





Abstract
Primary pigmented nodular adrenocortical disease (PPNAD) is a rare cause of ACTH-independent Cushing syndrome (CS) associated mostly with Carney complex (CNC), a rare autosomal dominant multiple neoplasia syndrome. More than two-thirds of familial cases and approximately one-third of sporadic cases of CNC harbor germline inactivating PRKAR1A defects. Increasingly sensitive technologies for the detection of genetic defects such as next-generation sequencing (NGS) have further highlighted the importance of mosaicism in human disease.
A 33-year-old woman was diagnosed with ACTH-independent CS with abdominal computed tomography showing bilateral micronodular adrenal hyperplasia with a left adrenal adenoma. She underwent left adrenalectomy with pathology demonstrating PPNAD with a 1.5-cm pigmented adenoma. DNA analysis by Sanger sequencing revealed 2 different PRKAR1A variants in the adenoma that were absent from DNA extracted from blood and saliva: c.682C > T and c.974-2A > G. “Deep” NGS revealed that 0.31% of DNA copies extracted from blood and saliva did in fact carry the c.682C > T variant, suggesting low-level mosaicism for this defect.
We present a case of PPNAD due to low-level mosaicism for a PRKAR1A defect which led to the formation of an adenoma due to a second, adrenal-specific, somatic PRKAR1A mutation. The identification of mosaicism for PRKAR1A, depending on the number and distribution of cells affected has implications for genetic counseling and tumor surveillance. This is the first recorded case of a patient with PRKAR1A mosaicism, PPNAD, and an adenoma forming due to complete inactivation of PRKAR1A in adrenal tissue from a second, somatic-only, PRKAR1A coding sequence mutation.
Primary pigmented nodular adrenocortical disease (PPNAD) is a rare cause of adrenocorticotropic hormone (ACTH)-independent Cushing syndrome (CS), characterized by normal-sized or enlarged hyperplastic adrenal glands with pigmented nodules predominantly less than 1 cm in diameter (micronodular) and atrophic internodular adrenal cortex [1]. More than 90% of cases of PPNAD are associated with Carney complex (CNC) (c-PPNAD); rarely, PPNAD may be an isolated finding (i-PPNAD) [2, 3]. CNC is a rare, autosomal dominant, multiple endocrine neoplasia and lentiginosis syndrome that is comprised of abnormal cutaneous and mucosal pigmentation, myxomas primarily of the heart, skin, and breast, psammomatous melanotic schwannomas, breast ductal adenomas, osteochondromyxomas, and other nonendocrine and endocrine tumors with or without hormone overproduction [3, 4]. In addition to PPNAD, endocrine tumors in CNC include pituitary adenomas or hyperplasia with primarily growth hormone and/or prolactin hypersecretion, thyroid tumors, and gonadal tumors, with PPNAD being the most common endocrine neoplasm, occurring in 35% to 60% of patients and found in almost all patients on autopsy [4-6].
More than 70% of patients with familial CNC and 37% of patients with sporadic CNC harbor germline inactivating defects in PRKAR1A, a tumor suppressor gene located on chromosome 17q22-24, that encodes the regulatory subunit type 1α (R1α) of protein kinase A (PKA) [2, 7, 8]. Approximately 80% of pathogenic PRKAR1A variants are subject to mRNA nonsense-mediated decay of the mutant sequence and predicted absence of mutant protein products in affected cells. [2, 9-11]. Tumors from patients with CNC exhibit germline mutations and subsequent loss of heterozygosity at the PRKAR1A locus through a “second-hit” mutation, although haploinsufficiency of PRKAR1A may be sufficient for the early development of certain tumors [12, 13]. Additionally, in patients with germline PRKAR1A defects and relatively large adrenocortical adenomas that developed in the background of PPNAD, somatic defects in the beta-catenin gene (CTNNB1) have been identified in the tumors only [14]. Somatic mutations in PRKAR1A have been described in other cortisol-producing adrenocortical tumors, including cortisol-producing adrenocortical adenomas and adrenocortical carcinoma (ACC), with ACC also being described in 2 patients with CNC and c-PPNAD due to germline PRKAR1A defects [15, 16].
PPNAD has been described as early as in the first 5 years of life in patients with CNC, but more frequently manifests in the second and third decades of life, and after puberty, it is more common in females [2, 17]. Patients may present with overt, cyclical, atypical, or subclinical CS. Hypercortisolemia may be detected with the measurement of 24-hour urine free cortisol (UFC), late-night salivary cortisol, midnight serum cortisol and/or the performance of the 1-mg overnight dexamethasone suppression test or the 6-day modified Liddle test (LT) [5, 18]. During the LT, patients with PPNAD may have a paradoxical increase of more than 50% in 24-hour UFC on the second day of high-dose dexamethasone administration when compared to baseline [19]. Computed tomography (CT) of the adrenal glands may demonstrate subtle adrenal contour abnormality and bilateral micronodules, that are small, round, well-delineated, and hypodense, and are best visualized when the CT slices are 3 mm or less in thickness [20]. CS due to PPNAD is most commonly treated with bilateral adrenalectomy, although rarely medical treatment with ketoconazole or mitotane has also been used [21, 22].
