






Abstract
Integrase inhibitors (INIs) are a key component of antiretroviral therapy for human immunodeficiency virus-1 (HIV-1) and HIV-2 infection. Although INI resistance pathways are well-defined for HIV-1, mutations that emerge in HIV-2 in response to INIs are incompletely characterized.
We performed systematic searches of GenBank and HIV-2 drug resistance literature to identify treatment-associated mutations for phenotypic evaluation. We then constructed a library of 95 mutants of HIV-2ROD9 that contained single or multiple amino acid changes in the integrase protein. Each variant was tested for susceptibility to raltegravir and dolutegravir using a single-cycle indicator cell assay.
We observed extensive cross-resistance between raltegravir and dolutegravir in HIV-2ROD9. HIV-2–specific integrase mutations Q91R, E92A, A153G, and H157Q/S, which have not been previously characterized, significantly increased the half maximum effective concentration (EC50) for raltegravir when introduced into 1 or more mutational backgrounds; mutations E92A/Q, T97A, and G140A/S conferred similar enhancements of dolutegravir resistance. HIV-2ROD9 variants encoding G118R alone, or insertions of residues SREGK or SREGR at position 231, were resistant to both INIs.
Our analysis demonstrates the contributions of novel INI-associated mutations to raltegravir and dolutegravir resistance in HIV-2. These findings should help to improve algorithms for genotypic drug resistance testing in HIV-2–infected individuals.
Public health campaigns to control human immunodeficiency virus (HIV) infection at the population level implicitly include HIV type 2 (HIV-2), which is endemic in West Africa and, like HIV-1, is a causative agent of AIDS [1–3]. Although HIV-2–infected patients can benefit from antiretroviral therapy (ART), the spectrum of clinically available drugs for HIV-2 treatment is limited by the intrinsic resistance of the virus to nonnucleoside reverse transcriptase inhibitors and the reduced sensitivity of HIV-2 (relative to HIV-1) to several protease inhibitors [4]. Historically, these differences in innate drug susceptibility have complicated patient care and programmatic ART deployment in areas where HIV-1 and HIV-2 cocirculate [2].
In December 2018, the World Health Organization issued new guidance regarding first- and second-line treatment of HIV infection [5]. According to these guidelines, which were reaffirmed in July 2021 [6], the preferred first-line regimen for all HIV-infected adults and adolescents initiating ART is composed of the nucleoside reverse transcriptase inhibitors tenofovir disoproxil fumarate and lamivudine (or emtricitabine) plus the integrase inhibitor (INI) dolutegravir. Dolutegravir is also recommended for second-line treatment in adult and adolescent patients for whom nondolutegravir-based regimens are failing. Regimens containing the first-generation INI raltegravir are recommended as first-line ART in neonates, and as an alternative first-line option in children when lopinavir/ritonavir solid formulations are unavailable [6]. Together, these recommendations solidify the role of INIs as crucial components of first- and second-line ART for all people living with HIV, including those infected with HIV-2.
The use of INI-based ART in HIV-2–infected individuals is supported by existing evidence. All of the INIs developed against HIV-1 are potent inhibitors of HIV-2 replication in culture [7–12], and single-arm clinical trials conducted in France [13] and Senegal [14] indicate that raltegravir- and elvitegravir-containing regimens, respectively, are effective in ART-naive, HIV-2–infected patients. Dolutegravir-based ART is also associated with favorable immunologic and virologic outcomes, as demonstrated in a retrospective study of HIV-2 patients in India (n = 62) [15]. However, the clinical benefits of INIs can be undermined by mutations in HIV-2 integrase that confer INI resistance [16–20]. As in HIV-1 infection, emergent drug resistance in HIV-2 can lead to treatment failure and the need to intensify therapy or switch to an alternative regimen [2, 4].
At present, most of the data regarding the genetic basis of INI resistance in HIV-2 are derived from studies of raltegravir-treated patients [8, 10, 13, 14, 16–29]. Two important features are apparent. First, HIV-2 shows some commonality with HIV-1 with regard to INI resistance pathways, as indicated by the frequent appearance of amino acid replacements at positions 92, 97, 140, 143, 148, and 155 of the HIV-2 integrase protein [8, 10, 13, 14, 16–29]. Second, HIV-2 integrase sequences from INI-treated patients often encode mutations that are not recognized as resistance-conferring changes in HIV-1; these include K46R, I84V, Q91R, A119T, V141I, A153G, A153S, H156R, H157Q, H157R, N160K, and insertions of up to 5 amino acids at position 231 [10, 17, 19, 20, 22, 23, 28]. The role of many INI-associated mutations in HIV-2 is uncertain, as only a small number of changes have been studied by site-directed mutagenesis (SDM) and culture-based or enzymatic drug susceptibility testing [11, 12, 25, 30].
To determine the phenotypic impact of mutations in HIV-2 that are associated with INI-based treatment, we constructed a library of 95 full-length, site-directed mutants of HIV-2ROD9, each containing up to 6 amino acid replacements in the integrase protein. Our choice of mutations was influenced in part by a recent statistical analysis of HIV-2 patient-derived integrase sequences [29] that identified 14 amino acid replacements that are significantly associated with INI-based ART. We also independently compiled and reviewed all publicly available sequence information from INI-treated, HIV-2–infected individuals to determine the specific genotypes (ie, combinations of mutations in integrase) that should be tested experimentally. After constructing the mutant library, we determined the susceptibility of each HIV-2ROD9 integrase variant to raltegravir and dolutegravir using a single-cycle indicator cell assay. Assays with INI-resistant, site-directed mutants of HIV-1NL4-3 were included for comparison.
METHODS
Cells, Plasmids, and Inhibitors
MAGIC-5A indicator cells and plasmid pROD9, a full-length molecular clone of HIV-2ROD (group A) were kindly provided by Michael Emerman, Fred Hutchinson Cancer Research Center, Seattle, WA. 293tsA1609neo clone 17 (293T/17) cells were obtained from the American Type Culture Collection (Manassas, VA). Full-length HIV-1 clone pNL4-3 (group M, subtype B) was obtained from Bruce Chesebro, NIH Rocky Mountain Laboratories, Hamilton, MT. Plasmids pROD9 and pNL4-3 and their derivatives were maintained in Escherichia coli strains XL10-Gold (Agilent Technologies) or TOP-10 (Invitrogen Life Technologies). Bacterial cells containing pROD9 and pNL4-3 were grown at 30°C to minimize the outgrowth of plasmids with deletions in the HIV insert [31]. Dolutegravir was purchased from Selleck Chemicals. Raltegravir monopotassium salt was obtained from the National Institutes of Health HIV Reagent Program (Manassas, VA) and from Cayman Chemical Co; preparations of raltegravir from these 2 suppliers showed comparable antiviral activity against HIV-1NL4-3 and HIV-2ROD9 (Supplementary Figure 1).
SDM and Virus Production
SDM was performed using full-length pROD9 or pNL4-3 as the plasmid template and synthetic oligonucleotides that were purchased from Integrated DNA Technologies (Supplementary Table 1). All mutants were constructed using the QuikChange II XL Site-directed Mutagenesis Kit (Agilent). For each mutant clone, the sequence of the integrase-encoding region was confirmed by automated Sanger DNA sequencing (Genewiz). Stocks of wild-type and mutant viruses were generated by transient transfection of 293T/17 cells as previously described [32, 33]; see Supplementary Methods for additional details.
