





Abstract
Constitutional delay of puberty (CDP) is highly heritable, but the genetic basis for CDP is largely unknown. Idiopathic hypogonadotropic hypogonadism (IHH) can be caused by rare genetic variants, but in about half of cases, no rare-variant cause is found.
To determine whether common genetic variants that influence pubertal timing contribute to CDP and IHH.
Case-control study.
80 individuals with CDP; 301 with normosmic IHH, and 348 with Kallmann syndrome (KS); control genotyping data from unrelated studies.
Polygenic scores (PGS) based on genome-wide association studies for timing of male pubertal hallmarks and age at menarche (AAM).
The CDP cohort had higher PGS for male pubertal hallmarks and for AAM compared to controls (for male hallmarks, Cohen's d = 0.67, P = 1 × 10−10; for AAM, d = 0.85, P = 1 × 10−16). The normosmic IHH cohort also had higher PGS for male hallmarks compared to controls, but the difference was smaller (male hallmarks d = 0.20, P = .003; AAM d = 0.10, P = .055). No differences were seen for the KS cohort compared to controls (male hallmarks d = 0.05, P = .45; AAM d = 0.03, P = .56).
Common genetic variants that influence pubertal timing in the general population contribute strongly to the genetics of CDP, weakly to normosmic IHH, and potentially not at all to KS. These findings demonstrate that the common-variant genetics of CDP and normosmic IHH are largely but not entirely distinct.
A patient presenting with delayed puberty with no cause identified on routine clinical evaluation may have constitutional delay of puberty (CDP) or idiopathic hypogonadotropic hypogonadism (IHH). Though these 2 conditions may be indistinguishable on initial presentation, CDP and IHH differ in prognosis and underlying physiology. CDP is a common, self-limited condition of unclear etiology and is often considered an extreme of normal variation in pubertal timing. In contrast, IHH is a rare, pathological, and longer lasting (usually lifelong) condition caused by impaired GnRH secretion or action. Individuals with IHH are classified as having Kallmann syndrome (KS) if they also have anosmia or hyposmia and as having normosmic IHH if the sense of smell is unaffected.
Approximately half of individuals with IHH have an identifiable genetic cause, with a rare genetic variant in one (or more) of over 40 genes associated with IHH (1). In contrast, the causes of CDP are less clear. While some cases of CDP appear to be due to rare variants in CDP-associated genes (some of which are also associated with IHH), most individuals with CDP do not carry such rare genetic variants (2-8). Nevertheless, CDP appears to be highly heritable, as 50% to 75% of individuals with CDP have a family member also affected by CDP (9). These findings prompted us to test the hypothesis that common genetic variants contribute to CDP as well as IHH.
Genome-wide association studies (GWAS) of age at voice-breaking and age at facial-hair growth (male pubertal hallmarks) and age at menarche (AAM; a female pubertal hallmark) in European cohorts have collectively identified more than 400 independent loci associated with pubertal timing at genome-wide significance (10, 11). The genome-wide significant loci for AAM account for ∼7.5% of the variation in menarchal timing. The identification of these common genetic variants allows the use of polygenic scores (PGS) to estimate the effects of common genetic variants on pubertal timing.
It has been long recognized that pubertal timing and the prevalence of CDP and IHH vary by sex. As a group, girls enter puberty earlier than boys, IHH is more common in males than in females, and boys are more likely than girls to be diagnosed with CDP (though referral bias contributes to this difference) (12-15). Most of the loci identified in the GWAS on pubertal timing have similar effects in females and males; however, some display differential effects (10, 11). This finding suggests that many of the mechanisms that determine pubertal timing are shared between females and males, but some mechanisms may differ.
In this study, we evaluate the contribution of these common genetic variants to CDP and IHH. For cohorts of patients with CDP, normosmic IHH, and KS, we calculated PGS based on the largest published GWAS meta-analyses of male pubertal hallmarks and of AAM, then compared these PGS between cases and controls in both sex-adjusted and sex-stratified analyses. We further analyzed how common genetic variants may work with or independently of rare genetic variants to contribute to the risk for normosmic IHH.
