





Abstract
Congenital hypothyroidism with gland-in-situ (CH-GIS) is usually attributed to mutations in the genes involved in thyroid hormone production. The diagnostic yield of targeted next-generation sequencing (NGS) varied widely between studies. We hypothesized that the molecular yield of targeted NGS would depend on the severity of CH.
Targeted NGS was performed in 103 CH-GIS patients from the French national screening program referred to the Reference Center for Rare Thyroid Diseases of Angers University Hospital. The custom targeted NGS panel contained 48 genes. Cases were classified as solved or probably solved depending on the known inheritance of the gene, the classification of the variants according to the American College of Medical Genetics and Genomics, the familial segregation, and published functional studies. Thyroid-stimulating hormone at CH screening and at diagnosis (TSHsc and TSHdg) and free T4 at diagnosis (FT4dg) were recorded.
NGS identified 95 variants in 10 genes in 73 of the 103 patients, resulting in 25 solved cases and 18 probably solved cases. They were mainly due to mutations in the TG (n = 20) and TPO (n = 15) genes. The molecular yield was, respectively, 73% and 25% if TSHsc was ≥ and < 80 mUI/L, 60% and 30% if TSHdg was ≥ and < 100 mUI/L, and 69% and 29% if FT4dg was ≤ and > 5 pmol/L.
NGS in patients with CH-GIS in France found a molecular explanation in 42% of the cases, increasing to 70% when TSHsc was ≥ 80 mUI/L or FT4dg was ≤ 5 pmol/L.
Congenital hypothyroidism (CH) is the most common neonatal endocrine disorder, with an incidence of 1/2500 to 1/3500 newborns (1, 2). Among patients with CH, 15% to 50% have a gland-in-situ (GIS) with or without goiter. In France, the CH screening program relies on thyroid-stimulating hormone (TSH) measurement on dried blood samples collected on filter paper from all newborns at 3 days of life. From the 2000s, an increased incidence of CH has been observed, up to 1/2419 newborns in 2019 (1, 3). This has been driven by an increase in CH-GIS, from 15% of the cases before the 2000s to 50% in 2018 (1). The annual incidence of CH-GIS increased by 5.1% in this period, whereas the incidence of thyroid dysgenesis remained constant (1).
CH-GIS is usually attributed to dyshormonogenesis resulting from mutations in the genes involved in thyroid hormone production or iodide metabolism, typically associated with goiter: thyroglobulin (TG), thyroid peroxidase (TPO), SLC26A4 also called pendrin, SLC5A5 also called Na+/I− symporter (NIS), dual oxidase 2 (DUOX2), dual oxidase maturation factor 2 (DUOXA2), dual oxidase 1 (DUOX1), dual oxidase maturation factor 1 (DUOXA1), iodotyrosine deiodinase (IYD) also called iodotyrosine dehalogenase 1 (DEHAL1), and the iodide transporter SLC26A7 (4–10). In addition, mutations in the genes formally involved in thyroid dysgenesis may also be responsible for CH-GIS, typically with thyroid hypoplasia: paired box 8 (PAX8), thyroid-stimulating hormone receptor (TSHR), Forkhead box E1 (FOXE1), and NKX2-1 genes (6, 11–14).
The proportion of patients with CH-GIS who receive a molecular diagnosis varies widely, from 10% to 93% in studies involving 10 to 290 patients (15–29). Factors contributing to this variability include differences in the clinical characterization of the patients (imaging techniques, perchlorate test), proportion of familial or sporadic cases, geographic origin, population inbreeding rate, and mainly variant classification. None of these studies has focused on the rate of molecular explanation of CH-GIS according to TSH at screening, TSH, and free T4 (FT4) at diagnosis, all usual markers of the disease severity (30).
Our hypothesis was that the molecular yield would be dependent on the severity of CH. We report 103 patients with CH-GIS referred to our Reference Center for Rare Thyroid Diseases at Angers University Hospital for custom targeted next-generation sequencing (NGS). We assessed the proportion of genetic resolution of patients with CH-GIS according to the TSH at screening, TSH at diagnosis, and FT4 at diagnosis. Cases were considered solved based on known recessive or dominant inheritance of the gene, classification of the variants in agreement with the American College of Medical Genetics and Genomics (ACMG) guidelines (31), the localization in the protein domain, allele frequency in GnomAD genomes (32), in silico prediction based on SIFT and Mutation Taster prediction software (and Polyphen-2 when the 2 previous ones gave discordant results), PhyloP100way conservation score (33), the same variant previously described in the literature in a patient with CH, an in vitro study confirming the pathogenicity of the variant when available, and segregation of variants with CH in multiple affected family members.