Drug Susceptibility Assays
Single-cycle assays were performed using MAGIC-5A cells as previously described [33], except that the viral titers were adjusted to 15 000 focus-forming units (FFU)/mL in complete Dulbecco’s Modified Eagle’s Medium prior to inoculation. For variants with titers <15 000 FFU/mL, undiluted virus stocks were used to inoculate the assay plates. After approximately 40 hours of infection, the MAGIC-5A monolayers were lysed using a nonionic detergent solution, and β-galactosidase activity was measured by treating the lysates with chlorophenol red-β-d-galactopyranoside (BioShop Canada) [33]; see Supplementary Methods for details. Substrate conversion was quantified by measuring absorbance at 570 nm using a Victor3 plate reader (PerkinElmer).
Data and Statistical Analyses
Half maximum effective concentration (EC50) values were calculated from semilog plots of the dose-response data using the sigmoidal regression function of Prism version 6.0h (GraphPad Software). All HIV-1NL4-3 and HIV-2ROD9 mutants were subjected to at least 3 independent determinations of raltegravir and dolutegravir susceptibility. Stocks produced from the parental wild-type clones were tested in each run of the single-cycle assay to monitor assay performance and ensure that drug potency was maintained throughout the course of the study. Log10-transformed EC50 values were subjected to an analysis of variance (ANOVA) with Tukey multiple comparisons test; separate ANOVAs were performed for the raltegravir and dolutegravir datasets. For wild-type HIV-2ROD9, the EC50 values for raltegravir (n = 59 determinations) and dolutegravir (n = 49 determinations) passed the D’Agostino-Pearson omnibus normality test (α = .05). All statistical analyses were performed using Prism version 6.0h.
RESULTS
To identify candidate genotypes for SDM and drug susceptibility testing, we compiled and reviewed all published reports that contained integrase sequence information from INI-treated, HIV-2–infected patients. Genotypes from 91 INI-treated individuals were extracted from the final collection of 19 research articles (Supplementary Table 2). Most of the HIV-2 patients in these studies were treated with a regimen that contained raltegravir (n = 80); 2 patients received dolutegravir-based ART without prior treatment with an INI, and 6 initially received raltegravir, but were switched to a dolutegravir-containing regimen prior to sample collection. The remaining patients received either elvitegravir (n = 1) or an unspecified INI (n = 2).
We also compiled a list of all integrase sequences from INI-treated, HIV-2–infected patients that were available in the National Center for Biotechnology Information GenBank database (see Supplementary Methods for search terms and inclusion criteria). This list partially overlaps the list of published genotypes (Supplementary Table 2), but also includes unpublished sequences, as well as changes at amino acid sites that were not assessed in the published studies. The resultant collection contained 80 group A, 24 group B, 1 CRF01_AB, and 2 group H sequences, including 6 group A sequences from samples that were obtained prior to the initiation of INI-based ART (Supplementary Table 3). The GenBank sequences were binned according to HIV-2 group and aligned to sequences from corresponding reference isolates of HIV-2 (specifically, HIV-2ROD9 for group A, HIV-2EHO for group B, and HIV-296FR12034 for group H). We then catalogued the amino acid differences present in each patient sequence (relative to the appropriate reference isolate) at each of 41 codon sites; 17 of these corresponded to the locations of known, group-specific polymorphisms. Additional details regarding the construction and analysis of the alignments are provided in the Supplementary Methods.
Based on these sources (Supplementary Tables 2 and 3), the aforementioned statistical analysis of HIV-2 patient sequences [29], and the known patterns of mutations that emerge in HIV-1 during INI-based treatment [34], we selected 31 amino acid changes in HIV-2 integrase for SDM and phenotypic evaluation (Table 1). We divided the mutations of interest into 3 categories. Substitutions Y143C/G/H/R (nomenclature, replacement of tyrosine 143 with cysteine, glycine, histidine, or arginine), Q148H/K/R, and N155H were classified as primary INI resistance changes. Nineteen additional substitutions were considered to be potential secondary INI resistance changes, as suggested by their frequent coappearance with replacements at positions 143, 148, or 155; 15 of these are unique to HIV-2 (K46R, I84V, Q91R, A119T, V141I, A153G/S, H156R, H157Q/R/S/Y, and N160K) or are infrequently observed in sequences from INI-treated HIV-1 patients (E92A and Y143G) [34].
Treatment-Associated Mutations in HIV-2 Integrase Examined in This Study
| Categorya | Mutation | HIV-2–Specific or Rarely Observed in HIV-1?b |
|---|---|---|
| Primary | Y143C/G/H/R | Yes (Y143G) |
| Q148H/K/R | No | |
| N155H | No | |
| Secondary | K46R | Yes |
| I84Vc | Yes | |
| Q91R | Yes | |
| E92A/G/Q | Yes (E92A) | |
| T97A | No | |
| A119Tc | Yesd | |
| G140A/S | No | |
| V141I | Yes | |
| A153G/Sc | Yese | |
| H156R | Yes | |
| H157Q/R/S/Y | Yesf | |
| N160K | Yes | |
| Alternative | G118R | No |
| 231ins SREGK/SREGRg | Yes | |
| R263K | No |
| Categorya | Mutation | HIV-2–Specific or Rarely Observed in HIV-1?b |
|---|---|---|
| Primary | Y143C/G/H/R | Yes (Y143G) |
| Q148H/K/R | No | |
| N155H | No | |
| Secondary | K46R | Yes |
| I84Vc | Yes | |
| Q91R | Yes | |
| E92A/G/Q | Yes (E92A) | |
| T97A | No | |
| A119Tc | Yesd | |
| G140A/S | No | |
| V141I | Yes | |
| A153G/Sc | Yese | |
| H156R | Yes | |
| H157Q/R/S/Y | Yesf | |
| N160K | Yes | |
| Alternative | G118R | No |
| 231ins SREGK/SREGRg | Yes | |
| R263K | No |
Mutations were assigned to categories as described in the text.
Based on the Stanford University HIV Drug Resistance Database—INSTI Resistance Notes [34].
Mutations I84V, A119T, and A153S are naturally occurring polymorphisms found in group A HIV-2 [29]. A153S is also polymorphic in group B HIV-2 [7].
A mutation at the corresponding site in HIV-1 integrase (S119R) is involved in integrase inhibitor resistance in HIV-1 [36].
The corresponding residue in HIV-1 integrase is S153. S153Y/F is a rare, nonpolymorphic mutation that is selected by dolutegravir in HIV-1 in vitro [35].
E157Q is a known polymorphism in HIV-1 integrase that confers modest (<2.0-fold) changes in the half maximum effective concentration (EC50) values for raltegravir and dolutegravir in HIV-1, as assessed by site-directed mutagenesis [37, 38].
Insertions of five amino acids at residue 231 of HIV-2 integrase. In the analysis performed by Tzou et al., insertions at position 231 were considered to be a single mutant form [29].