Methods
CDP and IHH Cohorts
Individuals with CDP were recruited at Boston Children's Hospital and Massachusetts General Hospital, Boston, Massachusetts, USA, Rothschild Hospital, Paris, France, the National Institutes of Health Clinical Center, Bethesda, Maryland, USA, and Hospital de Niños Ricardo Gutiérrez, Buenos Aires, Argentina. Individuals with IHH were enrolled in a longstanding genetics study at Massachusetts General Hospital (ClinicalTrials.gov no. NCT00494169; registered June 29, 2007). All protocols were approved by the institution's local ethics committee. All participants gave written informed consent or assent, and for participants under 18 years, a parent or guardian gave consent.
Criteria for delayed puberty, CDP, normosmic IHH, and KS are detailed in the Supplementary Materials (16). Briefly, individuals were classified as having delayed puberty if the SD score for breast development or testicular volume was −2.25 or less [based on nomograms from refs (17, 18)] or if serum testosterone was less than the 5th percentile for age [based on data from refs (19-21)]. Individuals with identifiable potential causes of delayed puberty were excluded. For this study, individuals with delayed puberty who exhibited pubertal onset before age 18 years and who did not later meet criteria for IHH were included in the CDP cohort. IHH was defined as age 18 years or older, serum sex-steroid concentrations below the adult reference range, and serum gonadotropin concentrations within or below the adult reference range (22). Individuals with subtypes of IHH associated with significant nonreproductive phenotypes aside from anosmia (eg, arhinia, ataxia, CHARGE syndrome, adrenal hypoplasia congenita, combined pituitary hormone deficiency) were excluded, as these syndromes may have unique pathophysiological mechanisms.
Genotyping of Common Genetic Variants
Participant DNA was extracted from saliva and blood samples. For the CDP cohort, genotyping of single-nucleotide polymorphisms (SNPs) was performed at the Broad Institute using the Illumina Infinium OmniExpress Exome 8-1v6 (n = 104) and Illumina HumanOmniExpress-12-v1 (n = 88) arrays. For the IHH cohort, genotyping of SNPs was performed at the Broad Institute using the Infinium® PsychArray v1.1 and B. Quality control and imputation were performed for all samples using standard methods [details in Supplementary Materials (16)].
Control datasets were selected to match the platforms used to genotype cases; thus, the CDP and IHH cohorts had different control groups. Control data for analysis of CDP genotypes were downloaded from dbGaP (Projects phs001315.v1.p1 and phs000963.v1.p1); the data were generated using the Illumina Infinium OmniExpress Exome 8-1v2 array on samples from healthy volunteers for the COLON and PGRN-RIKEN studies [4469 and 5891 individuals, respectively; further details are in the Supplementary Methods (16)]. Control data for analysis of IHH genotypes, shared with permission by the Schizophrenia Psychiatric GWAS Consortium (n = 1868), were genotyped on the Infinium® PsychArray (23).
Rare-variant Subgroups of the IHH Cohort
For the IHH cohort, rare variants in genes with strong evidence of association with IHH [listed in Supplementary Table S1 (16)] were identified through targeted gene sequencing and/or exome sequencing as described previously (24). Individuals with heterozygous rare (minor allele frequency <0.1%) variants in genes associated with autosomal dominant inheritance (eg, FGFR1), with 2 or more rare variants in genes associated with autosomal recessive inheritance (eg, GNRHR), and with hemizygous rare variants in X-linked recessive genes (eg, ANOS1) were classified as having an apparent rare-variant cause. Remaining individuals were classified as having no apparent rare-variant cause.