Methods
Patients
All clinical data and blood samples were collected in our Reference Center for Rare Thyroid Diseases, University Hospital of Angers, France, for targeted NGS testing between January 1, 2016, and May 1, 2020.
Patients were included if they had CH-GIS, with or without goiter, ascertained by thyroid ultrasound (100 cases) and/or thyroid scintigraphy (85 cases). All the families gave written informed consent for genetic testing. In France, the screening is performed in all newborns on day 3. Newborns with a neonatal filter paper TSH > 17 mIU/L with a genetic screening processor (PerkinElmer, Turku, Finland) or > 20 mIU/L with AutoDELFIA (PerkinElmer) underwent a diagnostic procedure consisting of serum TSH, FT4 and free T3 (FT3), and thyroglobulin measurements, thyroid ultrasound, and thyroid scintigraphy, and often a perchlorate discharge test (all performed locally).
CH severity was classified as severe if FT4 was < 5 pmol/L, moderate if FT4 was between 5 and < 10 pmol/L, and mild if FT4 was > 10 pmol/L (34).
For the perchlorate discharge test, partial iodide organification defect and total iodide organification defect were defined as a radioiodine washout of more than 10% and 90%, respectively (35). Reference values for thyroglobulin levels during the first 15 days of life were used (36).
Genetic Analysis by Next-Generation Sequencing
DNA was extracted from total blood samples with the EZ1 extraction kit (Qiagen, Hilden, Germany). High-throughput sequencing of genes was performed on an Ion Proton sequencer (Thermo Fisher Scientific, Waltham, MA, USA). An amplicon-based enrichment library that focused on 48 genes involved in thyroid dyshormonogenesis and thyroid dysgenesis was prepared using Ion AmpliSeq technology. It included 14 genes known to be involved in primary CH, 3 genes involved in central CH, and 31 in thyroid physiology [Supplementary Table S1 (37)]. The read depth was more than 20, with a high-quality score (Q30). Sanger sequencing was performed for variant confirmation.
Classification of Variants
We used the 2015 standards and guidelines for the interpretation of sequence variants from the ACMG and the Association for Molecular Pathology (31) and VarSome ACMG implementation (http://varsome.com) to classify variants. We also considered the pathogenicity according to the localization in the protein domain (in a mutational hot spot and/or critical and well-established functional domain), allele frequency in GnomAD genomes version 3.1.1 (32) (https://gnomad.broadinstitute.org/), in silico prediction of the pathogenicity of the missense variant based on SIFT version 4.1 (https://sift.bii.a-star.edu.sg/), and Mutation Taster version 4.2 (http://www.mutationtaster.org) prediction software (and Polyphen-2 when the 2 previous ones gave discordant results, (http://genetics.bwh.harvard.edu/pph2/), the PhyloP100way conservation score (33) (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/phyloP100way/), in vitro study confirming pathogenicity of the variant when available, the same variant previously described in the literature in a patient with CH, and familial segregation. This allowed us to classify the variants as “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” or “benign.”
Classification of Cases
From this classification of variants, we then classified the genetic results of each patient into 4 categories according to the association of variants, potential familial segregation, and the genes involved (according to the recessive or dominant inheritance of the disease): (1) genetically solved cases, (2) probably solved cases, (3) partially unsolved cases, or (4) unsolved cases (Fig. 1).
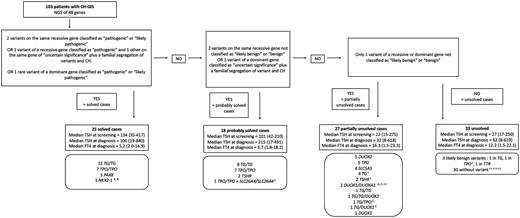
Classification of the cases, according to the genetic testing. See Supplementary Table S5 and Supplementary Table S6 and additional references (37) for a comprehensive classification of each case with variant descriptions. (A) Segregation of variants and CH in multiple affected family members. (B) The patient with NKX2-1 mutation had pulmonary involvement and neurological delay and a familial segregation of variant and CH. (C) Variants on 2 different genes were considered as partially unsolved because of the lack of evidence in the literature. t represents a patient with a transient form. Abbreviations: CH, congenital hypothyroidism.