Mosaicism describes the condition where an individual who arises from a single zygote has 2 or more populations of cells with different genotypes; it is caused by postzygotic de novo mutational events, although other mechanisms have also been postulated [23, 24]. The developmental timing and pathophysiological consequences of the mosaic mutation, as well as the cell lineage(s) affected, determine the pattern of distribution of this variant and whether it can be transmitted to offspring as a constitutional variant. Mosaicism may involve diploid germ cell precursors in the gonad (germline or gonadal mosaicism), non-germ cells (somatic mosaicism), or both (gonosomal mosaicism) [25]. The importance of mosaicism in human disease has become more evident with the introduction of increasingly sensitive technologies for the detection of genetic defects such as single-nucleotide polymorphism (SNP) microarrays and next-generation sequencing (NGS). Massively parallel NGS allows accurate detection of mutations in small subsets of cells that were not detected using Sanger sequencing methods.
We describe a case of PPNAD due to a mosaic PRKAR1A defect in a patient with CS who also had a left adrenocortical adenoma that developed in the background of hyperplasia. Our results demonstrated that there was very low-level mosaicism in DNA extracted from peripheral blood leukocytes for the PRKAR1A pathogenic variant c.682C > T which was also found in DNA extracted from the adrenocortical adenoma. The adenoma, however, also harbored an additional “second-hit” somatic c.974-2A > G PRKAR1A variant that has not been previously reported and is predicted to be pathogenic. This is the first time that a somatic, adrenal cortex–specific PRKAR1A defect arises in the context of PPNAD caused by a germline PRKAR1A defect, present in peripheral DNA albeit in the mosaic state.
Materials and Methods
Clinical Protocol
The patient was recruited by studies approved by the Institutional Review Boards of the National Institutes of Health (NIH) at the NIH Clinical Center. The patient signed an informed consent for all clinical and genetic studies and for the publication of this case.
Laboratory Studies
Peripheral and tumor DNA
DNA was extracted from 3 different aliquots of peripheral blood leukocytes, from 3 different aliquots of saliva and from 3 different 20- to 50-mg snap-frozen pieces of the adrenal adenoma using the DNeasy Blood and Tissue kit according to the manufacturer's protocol (Qiagen, Germantown, MD). Also, 3 nonrelated peripheral blood samples were run as experiment controls for normalization. The RNA from the adenoma was extracted using RNeasy Mini kit (Qiagen). Total RNA was reverse-transcribed into first-strand cDNA using SuperScript III Reverse Transcriptase (ThermoFisher, Waltham, MA) according to the manufacturer’s instructions.
Subsequently, 2 control groups were used for this study: (1) data from whole-exome sequencing performed on DNA extracted from lymphocytes from a cohort of 127 individuals without a family history of PPNAD (designated WES control group), and (2) data from PRKAR1A “deep” NGS performed on DNA extracted from paraffin blocks from a cohort of 74 individuals with ovarian cancer (designated FFPE control group). The complete PRKAR1A-coding and surrounding intronic sequence (canonical isoform NM_212471.2) of the blood DNA, saliva DNA, and the adrenal adenoma DNA was amplified using the conditions previously described [26]. The cDNA was used to identify possible alternative splice generated by the variant c.974-2A > G. The cDNA amplification was performed using the primers as follows: forward, 5′-TGCACTGCTTTAAGGAAATGTT-3′; reverse 5′-TTCGTTTGAGGATGTCTGAGC-3′. Each PCR product was amplified using BigDye Terminator V3.1 (Life Technologies, Grand Island, NY), purified using ZR DNA Sequencing Clean-up Kit (Zymo Research, Irvine, CA), and analyzed by classical bi-directional Sanger sequencing.
Mosaicism analysis by deep next-generation sequencing
To identify the presence of germline mosaicism for either of the variants, we amplified the specific regions with targeted flanking primers. Each primer was composed of 2 parts: (1) the 3′ end was complementary to the targeted genomic sequence; and (2) all primers had appended to their 5′ end either forward or reverse adaptor sequences to allow the addition of barcodes and Illumina sequencing adaptors. These appended sequences were: forward—5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG [locus-specific sequence]; and reverse—5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG [locus-specific sequence]. The locus-specific sequences used were: 974-2_F 5′-TGCACTGCTTTAAGGAAATGTT-3′ (forward), 974-2_R 5′-TTCGTTTGAGGATGTCTGAGC-3′ (reverse), 682_F 5′-GAGCAGCCACTGTCAAAGC-3′ (forward), 682_R 5′-AAGCTGGGCTTAATGCAAAGT-3′ (reverse). Amplicons generated from the sample DNA were then converted to sequenced libraries according to Illumina’s 16S Metagenomic Sequencing Library Preparation kit. Libraries were sequenced on an Illumina MiSeq using version 2 chemistry and generated 2 × 150-bp paired-end reads. Read pairs were trimmed with cutadapt, joined via “PEAR,” and aligned to expected amplicons with BWA. Variant frequencies were evaluated by counting bases at the variant position in aligned reads. Normalization was done using the wild-type (WT) sample result as “zero.” The experiment was done in technical triplicates and we tested 3 different samples from the nodule and 3 different DNA extractions for the germline DNA from the individual in the study. We also included a germline DNA sample from an unaffected individual (WT control).