Calculation and Comparison of PGS
Summary statistics for the following GWAS were used to calculate PGS: multitrait GWAS for male pubertal hallmarks (top 10 000 SNPs only) (11) and GWAS for AAM (10) and, as negative controls, GWAS for basal-cell carcinoma (25) and GWAS for anal carcinoma (26). Methods for calculating and comparing polygenic scores are detailed in the Supplementary Methods (16). Briefly, for each set of GWAS summary statistics, PRS-CS (27) was used to adjust SNP effect sizes based on the strength of associations and for linkage disequilibrium. PRSice-2 (28) was then used to calculate PGS based on these modified effect sizes and imputed genotypes; resulting scores were normalized to a mean of 0 and SD of 1 in control data. PRSice-2 was also used to compare PGS between cases and controls for CDP, normosmic IHH, and KS, with the first 10 genetic principal components and sex as covariates. Differences between groups were expressed using Cohen's d, with 0.2 or less interpreted as a small difference, greater than 0.2 to less than 0.8 as a moderate difference, and 0.8 or more as a large difference; P-values <.05 were considered statistically significant. We also conducted sex-stratified analyses and, for the normosmic IHH cohort, analyses based on rare-variant subgroups. Power calculations were performed using the PS program and used a significance threshold (α) of .05 and power of 80%.
Results
To determine whether CDP, normosmic IHH, and/or KS can be caused by the effects of common genetic variants, we compared PGS for pubertal timing—age at male pubertal hallmarks (voice-breaking and facial hair) (11) and AAM (10)—between cases and controls for CDP, normosmic IHH, and KS. We recruited 80 individuals with CDP (26 female, 54 male), 301 individuals with normosmic IHH (97 female, 204 male), and 348 individuals with KS (85 female, 263 male). We accessed data for 9222 controls (4171 female, 5450 male) genotyped on the same platform as the CDP cohort and 1868 controls (1032 female, 836 male) genotyped on the same platform as the normosmic IHH and KS cohorts (Table 1). We also calculated PGS for basal-cell carcinoma and for anal carcinoma, conditions unrelated to pubertal timing, to ensure that there were no systematic differences in genotype data between our cases and controls; these “negative control” PGS did not differ significantly between the CDP, normosmic IHH, and KS cohorts and the corresponding control cohorts [P all >.05; Supplementary Table S2 (16)].
Composition of cohorts
| Cohort | Females | Males | Total |
|---|---|---|---|
| CDP cases | 26 | 54 | 80 |
| Controls for CDP analysis | 4171 | 5450 | 9621 |
| Normosmic IHH | 97 | 204 | 301 |
| Kallmann syndrome | 85 | 263 | 348 |
| Controls for normosmic IHH and Kallmann syndrome analyses | 836 | 1032 | 1868 |
| Cohort | Females | Males | Total |
|---|---|---|---|
| CDP cases | 26 | 54 | 80 |
| Controls for CDP analysis | 4171 | 5450 | 9621 |
| Normosmic IHH | 97 | 204 | 301 |
| Kallmann syndrome | 85 | 263 | 348 |
| Controls for normosmic IHH and Kallmann syndrome analyses | 836 | 1032 | 1868 |
Abbreviations: CDP, constitutional delay of puberty; IHH, idiopathic hypogonadotropic hypogonadism.
Composition of cohorts
| Cohort | Females | Males | Total |
|---|---|---|---|
| CDP cases | 26 | 54 | 80 |
| Controls for CDP analysis | 4171 | 5450 | 9621 |
| Normosmic IHH | 97 | 204 | 301 |
| Kallmann syndrome | 85 | 263 | 348 |
| Controls for normosmic IHH and Kallmann syndrome analyses | 836 | 1032 | 1868 |
| Cohort | Females | Males | Total |
|---|---|---|---|
| CDP cases | 26 | 54 | 80 |
| Controls for CDP analysis | 4171 | 5450 | 9621 |
| Normosmic IHH | 97 | 204 | 301 |
| Kallmann syndrome | 85 | 263 | 348 |
| Controls for normosmic IHH and Kallmann syndrome analyses | 836 | 1032 | 1868 |
Abbreviations: CDP, constitutional delay of puberty; IHH, idiopathic hypogonadotropic hypogonadism.
For male pubertal hallmarks and for AAM, CDP cases had higher PGS compared to controls, and the differences were moderate to large (male pubertal hallmarks Cohen's d = 0.67, P = 1 × 10−10; AAM d = 0.86, P = 1 × 10−16; Fig. 1). Sex-stratified analyses revealed comparable results for both females and males with CDP (Table 2).