Genetic variants in the following genes are considered recessive disorders: TG, TPO, SLC26A4 (pendrin), SLC5A5 (NIS), FOXE1, and IYD (4, 12–14). The patients had homozygous or compound heterozygous variants in the same gene and were classified according to Supplementary Table S2 (37).
Genetic variants in PAX8 and NKX2-1 are considered dominant disorders, as cases have been described in a heterozygous state (4), and patients were classified according to Supplementary Table S3 (37).
Regarding both the DUOX gene and its corresponding maturation factor DUOXA, cases have been described in heterozygous, compound heterozygous, or homozygous states, in one or several genes of the DUOX system (4, 5). This is also the case for the TSHR gene.
Thus, for variants in a heterozygous state, we considered cases solved with only 1 variant in 1 gene if this variant had already been described in CH in a heterozygous state and had an in vitro study confirming its pathogenicity. Every other case was considered partially unsolved.
To study the genetic resolution, we only considered solved and probably solved cases. We did not include the partially unsolved cases, because their variants alone could not explain the CH of the patient.
Statistical Analysis
Continuous variables were expressed as the median (minimum and maximum). Discrete variables were expressed as percent and 95% CI. Significance was defined as P < .05 (SPSS Statistics v25; IBM Corp., Armonk, NY, USA). The receiver operating characteristic curves and the genetic yields according to TSH at screening, TSH at diagnosis, and FT4 at diagnosis were calculated for each hormonal value. Then the curves of the molecular yields were modeled using smoothing splines (GraphPad Prism 9; GraphPad Software Inc.).
Results
Clinical and Biological Characteristics of the Whole Group of Patients
One hundred three patients with CH-GIS identified through the French national screening program were referred to our center and underwent genetic testing with NGS: the median TSH at day 3 of the newborn screening program (TSHsc) was 60 mIU/L (15-417), the median TSH at diagnosis (TSHdg; between day 6 and day 10) was 73 mIU/L (8-840), and the median FT4 (FT4dg) was 9.4 pmol/L (1.5-26.7).
Thirty-two patients had a goiter (31%). Twenty-six patients had a familial history of CH in first-degree relatives (25%). Four patients were diagnosed with hearing loss later in the follow-up. Fifteen patients (14.5%) had various abnormalities, such as severe mental retardation, single kidney, or polydactyly.
Fifteen patients had proven transient CH, as their treatment could be stopped: their median TSHsc was 21.7 mIU/L (15-61), median TSHdg was 42 mIU/L (11-67), and median FT4dg was 12.2 pmol/L (8.4-23.3). Fourteen patients failed to stop reatment [median TSHsc = 36 mIU/L (17-189), median TSHdg = 18.9 mIU/L (8-558), median FT4dg = 14.4 pmol/L (1.5-24)], and the median levothyroxine dose was 2.9 μg/kg/day (0.8-7.5) at a median age of 7.2 years (2.7-21). Thirty-five patients 3 years old or older were considered by their attending physicians as permanent cases as TSH levels were sometimes above the upper limit while on levothyroxine treatment: the median levothyroxine dose was 2.7 μg/kg/day (0.88-9) at a median age of 11.2 years (3.7-37). Last, 15 patients younger than 3 years old were considered by their physician as too young to try stopping treatment: the median levothyroxine dose was 3.95 μg/kg/day (2.4-7.6) at a median age of 1.4 years (0.1-2.7).
Molecular Explanation of the Cases and Relationships Between Genotype and Phenotype
Genetic testing found 95 variants in 73 of 103 patients: 38 variants were classified as “pathogenic,” 14 as “likely pathogenic,” 28 of “uncertain significance,” and 15 as “likely benign” [Supplementary Table S4 (37)]. Variants classified as “benign” are not reported. After exclusion of “likely benign” variants, the 80 remaining variants were in 10 genes: TG (n = 28), TPO (n = 26), DUOX2 (n = 7), TSHR (n = 6), DUOX1 (n = 3), PAX8 (n = 3), SLC26A4 (n = 3), SLC5A5 (n = 2), DUOXA1 (n = 1), and NKX2-1 (n = 1).