Treatment-Associated Mutations in HIV-2 Integrase Examined in This Study
| Categorya | Mutation | HIV-2–Specific or Rarely Observed in HIV-1?b |
|---|---|---|
| Primary | Y143C/G/H/R | Yes (Y143G) |
| Q148H/K/R | No | |
| N155H | No | |
| Secondary | K46R | Yes |
| I84Vc | Yes | |
| Q91R | Yes | |
| E92A/G/Q | Yes (E92A) | |
| T97A | No | |
| A119Tc | Yesd | |
| G140A/S | No | |
| V141I | Yes | |
| A153G/Sc | Yese | |
| H156R | Yes | |
| H157Q/R/S/Y | Yesf | |
| N160K | Yes | |
| Alternative | G118R | No |
| 231ins SREGK/SREGRg | Yes | |
| R263K | No |
| Categorya | Mutation | HIV-2–Specific or Rarely Observed in HIV-1?b |
|---|---|---|
| Primary | Y143C/G/H/R | Yes (Y143G) |
| Q148H/K/R | No | |
| N155H | No | |
| Secondary | K46R | Yes |
| I84Vc | Yes | |
| Q91R | Yes | |
| E92A/G/Q | Yes (E92A) | |
| T97A | No | |
| A119Tc | Yesd | |
| G140A/S | No | |
| V141I | Yes | |
| A153G/Sc | Yese | |
| H156R | Yes | |
| H157Q/R/S/Y | Yesf | |
| N160K | Yes | |
| Alternative | G118R | No |
| 231ins SREGK/SREGRg | Yes | |
| R263K | No |
Mutations were assigned to categories as described in the text.
Based on the Stanford University HIV Drug Resistance Database—INSTI Resistance Notes [34].
Mutations I84V, A119T, and A153S are naturally occurring polymorphisms found in group A HIV-2 [29]. A153S is also polymorphic in group B HIV-2 [7].
A mutation at the corresponding site in HIV-1 integrase (S119R) is involved in integrase inhibitor resistance in HIV-1 [36].
The corresponding residue in HIV-1 integrase is S153. S153Y/F is a rare, nonpolymorphic mutation that is selected by dolutegravir in HIV-1 in vitro [35].
E157Q is a known polymorphism in HIV-1 integrase that confers modest (<2.0-fold) changes in the half maximum effective concentration (EC50) values for raltegravir and dolutegravir in HIV-1, as assessed by site-directed mutagenesis [37, 38].
Insertions of five amino acids at residue 231 of HIV-2 integrase. In the analysis performed by Tzou et al., insertions at position 231 were considered to be a single mutant form [29].
Other mutations in HIV-2 integrase were classified as possible alternative pathways to INI resistance (Table 1); these included G118R, R263K, and insertions of 5 amino acids at position 231 (231ins SREGK and SREGR) that were identified in a raltegravir-treated patient infected with group A HIV-2 [10]. We also considered combinations of primary INI resistance mutations as potential alternative paths to INI resistance. Although combinations of primary mutations have not been observed in HIV-2, we included them in our analysis based on reports that similar variants emerge in a small subset of HIV-1–infected patients and confer resistance to dolutegravir in HIV-1 in culture [39–41].
To determine the effects of primary, secondary, and alternative mutations in HIV-2 integrase on raltegravir and dolutegravir susceptibility, we constructed a library of 95 full-length plasmid clones of HIV-2ROD9 via SDM. Each clone was transfected into 293T/17 cells, and the resultant viruses (along with wild-type HIV-2ROD9) were individually tested against raltegravir and dolutegravir in the MAGIC-5A single-cycle assay.
We initially tested HIV-2ROD9 variants that contained either single, secondary amino acid changes alone, or combinations of secondary changes that have been observed in the absence of Y143C/G/H/R, Q148H/K/R, or N155H mutations in HIV-2 (Supplementary Tables 2 and 3). Nearly all of the single amino-acid variants were comparable to wild-type HIV-2ROD9 with respect to raltegravir and dolutegravir sensitivity (Figure 1). However, mutants encoding E92A and E92Q, as well as the combinations E92Q + T97A + A153G and I84V + E92Q + H157S, exhibited low-level resistance to dolutegravir (2.5- to 2.6-fold increase in EC50 relative to wild-type HIV-2ROD9; Figure 1B and Supplementary Table 4). In addition, all 7 of the multiply substituted mutants were resistant to raltegravir, with up to a 28-fold increase in the EC50 value, as observed for the E92Q + T97A + A153G mutant (Figure 1A and Supplementary Table 4).

Susceptibility of HIV-2ROD9 integrase variants that encode secondary integrase inhibitor-associated changes only to raltegravir (A) and dolutegravir (B). Bars indicate mean EC50 values ± 1 SD. Horizontal dotted lines indicate the wild-type EC50, and 10-, 100-, and 1000-fold increases in the EC50 value relative to wild-type HIV-2ROD9 (bottom to top). Asterisks indicate EC50 values that are significantly greater than that of wild type HIV-2ROD9 (P ≤ .05, ANOVA of log10-transformed EC50 values with Tukey multiple comparisons test). Abbreviations: EC50, half maximum effective concentration; HIV, human immunodeficiency virus; WT, wild type.
Next, we determined the effects of mutations at the 3 primary resistance-associated sites in HIV-2, both alone and in combination with candidate secondary INI resistance changes. HIV-2ROD9 variants in the Y143C/G/H/R, Q148H/K/R, and N155H subsets were broadly resistant to raltegravir (Figure 2A). The accumulation of secondary mutations in combination with Y143C, Y143G, and N155H resulted in stepwise increases in the EC50 value for raltegravir, and led to ≥50-fold resistance to the drug for several variants (Supplementary Figure 2). For dolutegravir, mutations Q148K and G140A/S + Q148H/K/R conferred 5.8- to 370-fold increases in the EC50 value relative to wild-type HIV-2ROD9 (Figure 2B). Notably, variants with mutations in the N155H pathway were also broadly resistant to dolutegravir (Figure 2B). Combinations E92A/Q + T97A + N155H and E92Q + N155H + H157Q, with or without additional secondary changes, yielded 16- to 33-fold resistance to dolutegravir in the single-cycle assay (n = 6 variants; Supplementary Table 7). Altogether, 21 of the 26 mutants in the N155H subset were cross-resistant to dolutegravir (Figure 2B). In contrast, nearly all of the Y143C/G/H/R-encoding mutants in our library were fully susceptible or slightly hypersusceptible to dolutegravir; the sole exception to this pattern, mutant Q91R + E92Q + T97A + Y143G + A153S, was 7.6-fold resistant to dolutegravir (Figure 2B and Supplementary Table 5).
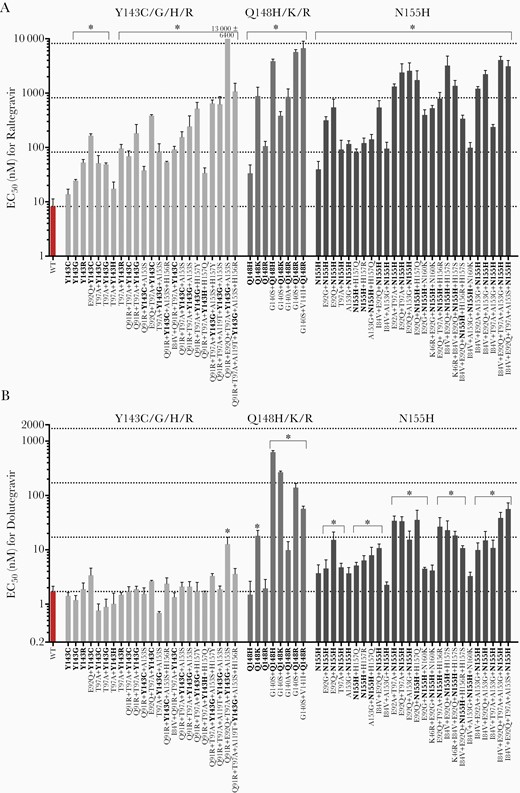
Susceptibility of HIV-2ROD9 integrase variants encoding mutations at sites Y143, Q148, and N155, with or without additional secondary treatment-associated mutations, to raltegravir (A) and dolutegravir (B). Primary mutations are indicated in bold type. Bars indicate mean EC50 values ± 1 SD. Horizontal dotted lines indicate the wild-type EC50, and 10-, 100-, and 1000-fold increases in the EC50 value relative to WT HIV-2ROD9 (bottom to top). Asterisks indicate EC50 values that are significantly greater than that of WT HIV-2ROD9 (P ≤ .05, ANOVA of log10-transformed EC50 values with Tukey multiple comparisons test). Abbreviations: EC50, half maximum effective concentration; HIV, human immunodeficiency virus; WT, wild type.