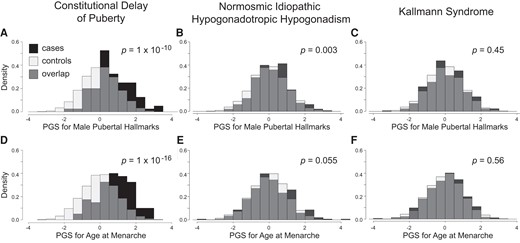
Contribution of common genetic variants to CDP and IHH. PGS for male pubertal hallmarks were significantly higher in CDP cases compared to controls (A), as well as in nIHH cases, though to a lesser extent (B), but not in KS cases (C). For age at menarche, CDP cases similarly had significantly higher PGS compared to controls (D), nIHH cases had nominally higher PGS (E), and again KS cases did not (F). Comparisons were adjusted for sex and the first 10 genetic principal components. PGS are expressed as normalized scores with a mean of 0 and a SD of 1. Density is the proportion of the cohort with PGS within the range indicated by the width of each bar.
Sex-stratified comparisons of polygenic scores for pubertal timing
| Cohort | Sex | Polygenic score | Cohen's d | P |
|---|---|---|---|---|
| CDP | Female | Age at male pubertal hallmarks | 0.73 | 1 × 10−4 |
| Age at menarche | 1.16 | 8 × 10−10 | ||
| Male | Age at male pubertal hallmarks | 0.66 | 2 × 10−7 | |
| Age at menarche | 0.71 | 4 × 10−9 | ||
| nIHH | Female | Age at male pubertal hallmarks | 0.19 | 0.21 |
| Age at menarche | −0.02 | 0.97 | ||
| Male | Age at male pubertal hallmarks | 0.20 | 5 × 10−3 | |
| Age at menarche | 0.16 | 8 × 10−3 |
| Cohort | Sex | Polygenic score | Cohen's d | P |
|---|---|---|---|---|
| CDP | Female | Age at male pubertal hallmarks | 0.73 | 1 × 10−4 |
| Age at menarche | 1.16 | 8 × 10−10 | ||
| Male | Age at male pubertal hallmarks | 0.66 | 2 × 10−7 | |
| Age at menarche | 0.71 | 4 × 10−9 | ||
| nIHH | Female | Age at male pubertal hallmarks | 0.19 | 0.21 |
| Age at menarche | −0.02 | 0.97 | ||
| Male | Age at male pubertal hallmarks | 0.20 | 5 × 10−3 | |
| Age at menarche | 0.16 | 8 × 10−3 |
Cohen's d of 0.2 or less was interpreted as a small difference, greater than 0.2 to less than 0.8 as a moderate difference, and 0.8 or more as a large difference.
Abbreviations: CDP, constitutional delay of puberty; nIHH, normosmic idiopathic hypogonadotropic hypogonadism.
Sex-stratified comparisons of polygenic scores for pubertal timing
| Cohort | Sex | Polygenic score | Cohen's d | P |
|---|---|---|---|---|
| CDP | Female | Age at male pubertal hallmarks | 0.73 | 1 × 10−4 |
| Age at menarche | 1.16 | 8 × 10−10 | ||
| Male | Age at male pubertal hallmarks | 0.66 | 2 × 10−7 | |
| Age at menarche | 0.71 | 4 × 10−9 | ||
| nIHH | Female | Age at male pubertal hallmarks | 0.19 | 0.21 |
| Age at menarche | −0.02 | 0.97 | ||
| Male | Age at male pubertal hallmarks | 0.20 | 5 × 10−3 | |
| Age at menarche | 0.16 | 8 × 10−3 |
| Cohort | Sex | Polygenic score | Cohen's d | P |
|---|---|---|---|---|
| CDP | Female | Age at male pubertal hallmarks | 0.73 | 1 × 10−4 |
| Age at menarche | 1.16 | 8 × 10−10 | ||
| Male | Age at male pubertal hallmarks | 0.66 | 2 × 10−7 | |
| Age at menarche | 0.71 | 4 × 10−9 | ||
| nIHH | Female | Age at male pubertal hallmarks | 0.19 | 0.21 |
| Age at menarche | −0.02 | 0.97 | ||
| Male | Age at male pubertal hallmarks | 0.20 | 5 × 10−3 | |
| Age at menarche | 0.16 | 8 × 10−3 |
Cohen's d of 0.2 or less was interpreted as a small difference, greater than 0.2 to less than 0.8 as a moderate difference, and 0.8 or more as a large difference.