According to the classification of variants, the potential familial segregation of the disease, and the involved genes (associated with either recessive or dominant disease inheritance), 25 cases were solved and 18 were probably solved [Supplementary Table S5; Supplementary Table S6; (37)]. A genetic explanation was found by NGS testing in 43/103 cases (42%; Fig. 1). CH-GIS was due to thyroglobulin mutations in 20 patients and to thyroid peroxidase mutations in 15 patients. Notably, 8 patients had heterozygous variants in genes first involved in thyroid dysgenesis: 5 with variants in the PAX8 gene (ID 61, 62, 85, 98, 99), 2 in TSHR (ID 1 and 12), and 1 in NKX2-1 (ID 75).
Family history increased the likelihood of genetic resolution, with 15/26 (58%) familial cases with a genetic explanation (8/15 families), compared with 28/77 (36%) sporadic cases.
The clinical characteristics of the patients with solved or probably solved genetic cases according to the mutated gene are indicated in Table 1.
Clinical characteristics of patients with solved or probably solved cases, according to the mutated gene
| Solved or probably solved cases | TG variants | TPO variants | DUOX2 variants | TSHR variants | PAX8 variants | NKX2-1 variants |
|---|---|---|---|---|---|---|
| Number of patients | 20 | 15 | 2 | 3 | 5 | 1 |
| Median TSH on screening (mIU/L; min-max) | 134 (42-210) (n = 14) | 113 (56-417) (n = 8) | 15 | 101 and 25 | 35, 38, and 186 | |
| Median TSH at diagnosis (mIU/L; min-max) | 150 (23-792) (n = 17) | 213 (100-840) (n = 8) | 14 and 100 | 17 and 25 | 38 (9-100) (n = 5) | |
| Median FT4 at diagnosis (pmol/L; min-max) | 5.7 (2-9.8) (n = 15) | 5.2 (1.8-8.9) (n = 9) | 8.9 and 16.3 | 18.2 et 20.2 | 12.35 (7-15) (n = 4) | |
| Goitre (clinical and/or radiological) | 70% | 80% | 50% | |||
| Other clinical signs | 30% | 13% | ||||
| Serum thyroglobulin | n = 14 | n = 5 | n = 2 | n = 2 | n = 1 | |
| Undetectable | 64% | 0 | ||||
| Low | 29% | |||||
| Normal | 7% | 2 | 1 | 2 | 1 | |
| High | 3 | 1 | ||||
| Thyroid scintigraphy | n = 16 | n = 10 | n = 2 | n = 1 | n = 4 | n = 1 |
| High level of uptakea | 94% | 100% | 2 | 1 | 3 | 1 |
| Low level of uptake or absenta | 6% | 1 | ||||
| Perchlorate discharge test | n = 6 | n = 7 | n = 1 | n = 1 | ||
| Normal | 2 | 3 | 1 | 1 | ||
| Partial iodide organification defect | 4 | 2 | ||||
| Total iodide organification defect | 2 | |||||
| Median treatment posology at last follow-up (μg/kg/day; min-max) | 3.2 (1.4-5.7) (n = 16) | 3 (1.4-5.4) (n = 10) | 1.5 | 3.6, 2.5, and 0.8 | 2.5, 3.5, and 5.8 | 1.6 |
| Median age at last follow-up (years; min-max) | 8.6 (1.4-17.4) (n = 16) | 14.5 (0.3-37) (n = 10) | 7.3 | 23.6, 8.2, and 13.5 | 5, 5, and 7 | 4.4 |
| Reevaluation of the thyroid axis | n = 2 | n = 2 | n = 2 | n = 1 | n = 2 | |
| Transient CH | 0 | 1 | 1 | 1 | 0 |
| Solved or probably solved cases | TG variants | TPO variants | DUOX2 variants | TSHR variants | PAX8 variants | NKX2-1 variants |
|---|---|---|---|---|---|---|
| Number of patients | 20 | 15 | 2 | 3 | 5 | 1 |
| Median TSH on screening (mIU/L; min-max) | 134 (42-210) (n = 14) | 113 (56-417) (n = 8) | 15 | 101 and 25 | 35, 38, and 186 | |
| Median TSH at diagnosis (mIU/L; min-max) | 150 (23-792) (n = 17) | 213 (100-840) (n = 8) | 14 and 100 | 17 and 25 | 38 (9-100) (n = 5) | |