In the category of mutations that potentially represent alternative pathways to dolutegravir resistance, changes G118R, 231ins SREGK, and 231ins SREGR individually conferred moderate resistance to raltegravir and dolutegravir in HIV-2ROD9 (6.6- to 22-fold), and the combined presence of 2 primary resistance mutations (Y143H + Q148H or Q148H/R + N155H) resulted in moderate to high-level resistance to both INIs (27- to >5000-fold; Figure 3 and Supplementary Table 8). In contrast, R263K, E92G + R263K, and E138K + R263K did not significantly impact the EC50 for either drug in HIV-2ROD9 (Figure 3 and Supplementary Figure 3). Similar results were obtained for R263K mutants of HIV-1NL4-3, whereas mutations 231ins YRDSR and H51Y + R263K resulted in low- to moderate-level resistance to raltegravir and dolutegravir in the HIV-1NL4-3 background (2.9- to 9.4-fold); the 231ins YREGR HIV-1NL4-3 mutant also showed low-level resistance to raltegravir (3.2-fold; Supplementary Table 9).
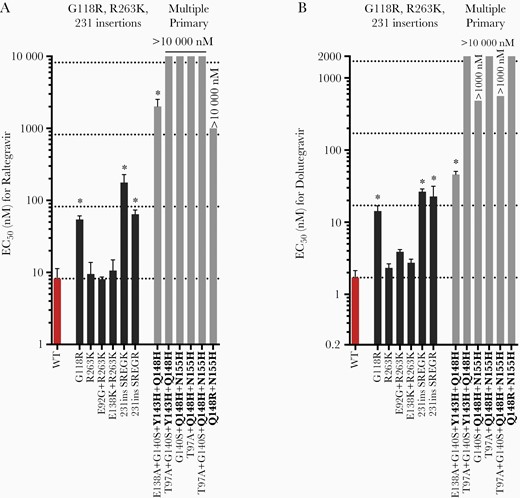
Raltegravir (A) and dolutegravir (B) susceptibility of HIV-2 integrase variants encoding mutations that represent potential alternative pathways to integrase inhibitor resistance. Primary mutations are indicated in bold type. Bars indicate mean EC50 values ± 1 SD. Horizontal dotted lines indicate the wild-type EC50, and 10-, 100-, and 1000-fold increases in the EC50 value relative to WT HIV-2ROD9 (bottom to top). Asterisks indicate EC50 values that are significantly greater than that of WT HIV-2ROD9 (P ≤ .05, ANOVA of log10-transformed EC50 values with Tukey multiple comparisons test). Abbreviations: EC50, half maximum effective concentration; HIV, human immunodeficiency virus; WT, wild type.
To examine the individual contributions of secondary INI-associated mutations to raltegravir and dolutegravir resistance, we compared the EC50 values obtained for pairs of HIV-2ROD9 variants that either lacked or contained each secondary change (Figure 4 and Figure 5). Replacements Q91R, E92A/G/Q, T97A, G140A/S, A153G, and H157Q/S individually conferred a statistically significant increase in the EC50 value for raltegravir in 1 or more mutational backgrounds (Figure 4); 5 of these mutations (Q91R, E92A, A153G, and H157Q/S) are unique to HIV-2 or are rarely observed in HIV-1 integrase sequences (Table 1). Similarly, replacements E92A/Q, T97A, and G140A/S increased the EC50 value for dolutegravir in 1 or more mutant clones (Figure 5). Mutations K46R, I84V, A119T, V141I, A153S, H156R, H157R/Y, and N160K did not confer a statistically significant increase in the EC50 for raltegravir or dolutegravir in any of the mutational combinations tested (Figure 4, Figure 5 and Supplementary Figure 4).
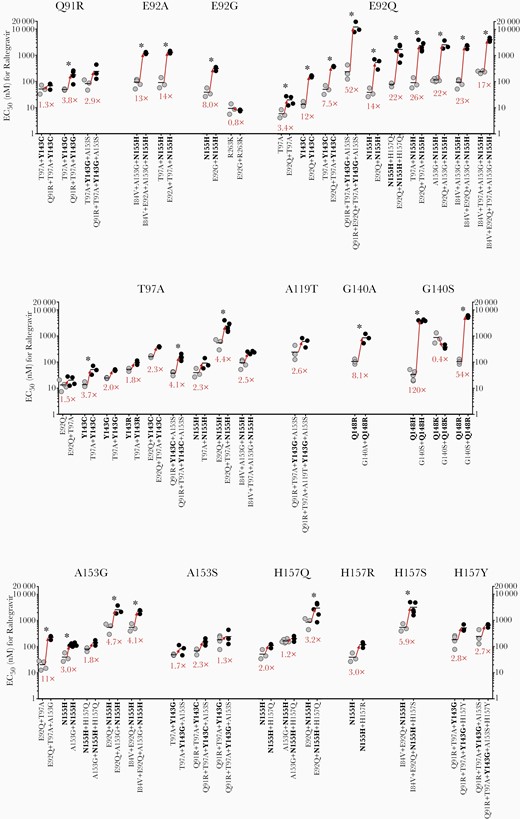
Effects of individual secondary integrase inhibitor-associated mutations in HIV-2ROD9 integrase on raltegravir susceptibility. Each point represents the result of a single assay run; horizontal lines indicate mean EC50 values. Values in red type indicate the fold change in mean EC50 that results from the addition of a single, secondary mutation. ∗P ≤ .05, ANOVA of log10-transformed EC50 values with Tukey multiple comparisons test. Abbreviations: EC50, half maximum effective concentration; HIV, human immunodeficiency virus.