Abbreviations: CDP, constitutional delay of puberty; nIHH, normosmic idiopathic hypogonadotropic hypogonadism.
The normosmic IHH cohort also had higher PGS for male pubertal hallmarks compared to controls, but the differences were small (male pubertal hallmarks d = 0.20, P = .003; AAM d = 0.10, P = .055; Fig. 1). Sex-stratified analyses demonstrated differences in males with normosmic IHH (male pubertal hallmarks d = 0.20, P = .005; AAM d = 0.16, P = .008) but not in females with normosmic IHH (male pubertal hallmarks d = 0.19, P = .21; AAM d = −0.03, P = .97; Table 2). No differences were seen between the KS cohort and controls (male pubertal hallmarks d = 0.05, P = .45; AAM d = 0.03, P = .56; Fig. 1); a post hoc power calculation showed that the cohort provided 80% statistical power to detect a difference of small magnitude [0.16 SD (16)].
We further examined a subcohort of 25 individuals with reversal of IHH, that is, who received a diagnosis of IHH but later achieved normal levels of reproductive endocrine function, a condition that may represent an extreme form of CDP, at least in some cases. We found no significant differences in PGS for this subcohort compared to controls (male pubertal hallmarks d = 0.09, P = .77; AAM d = 0.21, P = .3), but a post hoc power analysis revealed that we had less than 20% power to detect a statistically significant difference of this magnitude.
We next sought to determine whether common genetic variants may contribute to normosmic IHH independently of rare genetic variants or, alternatively or in addition, in conjunction with rare genetic variants. To test these possibilities, we further subdivided the normosmic IHH cohort into those who had an apparent rare-variant cause of normosmic IHH previously identified through exome or targeted-gene sequencing (n = 70) and those who did not (n = 231). The subcohort of those with an apparent rare-variant cause for normosmic IHH had no significant difference in PGS for pubertal timing compared to controls (male pubertal hallmarks d = 0.20, P = .15; AAM d = 0.03, P = .80; Fig. 2), but this subcohort provided sufficient power to detect only moderate size changes or larger (d > 0.34). In contrast, the subcohort without an apparent rare-variant cause showed higher PGS than controls (male pubertal hallmarks d = 0.20, P = .009; AAM d = 0.13, P = .04; Fig. 2).
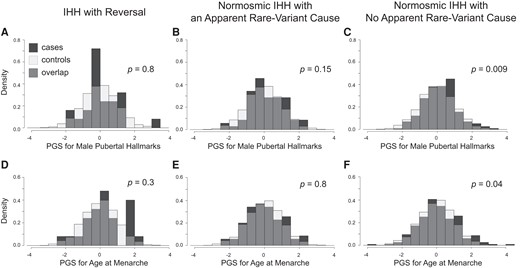
PGS for pubertal timing in IHH subcohorts. PGS for male pubertal hallmarks were not different between cases and controls for IHH with reversal (A) or for normosmic IHH with an apparent rare-variant cause (B) but were significantly higher for normosmic IHH with no apparent rare-variant cause (C). Similar results were found for PGS for age at menarche (D-F). Comparisons were adjusted for sex and the first 10 genetic principal components. PGS are expressed as normalized scores with a mean of 0 and a SD of 1. Density is the proportion of the cohort with PGS within the range indicated by the width of each bar.
Discussion
In this study of the contribution of common genetic variants to CDP and IHH, we found strong enrichment of puberty-delaying variants in both females and males with CDP (Cohen's d 0.66-1.16) and slight enrichment in males with normosmic IHH (Cohen's d 0.16-0.20) but no significant enrichment in individuals with KS. We further found that the enrichment in normosmic IHH was specifically present in those with no apparent rare-variant cause of IHH.