| Median FT4 at diagnosis (pmol/L; min-max) | 5.7 (2-9.8) (n = 15) | 5.2 (1.8-8.9) (n = 9) | 8.9 and 16.3 | 18.2 et 20.2 | 12.35 (7-15) (n = 4) | |
| Goitre (clinical and/or radiological) | 70% | 80% | 50% | |||
| Other clinical signs | 30% | 13% | ||||
| Serum thyroglobulin | n = 14 | n = 5 | n = 2 | n = 2 | n = 1 | |
| Undetectable | 64% | 0 | ||||
| Low | 29% | |||||
| Normal | 7% | 2 | 1 | 2 | 1 | |
| High | 3 | 1 | ||||
| Thyroid scintigraphy | n = 16 | n = 10 | n = 2 | n = 1 | n = 4 | n = 1 |
| High level of uptakea | 94% | 100% | 2 | 1 | 3 | 1 |
| Low level of uptake or absenta | 6% | 1 | ||||
| Perchlorate discharge test | n = 6 | n = 7 | n = 1 | n = 1 | ||
| Normal | 2 | 3 | 1 | 1 | ||
| Partial iodide organification defect | 4 | 2 | ||||
| Total iodide organification defect | 2 | |||||
| Median treatment posology at last follow-up (μg/kg/day; min-max) | 3.2 (1.4-5.7) (n = 16) | 3 (1.4-5.4) (n = 10) | 1.5 | 3.6, 2.5, and 0.8 | 2.5, 3.5, and 5.8 | 1.6 |
| Median age at last follow-up (years; min-max) | 8.6 (1.4-17.4) (n = 16) | 14.5 (0.3-37) (n = 10) | 7.3 | 23.6, 8.2, and 13.5 | 5, 5, and 7 | 4.4 |
| Reevaluation of the thyroid axis | n = 2 | n = 2 | n = 2 | n = 1 | n = 2 | |
| Transient CH | 0 | 1 | 1 | 1 | 0 |
Abbreviations: CH, congenital hypothyroidism; FT4, free T4.
aAccording to normal laboratory measure.
Clinical characteristics of patients with solved or probably solved cases, according to the mutated gene
| Solved or probably solved cases | TG variants | TPO variants | DUOX2 variants | TSHR variants | PAX8 variants | NKX2-1 variants |
|---|---|---|---|---|---|---|
| Number of patients | 20 | 15 | 2 | 3 | 5 | 1 |
| Median TSH on screening (mIU/L; min-max) | 134 (42-210) (n = 14) | 113 (56-417) (n = 8) | 15 | 101 and 25 | 35, 38, and 186 | |
| Median TSH at diagnosis (mIU/L; min-max) | 150 (23-792) (n = 17) | 213 (100-840) (n = 8) | 14 and 100 | 17 and 25 | 38 (9-100) (n = 5) | |
| Median FT4 at diagnosis (pmol/L; min-max) | 5.7 (2-9.8) (n = 15) | 5.2 (1.8-8.9) (n = 9) | 8.9 and 16.3 | 18.2 et 20.2 | 12.35 (7-15) (n = 4) | |
| Goitre (clinical and/or radiological) | 70% | 80% | 50% | |||
| Other clinical signs | 30% | 13% | ||||
| Serum thyroglobulin | n = 14 | n = 5 | n = 2 | n = 2 | n = 1 | |
| Undetectable | 64% | 0 | ||||
| Low | 29% | |||||
| Normal | 7% | 2 | 1 | 2 | 1 | |
| High | 3 | 1 | ||||
| Thyroid scintigraphy | n = 16 | n = 10 | n = 2 | n = 1 | n = 4 | n = 1 |
| High level of uptakea | 94% | 100% | 2 | 1 | 3 | 1 |
| Low level of uptake or absenta | 6% | 1 | ||||
| Perchlorate discharge test | n = 6 | n = 7 | n = 1 | n = 1 | ||
| Normal | 2 | 3 | 1 | 1 | ||
| Partial iodide organification defect | 4 | 2 | ||||
| Total iodide organification defect | 2 | |||||
| Median treatment posology at last follow-up (μg/kg/day; min-max) | 3.2 (1.4-5.7) (n = 16) | 3 (1.4-5.4) (n = 10) | 1.5 | 3.6, 2.5, and 0.8 | 2.5, 3.5, and 5.8 | 1.6 |
| Median age at last follow-up (years; min-max) | 8.6 (1.4-17.4) (n = 16) | 14.5 (0.3-37) (n = 10) | 7.3 | 23.6, 8.2, and 13.5 | 5, 5, and 7 | 4.4 |
| Reevaluation of the thyroid axis | n = 2 | n = 2 | n = 2 | n = 1 | n = 2 | |
| Transient CH | 0 | 1 | 1 | 1 | 0 |
