preparation of this manuscript.
Finanical support. This work was funded by the National Natural Science Foundation of China (Grant Nos. 81671534, 81370740, and 81560262); the National Institutes of Health (Grant No. R21NS083967); the Hubei Provincial Population and Family Planning Commission (Grant No. WJ2015MA011); and the Natural Science Foundation of Hubei Provincial Department of Education (Grant No. Q20152105).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.
References
Author notes
R. L. and C. W. contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}