CDP has long been recognized to be highly heritable, but rare genetic variants appear to account for only a small proportion of CDP (2-8). Our results suggest that CDP can be caused by the cumulative effect of common genetic variants that have been associated with pubertal timing in the general population. This finding is consistent with the hypothesis that CDP is an extreme of normal variation in pubertal timing, at least in some cases. Indeed, the magnitude of the difference that we observed in PGS between CDP cases and controls suggests that common genetic variants are a major contributor to the genetic basis for CDP. However, not all individuals with CDP exhibited an elevated puberty PGS, and other factors may be contributing to delayed puberty in these individuals; such factors potentially include common genetic variants not captured by the PGS, rare genetic variants (as noted previously), and environmental factors.
Our results also demonstrate that common genetic variants that influence pubertal timing make only a small contribution to normosmic IHH. The differences in the contribution of common genetic variants to CDP vs IHH found in this study, in combination with prior reports that only a small proportion of individuals with CDP harbor rare genetic variants in genes associated with IHH (2, 4), support the concept that CDP and IHH are largely distinct entities. However, the small contribution of common puberty-delaying variants to IHH and the slight overlap in rare-variant genetics between CDP and IHH indicate that the 2 conditions share some pathophysiologic pathways and raise the possibility that, in rare cases, IHH (and specifically, normosmic IHH) could represent an extreme form of constitutional delay. The phenomenon of reversal of IHH seems particularly likely to represent an extreme version of CDP (29, 30); however, we did not observe any significant difference in the PGS for pubertal timing between individuals with reversal of IHH and controls, but the statistical power for this comparison was limited by the modest size of the reversal subcohort.
One hypothetical model for how common genetic variants that affect pubertal timing could contribute to both CDP and IHH is that a moderate enrichment of puberty-delaying variants (reflected by a higher PGS) could result in CDP and that extreme enrichment of these variants could result in IHH. This model would predict that a subset of individuals with IHH would have markedly elevated PGS; however, we did not observe any such “outlier” individuals in the IHH cohort. This suggests that the common genetic variants represented by the PGS used in our analyses are not sufficient to cause IHH. Another hypothetical model is that common genetic variants and rare genetic variants could work in conjunction to cause IHH. This model would predict a higher PGS for the subcohort of those with apparent rare-variant causes of their IHH, but we did not observe such a difference (though we did not have sufficient sample sizes to test for associations with specific IHH variants, genes, and/or pathways). A remaining possibility is that other common genetic variants may provide a greater contribution to the pathogenesis of IHH, and future GWAS specifically for IHH may identify a distinct set of common genetic variants that uniquely contribute to the pathogenesis of this condition.
The differential contribution of common genetic variants to CDP vs IHH suggests a potential clinical application: using polygenic scores to prospectively distinguish CDP from IHH. However, despite the large difference in mean PGS between CDP cases and controls, there was substantial overlap in the distribution of PGS's for cases and controls, limiting prognostic power. Future studies using PGS's for pubertal timing in conjunction with clinical features may improve the ability to predict whether a patient with delayed puberty has CDP or IHH, analogous to efforts to combine PGS with other factors to predict risk of cardiovascular disease, breast cancer, all-cause mortality, and other outcomes (31-36). Results of future GWAS for normosmic IHH and KS may further improve the ability to distinguish between CDP and IHH. Another potential clinical application is to use a puberty PGS to predict when a patient with CDP will eventually enter puberty; additional research is needed to determine the accuracy of such predictions (ie, how closely the puberty PGS correlates with age at eventual pubertal entry).
Strengths of our study include the exclusion of causes of delayed puberty other than CDP and IHH and the size of the normosmic IHH and KS cohorts. However, some subgroups were small (eg, reversal of IHH), limiting statistical power. One limitation of our study is that analysis was restricted to individuals of European ancestry, necessitated by the fact that the GWAS on pubertal timing were done in individuals of European ancestry and that PGS have impaired performance when applied to different ancestral groups (37). Another limitation is that our cases and controls were not genotyped in the same batch; we attempted to mitigate this issue by performing joint quality control and imputation for cases and controls together, and the fact that no differences were seen for PGS for basal-cell carcinoma and anal carcinoma suggest that it is unlikely that there were systematic differences between cases and controls that caused the differences observed for the puberty PGS. Also, our CDP cohort was genotyped on a platform different from that used for the normosmic IHH and KS cohorts, precluding direct comparison between CDP and the other 2 cohorts. The power of the PGS for male pubertal hallmarks was limited by the fact that publicly available data are restricted to the 10 000 variants with the strongest associations (11); nevertheless, the male-puberty PGS had sufficient power to demonstrate significant differences.