In silico modeling
Somatic variants were evaluated by MutationTaster (mutationtaster.org) and Varsome (varsome.com) to predict the possible impact of the amino acid substitution on the structure and function of a human protein.
Results
Clinical Case
A 33-year-old woman presented with a 5-year history of progressive signs and symptoms suggestive of CS including fatigue, weight gain with central obesity, decreased concentration, irritability, insomnia, amenorrhea, facial rounding, alopecia, easy bruising, striae, and hypertension. She did not have a personal history of exogenous glucocorticoid use and family history was significant for hypertension, hyperlipidemia, obesity, coronary artery disease, and functional thyroid disease, but no endocrine or other tumors. Physical examination revealed stigmata of CS including bitemporal fat pads with facial rounding, dorsocervical and supraclavicular fat deposition, acne, lanugo, alopecia, wide though faint abdominal striae, central obesity with proximal muscle wasting, and Stage 2 hypertension (Fig. 1). Laboratory evaluation confirmed the diagnosis of CS with a midnight serum cortisol of 17.3 mcg/dL (<1.8; >7.5 indicative of hypercortisolemia), a 24-hour UFC of 196 mcg/24h (3.5-45), and 17% suppression of 24-hour UFC on day 6 of the LT when compared to baseline (>90% suppression). Plasma ACTH was undetectable (<5 pg/mL [5-46]) suggesting ACTH-independent CS and dehydroepiandrosterone sulfate (DHEAS) was <0.15 mcg/mL (0.35-4.30) (Table 1). Further laboratory evaluation showed impaired glucose tolerance, hypogonadotropic hypogonadism, and low bone mineral density. Abdominal CT demonstrated a 2 × 2 × 2.1-cm left adrenal adenoma, with a density of 33.4 Hounsfield units, and 66% absolute washout as well as bilateral micronodular adrenal hyperplasia (Fig. 2A, B). Fluorine-18 (18F)-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET)/CT showed an abnormally hypermetabolic left adrenal mass (Fig. 2C, 2D). Biochemical screening for primary aldosteronism and pheochromocytoma was negative (Table 1). The patient underwent left adrenalectomy with pathology consistent with PPNAD and a 1.5 × 1.3 × 1.3-cm oval yellowish-to-brown adenoma (Fig. 3).
Clinical Case Laboratory Evaluation
| Laboratory test | Result (reference range) |
|---|---|
| Serum midnight cortisol | 17.3 mcg/dL (<1.8; >7.5 indicative of hypercortisolemia) |
| 24-hour urine free cortisol | 196 mcg/24h (3.5-45) |
| Suppression in 24-hour urine free cortisol on day-6 of the Liddle test compared to baseline urine free cortisol | 17% (>90%) |
| ACTH | <5 pg/mL (5-46) |
| Plasma renin activity | 1.06 ng/mL/h (mean 1.9) |
| Plasma aldosterone concentration | 2 ng/dL (≤21) |
| 24-hour urine metanephrines | 101 mcg/24h (30-18 [normotensive]; <400 [hypertensive]) |
| 24-hour urine normetanephrine | 380 mcg/24h (111-419 [normotensive]; <900 [hypertensive]) |
| 24-hour urine total metanephrine | 481 mcg/24h (149-535 [normotensive]; <1300 [hypertensive]) |
| Laboratory test | Result (reference range) |
|---|---|
| Serum midnight cortisol | 17.3 mcg/dL (<1.8; >7.5 indicative of hypercortisolemia) |
| 24-hour urine free cortisol | 196 mcg/24h (3.5-45) |
| Suppression in 24-hour urine free cortisol on day-6 of the Liddle test compared to baseline urine free cortisol | 17% (>90%) |
| ACTH | <5 pg/mL (5-46) |
| Plasma renin activity | 1.06 ng/mL/h (mean 1.9) |
| Plasma aldosterone concentration | 2 ng/dL (≤21) |
| 24-hour urine metanephrines | 101 mcg/24h (30-18 [normotensive]; <400 [hypertensive]) |
| 24-hour urine normetanephrine | 380 mcg/24h (111-419 [normotensive]; <900 [hypertensive]) |
| 24-hour urine total metanephrine | 481 mcg/24h (149-535 [normotensive]; <1300 [hypertensive]) |
Clinical Case Laboratory Evaluation
| Laboratory test | Result (reference range) |