| Solved or probably solved cases | TG variants | TPO variants | DUOX2 variants | TSHR variants | PAX8 variants | NKX2-1 variants |
|---|---|---|---|---|---|---|
| Number of patients | 20 | 15 | 2 | 3 | 5 | 1 |
| Median TSH on screening (mIU/L; min-max) | 134 (42-210) (n = 14) | 113 (56-417) (n = 8) | 15 | 101 and 25 | 35, 38, and 186 | |
| Median TSH at diagnosis (mIU/L; min-max) | 150 (23-792) (n = 17) | 213 (100-840) (n = 8) | 14 and 100 | 17 and 25 | 38 (9-100) (n = 5) | |
| Median FT4 at diagnosis (pmol/L; min-max) | 5.7 (2-9.8) (n = 15) | 5.2 (1.8-8.9) (n = 9) | 8.9 and 16.3 | 18.2 et 20.2 | 12.35 (7-15) (n = 4) | |
| Goitre (clinical and/or radiological) | 70% | 80% | 50% | |||
| Other clinical signs | 30% | 13% | ||||
| Serum thyroglobulin | n = 14 | n = 5 | n = 2 | n = 2 | n = 1 | |
| Undetectable | 64% | 0 | ||||
| Low | 29% | |||||
| Normal | 7% | 2 | 1 | 2 | 1 | |
| High | 3 | 1 | ||||
| Thyroid scintigraphy | n = 16 | n = 10 | n = 2 | n = 1 | n = 4 | n = 1 |
| High level of uptakea | 94% | 100% | 2 | 1 | 3 | 1 |
| Low level of uptake or absenta | 6% | 1 | ||||
| Perchlorate discharge test | n = 6 | n = 7 | n = 1 | n = 1 | ||
| Normal | 2 | 3 | 1 | 1 | ||
| Partial iodide organification defect | 4 | 2 | ||||
| Total iodide organification defect | 2 | |||||
| Median treatment posology at last follow-up (μg/kg/day; min-max) | 3.2 (1.4-5.7) (n = 16) | 3 (1.4-5.4) (n = 10) | 1.5 | 3.6, 2.5, and 0.8 | 2.5, 3.5, and 5.8 | 1.6 |
| Median age at last follow-up (years; min-max) | 8.6 (1.4-17.4) (n = 16) | 14.5 (0.3-37) (n = 10) | 7.3 | 23.6, 8.2, and 13.5 | 5, 5, and 7 | 4.4 |
| Reevaluation of the thyroid axis | n = 2 | n = 2 | n = 2 | n = 1 | n = 2 | |
| Transient CH | 0 | 1 | 1 | 1 | 0 |
Abbreviations: CH, congenital hypothyroidism; FT4, free T4.
aAccording to normal laboratory measure.
Some phenotypic characteristics were remarkable:
Twenty-six of the 32 patients (81%) with goiter were solved or probably solved cases, mainly with TG and TPO variants (14 and 12 cases, respectively).
Two of the 4 patients with hearing loss were solved cases due to TPO mutations (other cases were not solved).
Among the 20 patients with probably solved or solved cases with TG variants, serum thyroglobulin measurements were available in 14 patients: it was undetectable in 9 patients, low in 4 patients, and in the normal range in 1 patient (ID 64). One other patient, ID 48, with a thyroid scintigraphy ascertaining GIS, had undetectable serum thyroglobulin but no variant found on NGS analysis. A mutation in a noncoding region altering gene expression has yet to be found.
Genetic Resolution According to Thyroid Function at Screening and at Diagnosis
We studied the proportion of genetic resolution according to the TSH level at neonatal screening and at diagnosis and the FT4 level at diagnosis for all cases and only sporadic cases (Fig. 2). Median TSHdg was 100 mIU/L (19-417) in solved cases, 215 mIU/L (17-491) in probably solved cases, 32 mIU/L (8-618) in partially unsolved cases, and 27 mIU/L (17-250) in unsolved cases. Among the partially unsolved or unsolved cases in our study, the median TSHsc was 29 mIU/L (17-250), 25 mIU/L (15-275), and 20 mIU/L (15-90) for patients carrying 0 (n = 30), 1 (n = 12), or ≥ 2 (n = 13) nonbenign variants, respectively.