One area for future research is to associate common genetic variants with subtypes of CDP. Our group and others have shown that some but not all children with CDP exhibit declining height percentiles across childhood (38-40). Associating these subtypes of CDP with subsets of the common genetic variants that affect pubertal timing may facilitate understanding of the genetic and molecular mechanisms that link pubertal timing to other processes such as childhood growth or, alternatively, that affect only pubertal timing.
Our results expand our understanding of the genetic architecture of CDP and IHH by demonstrating that common genetic variants that influence pubertal timing in the general population contribute strongly to the genetics of CDP but only weakly to IHH. These findings support the hypothesis that the common-variant genetics of CDP and IHH are largely distinct. Further research is needed to determine the proportion of individuals with IHH or CDP whose condition is caused by common variants, rare variants, or both and how this genetic architecture compares to other traits and conditions that appear to lie upon a continuum, such as age at menopause, early menopause, and premature ovarian insufficiency and serum lipids, dyslipidemia, and familial hyperlipidemia.
Acknowledgments
We thank the participants, their families, and their referring providers. We also thank members of the Massachusetts General Hospital Harvard Reproductive Sciences Center and the Division of Endocrinology at Boston Children's Hospital for their feedback on this work. Ryan Ciarlo, Elfa Jonsdottir-Lewis, Bhargava Morampalli Reddy, Jeremy Cuniff, Amalia Feld, Alexandria Shirey, and Anna Schmidt contributed to participant recruitment and sample collection.
Funding
This work was supported by grants R01 HD090071 and P50 HD104224 from the Eunice K. Shriver National Institute of Child Health and Human Development (NICHD). M.F.L. received support from K23 HD097296. Activities of the Delayed Puberty Genetics Consortium were supported by the National Institutes of Health (R01 HD090071, R01 HD048960, P50 HD104224, M01 RR002172, K23 RR15544, M01 RR000080, ZIA HD008919, and ES103322) and by the Doris Duke Charitable Foundation (2013110).
Author Contributions
Y.-M.C., M.F.L., and R.M.S. conceived the study. M.F.L., E.S., A.A.H., and W.H. conducted analyses. M.F.L., R.B., A.D., R.G., J.E.H., J.N.H., K.M., M.R.P., R.R., S.B.S., and Y.-M.C. recruited participants, handled DNA samples, and/or managed genetic data. Y.-M.C., M.F.L., and E.S. wrote the first draft of the manuscript, and all authors reviewed and approved the final version of the manuscript.
Disclosures
Y.-M.C. and M.R.P. receive royalties from UpToDate on topics related to puberty. M.F.L. serves as an expert consultant for the Endocrine Society Endocrine Board Review. Rodolfo A. Rey is an editorial board member for The Journal of Clinical Endocrinology & Metabolism and played no role in the journal's evaluation of the manuscript. Verónica Mericq (included in the banner authorship for the Delayed Puberty Genetics Consortium) is an associate editor for The Journal of Clinical Endocrinology & Metabolism and played no role in the journal's evaluation of the manuscript.
Data Availability
Some or all datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References
Author notes
Additional members of the Delayed Puberty Genetics Consortium are Sasha R. Howard, Leo Dunkel, Centre for Endocrinology, William Harvey Research Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London UK; Ana Claudia Latronico, Alexander A. de Lima Jorge, Raíssa Carneiro Rezende, Universidade de São Paulo Faculdade de Medicina, São Paulo, Brazil; Aristeides Giannakopoulos, University Hospital of Patras, Rio, 26504 Patras Greece; Verónica Mericq, Paulina Merino, Institute of Maternal and Child Research, Faculty of Medicine, University of Chile, Santiago, Chile.

{kind=link}
{kind=link}