|---|---|
| Serum midnight cortisol | 17.3 mcg/dL (<1.8; >7.5 indicative of hypercortisolemia) |
| 24-hour urine free cortisol | 196 mcg/24h (3.5-45) |
| Suppression in 24-hour urine free cortisol on day-6 of the Liddle test compared to baseline urine free cortisol | 17% (>90%) |
| ACTH | <5 pg/mL (5-46) |
| Plasma renin activity | 1.06 ng/mL/h (mean 1.9) |
| Plasma aldosterone concentration | 2 ng/dL (≤21) |
| 24-hour urine metanephrines | 101 mcg/24h (30-18 [normotensive]; <400 [hypertensive]) |
| 24-hour urine normetanephrine | 380 mcg/24h (111-419 [normotensive]; <900 [hypertensive]) |
| 24-hour urine total metanephrine | 481 mcg/24h (149-535 [normotensive]; <1300 [hypertensive]) |
| Laboratory test | Result (reference range) |
|---|---|
| Serum midnight cortisol | 17.3 mcg/dL (<1.8; >7.5 indicative of hypercortisolemia) |
| 24-hour urine free cortisol | 196 mcg/24h (3.5-45) |
| Suppression in 24-hour urine free cortisol on day-6 of the Liddle test compared to baseline urine free cortisol | 17% (>90%) |
| ACTH | <5 pg/mL (5-46) |
| Plasma renin activity | 1.06 ng/mL/h (mean 1.9) |
| Plasma aldosterone concentration | 2 ng/dL (≤21) |
| 24-hour urine metanephrines | 101 mcg/24h (30-18 [normotensive]; <400 [hypertensive]) |
| 24-hour urine normetanephrine | 380 mcg/24h (111-419 [normotensive]; <900 [hypertensive]) |
| 24-hour urine total metanephrine | 481 mcg/24h (149-535 [normotensive]; <1300 [hypertensive]) |
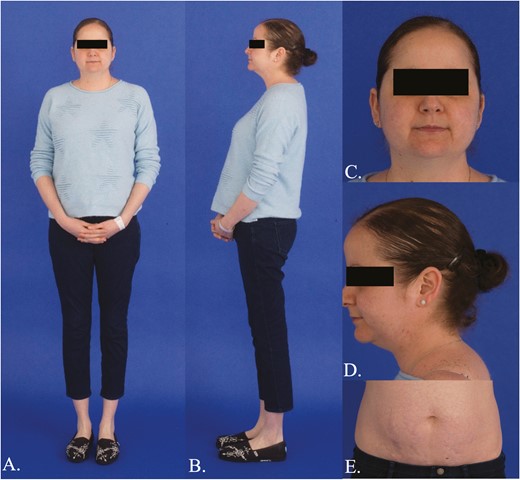
Clinical features of Cushing syndrome. A, B, Central obesity with proximal muscle wasting; C, Bitemporal fat pads, facial rounding, acne, lanugo; D, Dorsocervical and supraclavicular fat deposition, alopecia; E, Wide, though faint, abdominal striae.
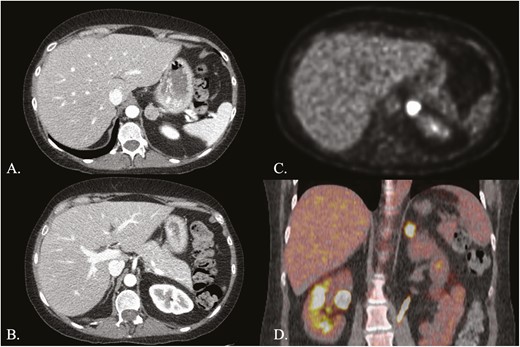
Adrenal imaging. A, Computed tomography (CT) of the adrenal glands with intravenous contrast showing left micronodular adrenal hyperplasia with an adenoma; B, CT adrenal glands with intravenous contrast demonstrating right micronodular adrenal hyperplasia; C, Axial view of 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET) showing an abnormally hypermetabolic left adrenal mass; D, Coronal view of 18F-FDG PET fused to CT without intravenous contrast, showing the hypermetabolic left adrenal mass.
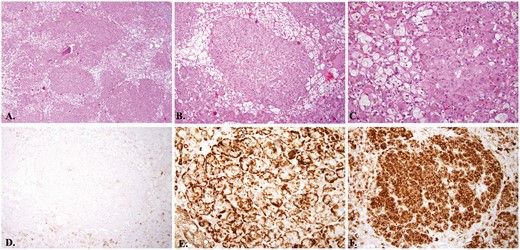
Left adrenal gland pathology consistent with primary pigmented nodular adrenocortical hyperplasia with a macronodule. A, Left adrenal gland. Scanning magnification view shows multiple well-circumscribed unencapsulated hyperplastic adrenocortical nodules composed of large epithelioid cells with abundant eosinophilic cytoplasm partially separated by adrenocortical cells with pale staining cytoplasm (hematoxylin and eosin staining, 4× original magnification); B, Left adrenal gland hematoxylin. Intermediate magnification view shows one of the hyperplastic nodules composed of large epithelioid cells with abundant eosinophilic cytoplasm and surrounding adrenocortical cells with pale staining cytoplasm (hematoxylin and eosin staining, 10× original magnification); C, Left adrenal gland. High magnification view revealed intracytoplasmic melanin pigments within large epithelioid cells of hyperplastic nodule (hematoxylin and eosin staining, 4× original magnification); D, Left adrenal gland. Lesional cells of the hyperplastic nodule are mostly negative for HMB-45. The adjacent adrenal cortical epithelium shows focal weak staining by HMB-45 immunohistochemistry, ×10 magnification; E, Left adrenal gland. Lesional cells of the hyperplastic nodule show strong cytoplasmic staining by melan-A immunohistochemistry, 10× original magnification; F, Left adrenal gland. Lesional cells of the hyperplastic nodule show strong and diffuse staining by synaptophysin immunohistochemistry, 10× original magnification.