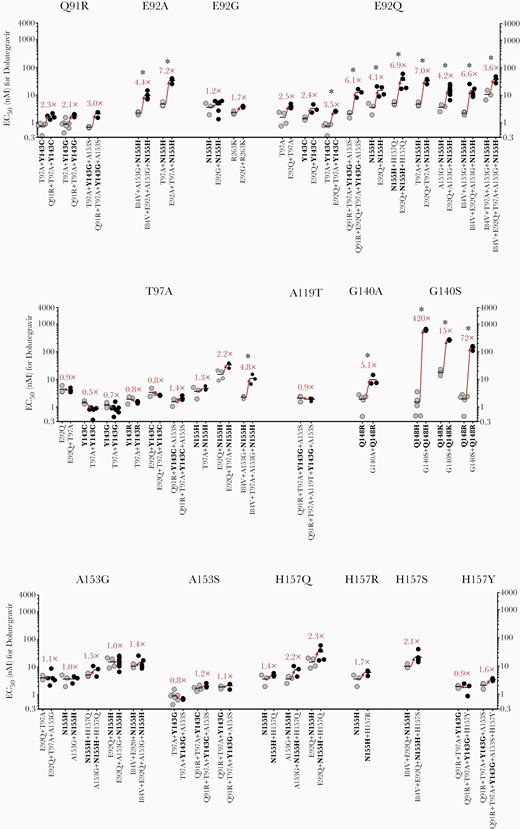
Effects of individual secondary integrase inhibitor-associated mutations in HIV-2ROD9 integrase on dolutegravir susceptibility. Each point represents the result of a single assay run; horizontal lines indicate mean EC50 values. Values in red type indicate the fold change in mean EC50 that results from the addition of a single, secondary mutation. ∗P ≤ .05, ANOVA of log10-transformed EC50 values with Tukey multiple comparisons test. Abbreviations: EC50, half maximum effective concentration; HIV, human immunodeficiency virus.
DISCUSSION
In this study, we examined the contributions of INI treatment-associated amino acid changes in HIV-2 integrase to raltegravir and dolutegravir resistance using an extensive collection of site-directed mutants. Our analysis is an experimental test of the findings published by Tzou and colleagues [29], who identified 14 amino acid changes in HIV-2 integrase that are significantly associated with ART (codon 231 insertions were grouped by Tzou et al as a single mutant form). Of these 14 mutations, 10 conferred a statistically significant increase in the EC50 for raltegravir, and 8 conferred a significant increase in the EC50 for dolutegravir when introduced into 1 or more wild-type or mutant backgrounds (Table 2, bold type). We also tested the effects of 16 other mutations that are present in sequences from INI-treated HIV-2 patients (Table 2, not bold type). Nine of these 16 replacements contributed to or directly conferred resistance to raltegravir in the single-cycle assay, and 4 of these (G118R, G140A, and Q148H/K) also led to reductions in dolutegravir susceptibility. Altogether, 21 of the 31 mutations evaluated in our study reduced the susceptibility of HIV-2ROD9 to 1 or both INIs in at least 1 genetic context (Table 2). Replacements I84V, A119T, V141I, and H156R, which were identified as INI treatment-associated mutations by Tzou et al [29], had no statistically significant effect on raltegravir or dolutegravir sensitivity; these substitutions might contribute to INI resistance in mutational backgrounds other than the ones tested here, or may impart levels of resistance that are too low to measure in the single-cycle assay. These replacements might also act as compensatory mutations that improve the replicative fitness of INI-resistant variants. Additional in vitro experiments are needed to explore these possibilities.
Summary of the Effects of Site-Directed Mutations in HIV-2ROD9 Integrase on Raltegravir and Dolutegravir Susceptibility in the MAGIC-5A Assay
| Categorya | Mutationb | Contributes to Raltegravir Resistance?c | Fold Change in EC50 for Raltegravird | Fold Enhancement of Raltegravir Resistancee | Contributes to Dolutegravir Resistance?c | Fold Change in EC50 for Dolutegravird | Fold Enhancement of Dolutegravir Resistancee |
|---|---|---|---|---|---|---|---|
| Primary ± Secondary | Y143C | Yes | 1.7–48 | No | 0.45–2.0 | ||
| Y143G | Yes | 3.0–1600 | Yesf | 0.41–7.6 | |||
| Y143H | Yes | 2.1–4.1 | No | 0.59–0.94 | |||
| Y143R | Yes | 6.5–12 | No | 0.88–1.1 | |||
| Q148H | Yes | 4.0–480 | Yes | 0.88–370 | |||
| Q148K | Yes | 46–110 | Yes | 11–160 | |||
| Q148R | Yes | 13–820 | Yes | 1.2–82 | |||
| N155H | Yes | 4.9–500 | Yes | 1.4–33 | |||
| Secondary | K46R | No | 1.6 | 0.4–1.4 | No | 0.65 | 0.7–0.9 |
| I84V | No | 1.1 | 0.8–1.3 | No | 1.6 | 0.6–1.0 | |
| Q91R | Yes | 0.89 | 1.3–3.8 | No | 1.6 | 2.1–3.0 | |
| E92A | Yes | 0.83 | 13–14 | Yesg | 2.5 | 4.4–7.2 | |
| E92G | Yes | 1.6 | 0.8–8.0 | No | 2.3 | 1.2–1.7 | |
| E92Q | Yes | 1.6 | 3.4–52 | Yesg | 2.6 | 2.4–6.9 | |
| T97A | Yes | 0.73 | 1.5–4.4 | Yes | 0.94 | 0.5–4.8 | |
| A119T | No | 1.3 | 2.6 | No | 1.2 | 0.9 | |
| G140A | Yes | 1.7 | 8.1 | Yes | 1.1 | 5.1 | |
| G140S | Yes | 1.0 | 0.4–120 | Yes | 1.5 | 15–420 | |
| V141I | No | 1.0 | 1.2 | No | 0.88 | 0.4 | |
| A153G | Yes | 1.7 | 1.8–11 | No | 1.5 | 1.0–1.5 | |
| A153S | No | 1.5 | 1.3–2.3 | No | 1.1 | 0.8–1.2 | |
| H156R | No | 1.6 | 0.1–1.7 | No | 1.4 | 0.5–1.9 | |
| H157Q | Yes | 0.73 | 1.2–3.2 | No | 1.4 | 1.4–2.3 | |
| H157R | No | 1.6 | 3.0 | No | 1.2 | 1.7 | |
| H157S | Yes | 1.6 | 5.9 | No | 1.1 | 2.1 | |
| H157Y | No | 1.3 | 2.7–2.8 | No | 1.6 | 0.9–1.6 | |
| N160K | No | 1.5 | 1.0–2.5 | No | 1.2 | 1.0–1.5 | |
| Alternative | G118R | Yes | 6.6 | Yes | 8.2 | ||
| 231ins SREGKh | Yes | 22 | Yes | 16 | |||
| 231ins SREGRh | Yes | 7.8 | Yes | 14 | |||
| R263K | No | 1.0–1.3 | No | 1.4–2.3 |
| Categorya | Mutationb | Contributes to Raltegravir Resistance?c | Fold Change in EC50 for Raltegravird | Fold Enhancement of Raltegravir Resistancee | Contributes to Dolutegravir Resistance?c | Fold Change in EC50 for Dolutegravird | Fold Enhancement of Dolutegravir Resistancee |
|---|---|---|---|---|---|---|---|
| Primary ± Secondary | Y143C | Yes | 1.7–48 | No | 0.45–2.0 | ||
| Y143G | Yes | 3.0–1600 | Yesf | 0.41–7.6 | |||
| Y143H | Yes | 2.1–4.1 | No | 0.59–0.94 | |||
| Y143R | Yes | 6.5–12 | No | 0.88–1.1 | |||
| Q148H | Yes | 4.0–480 | Yes | 0.88–370 | |||
| Q148K | Yes | 46–110 | Yes | 11–160 | |||
| Q148R | Yes | 13–820 | Yes | 1.2–82 | |||
| N155H | Yes | 4.9–500 | Yes | 1.4–33 | |||
| Secondary | K46R | No | 1.6 | 0.4–1.4 | No | 0.65 | 0.7–0.9 |
| I84V | No | 1.1 | 0.8–1.3 | No | 1.6 | 0.6–1.0 | |
| Q91R | Yes | 0.89 | 1.3–3.8 | No | 1.6 | 2.1–3.0 | |
| E92A | Yes | 0.83 | 13–14 | Yesg | 2.5 | 4.4–7.2 | |