Evaluation for other tumors associated with CNC was also performed: pituitary magnetic resonance imaging (MRI) demonstrated a pituitary microadenoma that was nonfunctional per biochemical evaluation and abdominal MRI demonstrated a 1-cm arterial enhancing hepatic lesion with differential diagnosis, including a hepatic adenoma or focal nodular hyperplasia. Echocardiogram, thyroid ultrasound, pelvic ultrasound, and MRI brain and spine did not reveal cardiac myxoma or additional tumors. Dermatological evaluation did not reveal any spotty skin pigmentation, cutaneous or mucosal myxomas, blue nevi, or epithelioid blue nevi. Postoperative ACTH-stimulation test was consistent with adrenal insufficiency with a serum cortisol of 2.0 and 2.9 mcg/dL at 30 and 60 minutes after administration of 0.25 mg of cosyntropin intravenously, respectively. The patient had significant clinical improvement on 6-month postoperative evaluation.
PRKAR1A DNA and RNA Splice Analysis
The patient was found to have 2 different pathogenic PRKAR1A (OMIM: 188830, NM_212471.2) variants that were present in the DNA extracted from the adrenocortical adenoma and absent in the DNA extracted from both blood and saliva: c.682C > T / p.Arg228Ter (rs281864784) and c.974-2A > G (previously unreported) (Fig. 4).
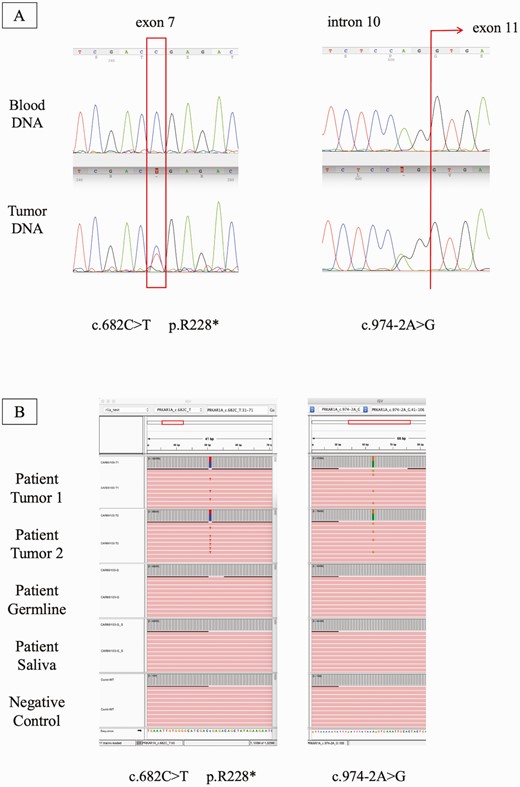
PRKAR1A DNA analysis. A, Electropherogram showing the 2 PRKAR1A variants found in the DNA extracted from the patient’s adrenocortical adenoma. Red rectangles show the location of the mutation; B, Integrative genome viewer (IGV) raw data analysis showing the presence of the variant in high frequency only at the tumor level.
In silico analysis using MutationTaster software predicted the variant c.974-2A > G as pathogenic. Varsome software performed analysis using 6 different tools and the pathogenic computational verdict was defined based on 6 pathogenic predictions vs no benign predictions. The variant is also classified as pathogenic according to the American College of Medical Genetics and Genomics (ACMG) guidelines [27].
The variant c.974-2A>G was further investigated to clarify if this defect led to the abolition of the regular exon 11 splice acceptor site or the creation of an alternative acceptor splice site. At the cDNA level it was identified that a new acceptor site was created before the regular one. The presence of the variant c.974-2A>G generated a new acceptor site 67 bases before the beginning of the normal exon 11 (Fig. 5A). Also, based on this new sequence, a new donor and acceptor site were created at the beginning of exon 11, creating a new exon 12 (Fig. 5B). In the 2 new isoforms generated by this variant there was a new stop codon positioned at what would be the start of the original exon 11.
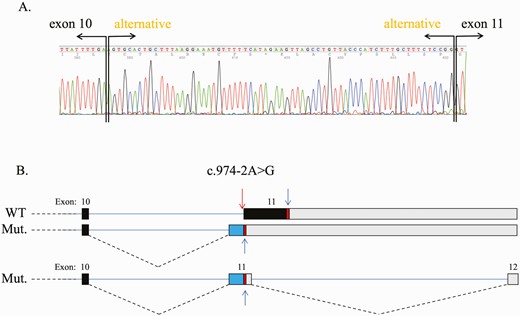
PRKAR1A RNA splice analysis. A, c.974-2A>G was identified at the cDNA level where a new acceptor site was created before the regular one. The presence of this change generated a new acceptor site 67 bases before the beginning of the normal exon 11; B, This new sequence created a new exon 12.
Based on the cDNA analysis it could be implied that both variants are not expressed into protein due to nonsense-mediated decay, like most PPNAD mutations leading to a frame shift, since the cDNA sequence showed just 1 WT allele at this position. Additionally, the 2 variants identified in this patient are in trans, as the variant c.682C>T was not found in the RNA analysis where the alternative splice site was noted. This c.682C>T absence not only indicates that the variants are in trans but also supports that nonsense-mediated decay is triggered due to the presence of the premature stop codon, possibly leading to mRNA degradation.
Considering the presence of both variants in the DNA extracted from the adrenocortical tumor and no detectable changes in the genomic DNA through standard Sanger sequencing, we sought to determine if the presence of one of the variants could be due to low-level mosaicism for a PRKAR1A defect. We performed “deep” NGS of DNA from tumor tissue, saliva, and peripheral blood leukocytes. For the variant c.682C>T the frequency of the mutant allele in the sample that was tested from the adenoma was approximately 43%, while in peripheral blood leukocytes and saliva DNA, we found an average frequency of 0.31% (ranging from 0.0025 to 0.0037) after normalization with control samples (original frequencies). For the variant c.974-2A>G, the frequency of the mutant allele at the tumor level was approximately 41% and its frequency in the peripheral blood leukocytes was between 0.01% and 0.05%. These results suggested the presence of very low levels of c.682C>T PRKAR1A mosaicism in this patient. The average number of reads per sample was 614 016. The control group analysis was performed to identify the frequency of false positives. In the WES control group, the average frequency of this variant was 0.08% (ranging from 0.000 to 0.0012 in a total of 24 638 reads). In the FFPE control group, the average frequency of this variant was 0.1% (ranging from 0.000 to 0.0019 in a total of 2 442 638 reads). FFPE samples are more prone to cytosine deamination, which may have led to the higher frequency of false positive results in this group.
Discussion
To our knowledge, this is the first report of PPNAD due to mosaicism for a PRKAR1A defect supported by in vitro data demonstrating low-level mosaicism for this defect in the affected individual; in addition, complete loss of PRKAR1A in the adrenocortical adenoma of this patient was caused by a second, adrenal-only somatic PRKAR1A defect on the other allele. Previously, a case of 2 siblings with CNC due to an apparently de novo large germline deletion at 17q24.2 that included the PRKAR1A gene was reported, with 1 sibling manifesting with PPNAD among other tumors. Both asymptomatic parents were not found to have the deletion on comparative genomic hybridization array and it was assumed that one of the parents had germ cell mosaicism for the 17q24.2 deletion [28].
The previously unreported c.974-2A>G somatic PRKAR1A variant which was found only in the adenoma is predicted to be pathogenic by in silico analysis, with individuals from our cohort with germline inactivating defects affecting the same PRKAR1A exon presenting with CNC, although functional studies could further support our finding. This is the second time that any PRKAR1A second defect has been identified as a means of complete inactivation of the PRKAR1A gene in any tissue of a patient with CNC: a “second-hit” somatic mutation was described previously in DNA sequenced from a growth hormone–producing pituitary adenoma in a patient with CNC due to a large germline PRKAR1A deletion [29]. Somatic CTNNB1 mutations, not PRKAR1A defects, have previously been reported in relatively large adrenocortical adenomas that developed in the background of PPNAD caused by germline PRKAR1A defects [14, 30].
Mosaicism has been implicated in the development of multiple tumor syndromes, including syndromes with endocrine neoplasms such as Von Hippel-Lindau disease (due to mutations in the VHL tumor suppressor gene); McCune-Albright syndrome (due to activating defects of the GNAS gene leading to aberrant cAMP–PKA signaling), which manifests only as mosaicism and is associated with CS due to a form of bilateral macronodular adrenocortical hyperplasia; and the syndrome of paraganglioma and somatostatinoma associated with polycythemia (due to gain-of-function somatic pathogenic variants in the HIF2A gene that encodes a hypoxia-inducible transcription factor) [31-35]. The identification of mosaicism for causative variants of tumor predisposition syndromes has significant implications for genetic counseling for patients and their families as well as for tumor surveillance which may affect the prognosis of the respective disorders.