| E92G | Yes | 1.6 | 0.8–8.0 | No | 2.3 | 1.2–1.7 | |
| E92Q | Yes | 1.6 | 3.4–52 | Yesg | 2.6 | 2.4–6.9 | |
| T97A | Yes | 0.73 | 1.5–4.4 | Yes | 0.94 | 0.5–4.8 | |
| A119T | No | 1.3 | 2.6 | No | 1.2 | 0.9 | |
| G140A | Yes | 1.7 | 8.1 | Yes | 1.1 | 5.1 | |
| G140S | Yes | 1.0 | 0.4–120 | Yes | 1.5 | 15–420 | |
| V141I | No | 1.0 | 1.2 | No | 0.88 | 0.4 | |
| A153G | Yes | 1.7 | 1.8–11 | No | 1.5 | 1.0–1.5 | |
| A153S | No | 1.5 | 1.3–2.3 | No | 1.1 | 0.8–1.2 | |
| H156R | No | 1.6 | 0.1–1.7 | No | 1.4 | 0.5–1.9 | |
| H157Q | Yes | 0.73 | 1.2–3.2 | No | 1.4 | 1.4–2.3 | |
| H157R | No | 1.6 | 3.0 | No | 1.2 | 1.7 | |
| H157S | Yes | 1.6 | 5.9 | No | 1.1 | 2.1 | |
| H157Y | No | 1.3 | 2.7–2.8 | No | 1.6 | 0.9–1.6 | |
| N160K | No | 1.5 | 1.0–2.5 | No | 1.2 | 1.0–1.5 | |
| Alternative | G118R | Yes | 6.6 | Yes | 8.2 | ||
| 231ins SREGKh | Yes | 22 | Yes | 16 | |||
| 231ins SREGRh | Yes | 7.8 | Yes | 14 | |||
| R263K | No | 1.0–1.3 | No | 1.4–2.3 |
Abbreviation: EC50, half maximum effective concentration.
Mutations are categorized as described in the main text.
Mutations listed in bold type were shown to be significantly associated with INSTI-based therapy by Tzou and colleagues [29]. Additional mutations evaluated in the present study are shown in not bold type.
Primary and alternative mutations were considered to be contributors to resistance if they conferred a statistically significant increase in the EC50 value for raltegravir or dolutegravir as a single amino acid replacement or insertion alone (ie, in an otherwise wild-type background), or when combined with 1 or more secondary integrase inhibitor-associated changes (see Figures 1–3). Secondary mutations were classified as contributors to resistance if they resulted in a statistically significant increase in the EC50 for raltegravir or dolutegravir in 1 or more mutational backgrounds (Figure 4 and Figure 5), or in wild-type HIV-2ROD9 (Figure 1). In all cases, significance was determined as P ≤ .05 in an ANOVA of log10-transformed EC50 values with Tukey multiple comparisons test.
For primary treatment-associated mutations, the fold change in EC50 value (relative to wild-type HIV-2 ROD9) is listed as the range of fold-change values that were observed for all variants containing the indicated amino acid change (ie, with or without additional secondary mutations in integrase; Supplementary Tables 5–7). Combinations of primary mutations (Supplementary Table 8) were not included in this summation because these combinations have not been observed in HIV-2–infected individuals. For the secondary integrase inhibitor-associated mutations, as well as mutations G118R, 231ins SREGK, and 231ins SREGR, fold change is listed as effect of introducing the indicated single replacement or insertion alone into wild-type HIV-2 ROD9. For R263K, the range of fold-change values observed for mutants R263K, E92G + R263K, and E138K + R263K (relative to wild-type HIV-2 ROD9) is reported.
Fold change in EC50 value that resulted from the introduction of each secondary mutation into 1 or more mutational backgrounds (ie, in combination with other primary and secondary mutations; Figure 4, Figure 5, and Supplementary Figure 4).
The combination of Q91R + E92Q + T97A + Y143G + A153S resulted in moderate dolutegravir resistance (7.6-fold). However, 8 other Y143G-encoding variants were fully susceptible to dolutegravir.
Mutations E92A and E92Q conferred low-level dolutegravir resistance in an otherwise wild-type background (2.5- and 2.6-fold increase in EC50, respectively; Figure 1B). E92A and E92G also increased the EC50 for dolutegravir by a factor of 2.4- to 6.9-fold when combined with other primary and secondary integrase inhibitor-associated mutations (Figure 5).
Insertions of 5 amino acids at residue 231 of HIV-2 integrase. In the analysis performed by Tzou et al, insertions at position 231 were considered to be a single mutant form [29].
Summary of the Effects of Site-Directed Mutations in HIV-2ROD9 Integrase on Raltegravir and Dolutegravir Susceptibility in the MAGIC-5A Assay
| Categorya | Mutationb | Contributes to Raltegravir Resistance?c | Fold Change in EC50 for Raltegravird | Fold Enhancement of Raltegravir Resistancee | Contributes to Dolutegravir Resistance?c | Fold Change in EC50 for Dolutegravird | Fold Enhancement of Dolutegravir Resistancee |
|---|---|---|---|---|---|---|---|
| Primary ± Secondary | Y143C | Yes | 1.7–48 | No | 0.45–2.0 | ||
| Y143G | Yes | 3.0–1600 | Yesf | 0.41–7.6 | |||
| Y143H | Yes | 2.1–4.1 | No | 0.59–0.94 | |||
| Y143R | Yes | 6.5–12 | No | 0.88–1.1 | |||
| Q148H | Yes | 4.0–480 | Yes | 0.88–370 | |||
| Q148K | Yes | 46–110 | Yes | 11–160 | |||
| Q148R | Yes | 13–820 | Yes | 1.2–82 | |||
| N155H | Yes | 4.9–500 | Yes | 1.4–33 | |||
| Secondary | K46R | No | 1.6 | 0.4–1.4 | No | 0.65 | 0.7–0.9 |
| I84V | No | 1.1 | 0.8–1.3 | No | 1.6 | 0.6–1.0 | |
| Q91R | Yes | 0.89 | 1.3–3.8 | No | 1.6 | 2.1–3.0 | |
| E92A | Yes | 0.83 | 13–14 | Yesg | 2.5 | 4.4–7.2 | |
| E92G | Yes | 1.6 | 0.8–8.0 | No | 2.3 | 1.2–1.7 | |
| E92Q | Yes | 1.6 | 3.4–52 | Yesg | 2.6 | 2.4–6.9 | |
| T97A | Yes | 0.73 | 1.5–4.4 | Yes | 0.94 | 0.5–4.8 | |
| A119T | No | 1.3 | 2.6 | No | 1.2 | 0.9 | |
| G140A | Yes | 1.7 | 8.1 | Yes | 1.1 | 5.1 | |
| G140S | Yes | 1.0 | 0.4–120 | Yes | 1.5 | 15–420 | |
| V141I | No | 1.0 | 1.2 | No | 0.88 | 0.4 | |
| A153G | Yes | 1.7 | 1.8–11 | No | 1.5 | 1.0–1.5 | |