Given that the extent of PRKAR1A mosaicism in our patient and the tissues affected are unknown, the patient was evaluated for other manifestations of CNC in addition to PPNAD, to determine if she met diagnostic criteria for CNC, and surveillance was performed to identify possible additional tumors. The diagnosis of CNC is made when 2 major manifestations or criteria, verified by histological evaluation, biochemical testing, or imaging, are present, or when 1 major manifestation or criterion is present and the patient has an inactivating mutation in the PRKAR1A gene or an affected first-degree relative (Table 2) [6]. Manifestations of CNC develop over years and may be evident as early as the neonatal period, with peak penetrance in young adulthood. While most patients with CNC, particularly those involved in active surveillance, have a normal lifespan, early death in these patients may be a consequence of complications of cardiac myxomas, metastatic or intracranial psammomatous melanotic schwannomas, thyroid carcinoma, and metastatic pancreatic and testicular tumors [6]. Clinical surveillance for early detection of manifestations of CNC differs according to age group. In prepubertal children, evaluation with echocardiogram annually, or biannually for patients with a prior history of excised cardiac myxoma, should begin at 6 months of age, monitoring of the growth rate and annual pubertal staging is recommended to detect abnormalities that may indicate hormone excess, and boys should undergo annual testicular ultrasonography for the detection of large-cell calcifying Sertoli cell tumors. Postpubertal pediatric patients and adult patients should have at least annual echocardiograms for the detection of cardiac myxomas and annual thyroid ultrasound and measurement of 24-hour UFC and serum IGF-1. Transabdominal pelvic ultrasound is recommended during initial evaluation for women with CNC and should be repeated if an abnormality is detected. Additional evaluation for all age groups may include midnight cortisol, the LT, and adrenal CT for those suspected of having PPNAD, a 3-hour oral glucose tolerance test and MRI of the pituitary for patients suspected of having acromegaly or gigantism, and MRI of the brain, spine, chest, abdomen, retroperitoneum, and/or pelvis for evaluation of possible psammomatous melanotic schwannomas [36]. Our patient was evaluated according to these surveillance recommendations under our research protocol and was found to have a nonfunctional pituitary microadenoma and a benign-appearing hepatic lesion. A higher rate of liver lesions on imaging has been noted in patients with CNC, particularly those with PRKAR1A defects, although liver abnormalities are not currently included in the diagnostic criteria for CNC [37]. At the time of evaluation, our patient did not meet the current diagnostic criteria for CNC as she had only 1 major manifestation of CNC (PPNAD) and mosaicism for a PRKAR1A defect is not included in the supplemental molecular criteria. It is unclear if PRKAR1A mosaicism may comprise its own independent clinical entity or whether this genetic abnormality is a cause of CNC. Long-term monitoring may determine if she develops manifestations that meet additional major criteria for the disease and she should be monitored for the development of hypercortisolemia in the future given the micronodular appearance of her right adrenal gland on imaging as well as for hormone hypersecretion from the anterior pituitary gland.
Diagnostic Criteria for Carney Complex*
| Major criteria: |
|---|
| 1. Spotty skin pigmentation with a typical distribution (lips, conjunctiva and inner or outer canthi, vaginal and penile mucosa) |
| 2. Myxoma (cutaneous and mucosal)† |
| 3. Cardiac myxoma† |
| 4. Breast myxomatosis† or fat-suppressed magnetic resonance imaging findings suggestive of this diagnosis |
| 5. PPNAD† or paradoxical positive response of urinary glucocorticosteroids to dexamethasone administration during Liddle test |
| 6. Acromegaly due to GH-producing adenoma† |
| 7. Large-cell calcifying Sertoli cell tumor† or characteristic calcification on testicular ultrasonography |
| 8. Thyroid carcinoma† or multiple, hypoechoic nodules on thyroid ultrasonography |
| 9. Psammomatous melanotic schwannoma† |
| 10. Blue nevus, epithelioid blue nevus (multiple)† |
| 11. Breast ductal adenoma (multiple)† |
| 12. Osteochondromyxoma of bone† |
| Supplemental criteria: |
| 1. Affected first-degree relative |
| 2. Inactivating mutation of the PRKAR1A gene |
| Major criteria: |
|---|
| 1. Spotty skin pigmentation with a typical distribution (lips, conjunctiva and inner or outer canthi, vaginal and penile mucosa) |
| 2. Myxoma (cutaneous and mucosal)† |
| 3. Cardiac myxoma† |
| 4. Breast myxomatosis† or fat-suppressed magnetic resonance imaging findings suggestive of this diagnosis |
| 5. PPNAD† or paradoxical positive response of urinary glucocorticosteroids to dexamethasone administration during Liddle test |
| 6. Acromegaly due to GH-producing adenoma† |
| 7. Large-cell calcifying Sertoli cell tumor† or characteristic calcification on testicular ultrasonography |
| 8. Thyroid carcinoma† or multiple, hypoechoic nodules on thyroid ultrasonography |
| 9. Psammomatous melanotic schwannoma† |
| 10. Blue nevus, epithelioid blue nevus (multiple)† |
| 11. Breast ductal adenoma (multiple)† |
| 12. Osteochondromyxoma of bone† |
| Supplemental criteria: |
| 1. Affected first-degree relative |
| 2. Inactivating mutation of the PRKAR1A gene |
*To make a diagnosis of Carney complex, a patient must either: (A) exhibit 2 of the major criteria of the disease listed above, or (B) exhibit 1 of the major criteria and meet 1 of the supplemental criteria (an affected first-degree relative or an inactivating mutation of the PRKAR1A gene).
†with histologic confirmation
Diagnostic Criteria for Carney Complex*
| Major criteria: |
|---|
| 1. Spotty skin pigmentation with a typical distribution (lips, conjunctiva and inner or outer canthi, vaginal and penile mucosa) |
| 2. Myxoma (cutaneous and mucosal)† |
| 3. Cardiac myxoma† |
| 4. Breast myxomatosis† or fat-suppressed magnetic resonance imaging findings suggestive of this diagnosis |
| 5. PPNAD† or paradoxical positive response of urinary glucocorticosteroids to dexamethasone administration during Liddle test |
| 6. Acromegaly due to GH-producing adenoma† |
| 7. Large-cell calcifying Sertoli cell tumor† or characteristic calcification on testicular ultrasonography |
| 8. Thyroid carcinoma† or multiple, hypoechoic nodules on thyroid ultrasonography |
| 9. Psammomatous melanotic schwannoma† |
| 10. Blue nevus, epithelioid blue nevus (multiple)† |
| 11. Breast ductal adenoma (multiple)† |
| 12. Osteochondromyxoma of bone† |
| Supplemental criteria: |
| 1. Affected first-degree relative |
| 2. Inactivating mutation of the PRKAR1A gene |
| Major criteria: |
|---|
| 1. Spotty skin pigmentation with a typical distribution (lips, conjunctiva and inner or outer canthi, vaginal and penile mucosa) |
| 2. Myxoma (cutaneous and mucosal)† |
| 3. Cardiac myxoma† |
| 4. Breast myxomatosis† or fat-suppressed magnetic resonance imaging findings suggestive of this diagnosis |
| 5. PPNAD† or paradoxical positive response of urinary glucocorticosteroids to dexamethasone administration during Liddle test |
| 6. Acromegaly due to GH-producing adenoma† |
| 7. Large-cell calcifying Sertoli cell tumor† or characteristic calcification on testicular ultrasonography |
| 8. Thyroid carcinoma† or multiple, hypoechoic nodules on thyroid ultrasonography |
| 9. Psammomatous melanotic schwannoma† |
| 10. Blue nevus, epithelioid blue nevus (multiple)† |
| 11. Breast ductal adenoma (multiple)† |
| 12. Osteochondromyxoma of bone† |
| Supplemental criteria: |
| 1. Affected first-degree relative |
| 2. Inactivating mutation of the PRKAR1A gene |
*To make a diagnosis of Carney complex, a patient must either: (A) exhibit 2 of the major criteria of the disease listed above, or (B) exhibit 1 of the major criteria and meet 1 of the supplemental criteria (an affected first-degree relative or an inactivating mutation of the PRKAR1A gene).
†with histologic confirmation
Additionally the patient may benefit from periodic surveillance for complications of CNC, particularly those that may lead to excess morbidity or mortality, and was counseled on the possibility of transmission of the PRKAR1A defect to offspring if germ cells are affected. Screening of offspring for the identified c.682C>T PRKAR1A variant in the germline may be considered to determine if surveillance may be beneficial. The full clinical phenotype of PRKAR1A mosaicism, as well as the appropriate frequency of surveillance for the development of tumors or hormone excess, and the risk of transmission of the PRKAR1A defect to offspring are yet to be determined. Lastly, consideration should be given for evaluation for low-level PRKAR1A mosaicism with sensitive techniques such as NGS in patients manifesting with isolated tumors associated with CNC and PRKAR1A loss of heterozygosity in DNA from tumor tissue when sequencing of DNA extracted from “normal” tissues is unrevealing, as there are potential clinical and genetic implications.
Abbreviations
- ACC
adrenocortical carcinoma
- ACTH
adrenocorticotropic hormone
- CNC
Carney complex
- CS
Cushing syndrome
- CT
computed tomography
- 18F-FDG
18F-fluorodeoxyglucose
- LT
Liddle test
- MRI
magnetic resonance imaging
- NGS
next-generation sequencing
- PET
positron emission tomography
- PKA
protein kinase A
- PPNAD
primary pigmented nodular adrenocortical disease
- UFC
urine free cortisol
- WT
wild-type
Acknowledgments
We thank our patient for participating in our study as well as Drs. Elena Belyavskaya and Charalampos Lyssikatos and Lola Saidkhodjaeva for coordinating the care of this patient (and other patients), and our endocrinology fellows and nursing staff participating in the care of this patient at the National Institutes of Health Clinical Center. The authors also would like to thank the NICHD’s IRP Molecular Genomics Core (Dr. Steven Coon, Dr. James Iben, and Dr. Li Tianwei) for its support.
Financial Support: This work was supported by the research project Z01-HD008920 (Principal Investigator: Constantine A. Stratakis) of the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health (NIH), Bethesda, Maryland, USA.
Additional Information
Disclosures: Constantine A. Stratakis holds patents on the PRKAR1A, PDE11A, and GPR101 genes and/or their function and has received research funding from Pfizer Inc. on the genetics and treatment of abnormalities of growth hormone secretion. Fabio R. Faucz holds a patent on the GPR101 gene and/or its function. The other authors have nothing to disclose.
Data Availability
All data generated during this article will be available at database of Genotypes and Phenotypes (dbGaP) (https://www.ncbi.nlm.nih.gov/gap/). The accession code will be available by request.
References
Author notes
Co-first authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}