| A153S | No | 1.5 | 1.3–2.3 | No | 1.1 | 0.8–1.2 | |
| H156R | No | 1.6 | 0.1–1.7 | No | 1.4 | 0.5–1.9 | |
| H157Q | Yes | 0.73 | 1.2–3.2 | No | 1.4 | 1.4–2.3 | |
| H157R | No | 1.6 | 3.0 | No | 1.2 | 1.7 | |
| H157S | Yes | 1.6 | 5.9 | No | 1.1 | 2.1 | |
| H157Y | No | 1.3 | 2.7–2.8 | No | 1.6 | 0.9–1.6 | |
| N160K | No | 1.5 | 1.0–2.5 | No | 1.2 | 1.0–1.5 | |
| Alternative | G118R | Yes | 6.6 | Yes | 8.2 | ||
| 231ins SREGKh | Yes | 22 | Yes | 16 | |||
| 231ins SREGRh | Yes | 7.8 | Yes | 14 | |||
| R263K | No | 1.0–1.3 | No | 1.4–2.3 |
| Categorya | Mutationb | Contributes to Raltegravir Resistance?c | Fold Change in EC50 for Raltegravird | Fold Enhancement of Raltegravir Resistancee | Contributes to Dolutegravir Resistance?c | Fold Change in EC50 for Dolutegravird | Fold Enhancement of Dolutegravir Resistancee |
|---|---|---|---|---|---|---|---|
| Primary ± Secondary | Y143C | Yes | 1.7–48 | No | 0.45–2.0 | ||
| Y143G | Yes | 3.0–1600 | Yesf | 0.41–7.6 | |||
| Y143H | Yes | 2.1–4.1 | No | 0.59–0.94 | |||
| Y143R | Yes | 6.5–12 | No | 0.88–1.1 | |||
| Q148H | Yes | 4.0–480 | Yes | 0.88–370 | |||
| Q148K | Yes | 46–110 | Yes | 11–160 | |||
| Q148R | Yes | 13–820 | Yes | 1.2–82 | |||
| N155H | Yes | 4.9–500 | Yes | 1.4–33 | |||
| Secondary | K46R | No | 1.6 | 0.4–1.4 | No | 0.65 | 0.7–0.9 |
| I84V | No | 1.1 | 0.8–1.3 | No | 1.6 | 0.6–1.0 | |
| Q91R | Yes | 0.89 | 1.3–3.8 | No | 1.6 | 2.1–3.0 | |
| E92A | Yes | 0.83 | 13–14 | Yesg | 2.5 | 4.4–7.2 | |
| E92G | Yes | 1.6 | 0.8–8.0 | No | 2.3 | 1.2–1.7 | |
| E92Q | Yes | 1.6 | 3.4–52 | Yesg | 2.6 | 2.4–6.9 | |
| T97A | Yes | 0.73 | 1.5–4.4 | Yes | 0.94 | 0.5–4.8 | |
| A119T | No | 1.3 | 2.6 | No | 1.2 | 0.9 | |
| G140A | Yes | 1.7 | 8.1 | Yes | 1.1 | 5.1 | |
| G140S | Yes | 1.0 | 0.4–120 | Yes | 1.5 | 15–420 | |
| V141I | No | 1.0 | 1.2 | No | 0.88 | 0.4 | |
| A153G | Yes | 1.7 | 1.8–11 | No | 1.5 | 1.0–1.5 | |
| A153S | No | 1.5 | 1.3–2.3 | No | 1.1 | 0.8–1.2 | |
| H156R | No | 1.6 | 0.1–1.7 | No | 1.4 | 0.5–1.9 | |
| H157Q | Yes | 0.73 | 1.2–3.2 | No | 1.4 | 1.4–2.3 | |
| H157R | No | 1.6 | 3.0 | No | 1.2 | 1.7 | |
| H157S | Yes | 1.6 | 5.9 | No | 1.1 | 2.1 | |
| H157Y | No | 1.3 | 2.7–2.8 | No | 1.6 | 0.9–1.6 | |
| N160K | No | 1.5 | 1.0–2.5 | No | 1.2 | 1.0–1.5 | |
| Alternative | G118R | Yes | 6.6 | Yes | 8.2 | ||
| 231ins SREGKh | Yes | 22 | Yes | 16 | |||
| 231ins SREGRh | Yes | 7.8 | Yes | 14 | |||
| R263K | No | 1.0–1.3 | No | 1.4–2.3 |
Abbreviation: EC50, half maximum effective concentration.
Mutations are categorized as described in the main text.
Mutations listed in bold type were shown to be significantly associated with INSTI-based therapy by Tzou and colleagues [29]. Additional mutations evaluated in the present study are shown in not bold type.
Primary and alternative mutations were considered to be contributors to resistance if they conferred a statistically significant increase in the EC50 value for raltegravir or dolutegravir as a single amino acid replacement or insertion alone (ie, in an otherwise wild-type background), or when combined with 1 or more secondary integrase inhibitor-associated changes (see Figures 1–3). Secondary mutations were classified as contributors to resistance if they resulted in a statistically significant increase in the EC50 for raltegravir or dolutegravir in 1 or more mutational backgrounds (Figure 4 and Figure 5), or in wild-type HIV-2ROD9 (Figure 1). In all cases, significance was determined as P ≤ .05 in an ANOVA of log10-transformed EC50 values with Tukey multiple comparisons test.
For primary treatment-associated mutations, the fold change in EC50 value (relative to wild-type HIV-2 ROD9) is listed as the range of fold-change values that were observed for all variants containing the indicated amino acid change (ie, with or without additional secondary mutations in integrase; Supplementary Tables 5–7). Combinations of primary mutations (Supplementary Table 8) were not included in this summation because these combinations have not been observed in HIV-2–infected individuals. For the secondary integrase inhibitor-associated mutations, as well as mutations G118R, 231ins SREGK, and 231ins SREGR, fold change is listed as effect of introducing the indicated single replacement or insertion alone into wild-type HIV-2 ROD9. For R263K, the range of fold-change values observed for mutants R263K, E92G + R263K, and E138K + R263K (relative to wild-type HIV-2 ROD9) is reported.
Fold change in EC50 value that resulted from the introduction of each secondary mutation into 1 or more mutational backgrounds (ie, in combination with other primary and secondary mutations; Figure 4, Figure 5, and Supplementary Figure 4).
The combination of Q91R + E92Q + T97A + Y143G + A153S resulted in moderate dolutegravir resistance (7.6-fold). However, 8 other Y143G-encoding variants were fully susceptible to dolutegravir.
Mutations E92A and E92Q conferred low-level dolutegravir resistance in an otherwise wild-type background (2.5- and 2.6-fold increase in EC50, respectively; Figure 1B). E92A and E92G also increased the EC50 for dolutegravir by a factor of 2.4- to 6.9-fold when combined with other primary and secondary integrase inhibitor-associated mutations (Figure 5).
Insertions of 5 amino acids at residue 231 of HIV-2 integrase. In the analysis performed by Tzou et al, insertions at position 231 were considered to be a single mutant form [29].
Our findings for HIV-2 variants representing the Y143C/G/H/R and Q148H/K/R pathways (Figure 2) are consistent with those seen in previous studies of INI-resistant HIV-1 mutants [35, 42, 43]. However, our analysis of N155H pathway yielded an unexpected result—15 of the 26 HIV-2 variants within this subset were moderately resistant to dolutegravir (5- to 33-fold increase in the EC50; Figure 2B and Supplementary Table 7). In contrast, N155H-pathway mutants of HIV-1 tend to show negligible resistance to dolutegravir in culture, although a few exceptions to this trend have been reported (Supplementary Table 10). While this outcome might be attributable to more frequent drug resistance testing (and therefore, prompter identification of INI resistance) in HIV-1–infected individuals, our data suggest that the N155H pathway could potentially lead to clinical resistance to dolutegravir in HIV-2–infected patients. Further genotypic and phenotypic studies are needed to clarify the role of the N155H pathway in emergent dolutegravir resistance.
In a previous study performed by our group [12], dose-response curves for dolutegravir for the T97A + Y143C mutant of HIV-2ROD9 failed to drop below 50% of the no-drug controls at drug concentrations equaling 10 µM, although modest dose-dependent inhibition was observed at 100 nM dolutegravir and higher. Accordingly, the EC50 value for T97A + Y143C HIV-2ROD9 was reported as >10 µM [12]. These data were obtained via X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining and manual counting of β-galactosidase–positive (Lac+) foci in MAGIC-5A monolayers. In the present study, using spectrophotometric measurements of CPRG (chlorophenol red-β-d-galactopyranoside) substrate conversion to quantify HIV-2 infection, dose-response curves for the T97A + Y143C mutant showed a similar plateau at high concentrations of drug (Supplementary Figure 3); in 5 independent assay runs, the inhibition curves reached a minimum of 52% (SD 9.3%) of no-drug controls. However, the position of the downward slopes of the curves for T97A + Y143C HIV-2ROD9 was comparable to that observed for wild-type HIV-2ROD9, resulting in an EC50 value of 0.76 nM (SD 0.32 nM) for the double mutant. The discordance between our current data and the previous study is attributable to difficulties in accurately identifying Lac+ foci induced by T97A + Y143C HIV-2ROD9, which shows poor replication capacity in single-round infections [44], and fails to produce detectable levels of virus in spreading infections of CEM-ss cells [12]. In addition, HIV-2ROD9 mutants Q91R + T97A + Y143C and Q91R + T97A + Y143C + A153S yielded dose-response curves and EC50 values for dolutegravir that were comparable to those obtained with wild-type HIV-2ROD9 (Supplementary Figure 3 and Supplementary Table 5). Based on these findings, we conclude that the T97A + Y143C mutant of HIV-2ROD9 is not resistant to dolutegravir in a single round of infection.
There are some inherent limitations to our present work. SDM is necessary to empirically test the contributions of individual amino acid changes or groups of changes to antiretroviral drug resistance. However, this approach is limited by the resources needed to generate and test large numbers of HIV clones; it would be very difficult to assess all of the combinations of mutations and polymorphisms that can potentially influence drug susceptibility. We constructed our HIV-2 mutant library using a single clone of a prototypic group A isolate (HIV-2ROD9); the degree to which our findings are applicable to a broad range of HIV-2 isolates, including group B HIV-2, remains to be determined. Furthermore, the sequences used to guide our choice of genotypes for drug susceptibility testing were primarily obtained from patients who received raltegravir-containing ART (Supplementary Tables 2 and 3). We cannot exclude the possibility that dolutegravir-based regimens select for other, as-yet undiscovered mutations in HIV-2, including mutations outside the integrase-encoding region of pol [45–47].
Despite these limitations, our analysis provides several insights into the genetic basis of INI resistance in HIV-2. We show that mutations E92A and E92Q alone confer low-level resistance to dolutegravir, and that combinations of secondary mutations in HIV-2ROD9 integrase can confer resistance to both raltegravir and dolutegravir in the absence of primary INI-associated changes (Figure 1). All 3 canonical pathways (Y143C/G/H/R, Q148H/K/R, and N155H) lead to high-level raltegravir resistance in HIV-2ROD9, whereas most Y143C/G/H/R mutants are susceptible to dolutegravir (Figure 2). In addition, mutations G118R, 231ins SREGK, and 231ins SREGR confer resistance to raltegravir and dolutegravir in HIV-2ROD9 in the single cycle assay (Figure 3). Our results for the 231ins variants are concordant with recently published data from Le Hingrat et al [30]. Lastly, we demonstrate that secondary mutations that are unique to HIV-2 can increase the level of resistance to raltegravir and (in the case of E92A) dolutegravir in HIV-2ROD9 when introduced into 1 or more mutational backgrounds (Figure 4 and Figure 5).
Altogether, our study substantially expands the available body of data regarding the phenotypic effects of INI-associated mutations in HIV-2. These findings should help to improve algorithms for interpreting sequence information from HIV-2–infected patients [48–50] in settings where genotypic drug resistance testing is available. In West Africa and other resource-constrained areas, our data will inform cohort-level studies of drug resistance in HIV-2 patients receiving dolutegravir-based ART. This information will be increasingly important as routine clinical care and programmatic efforts return to full strength in the wake of the coronavirus disease 2019 (COVID-19) pandemic.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank undergraduate students Pallas Burhen and Robbie Nixon for their technical assistance. We also thank Robert Shafer and Philip Tzou (Stanford University) for helpful discussions regarding HIV-2 drug resistance. Additional University of Washington-Senegal HIV-2 Study Group members (http://www.uwsenegalresearch.com) are: Fatima Sall, Mouhamadou Baïla Diallo, Khadim Faye, Samba Cisse, Marie Pierre Sy, Binetou Diaw, Ousseynou Ndiaye, Babacar Faye, Ndeye Astou Diop, Amadou Bale Diop, and Marianne Fadam Diome, Clinique des Maladies Infectieuses Ibrahima DIOP Mar, Centre Hospitalier Universitaire de Fann, Universite’ Cheikh Anta Diop de Dakar, Dakar, Senegal; Jean Jacques Malomar, ElHadji Ibrahima Sall, Ousseynou Cisse, Ibrahima Tito Tamba, Dominique Faye, Jean Philippe Diatta, Raphael Bakhoum, Jacques Francois Sambou, Juliette Gomis, and Therese Dieye, Région Médicale de Ziguinchor, Ziguinchor, Casamance, Senegal; and Stephen Hawes, Noelle Benzekri, John Lin, Ming Chang, Robert Coombs, James Mullins, Papa Salif Sow, and Nancy Kiviat, University of Washington, Seattle, Washington.
Financial support. This work was supported the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (grant number R01 AI120765 to G. S. G); the University of Washington Center for AIDS Research, a National Institutes of Health-funded program (grant number P30 AI027757); and the University of Washington Royalty Research Fund (grant number A92723). G. S. G is a collaborator on a grant from the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (grant number R24 AI136618) which funds, in part, the Stanford HIV Drug Resistance Database Program.
Potential conflicts of interest. G. S. G. has received research grants and support from the Bill and Melinda Gates Foundation, Gilead Sciences, Alere Technologies, Merck & Co, Inc, Janssen Pharmaceuticals, Cerus Corporation, ViiV Healthcare, Bristol-Myers Squibb, Roche Molecular Systems, Abbott Molecular Diagnostics, and THERA Technologies/TaiMed Biologics, Inc. M. S. has received support from Gilead Sciences. All other authors: No reported conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}