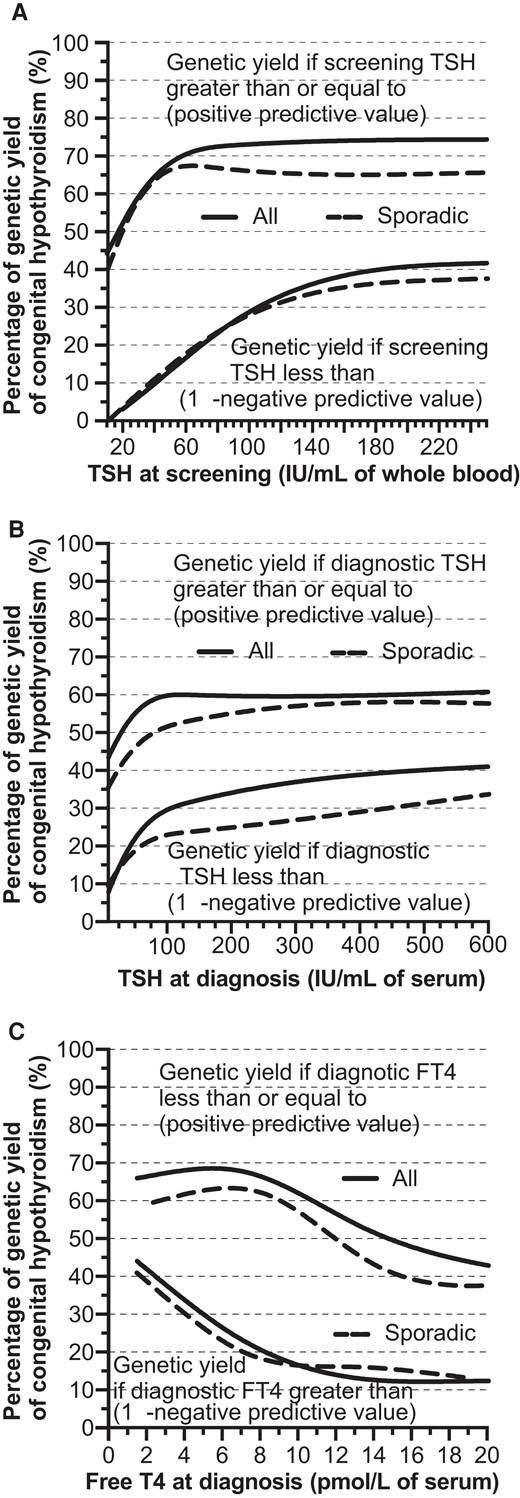
Proportion of genetic resolution according to the TSH level at neonatal screening (A) and at diagnosis (B), and the free T4 level at diagnosis (C) for all cases and only sporadic cases.
Considering the trade-off between sensitivity (population of solved or probably solved cases) and specificity (population of partially unsolved and unsolved cases), the area under the receiver operator characteristic curve (AUC) was 0.84 (CI95 0.74-0.94) for TSH at screening. A whole blood TSH at screening (TSHsc) ≥ and < 80 mIU/L resulted in 73% (CI95 58-92%; cut-off corresponding to the highest positive predictive value) and 25% (CI95 15-45%; 1—negative predictive value) of the molecular yield of the cases, respectively (Fig. 2A).
The AUC was 0.71 (CI95 0.60-0.83) for TSH at diagnosis. A serum TSH at diagnosis (TSHdg) ≥ and < 100 mIU/L resulted in 60% (CI95 44-81%; cut-off corresponding to the highest positive predictive value) and 30% (CI95 20-46%) of the molecular yield of the cases, respectively (Fig. 2B).
The AUC was 0.81 (CI95 0.70-0.91) for FT4 at diagnosis. A serum FT4 at diagnosis (FT4dg) ≤ and > 10 pmol/L resulted in 60% (CI95 47-77%) and 15% (CI95 7-33%) of the molecular yield of the cases, respectively, with ≤ and > 5 pmol/L resulting in 69% (CI95 50-93%) and 29% (CI95 19-44%) of the molecular yield, respectively. The highest positive predictive values were observed for the lowest FT4 levels (Fig. 2C). Slightly lower positive predictive values, but with similar trends, were found when studying only sporadic cases (Fig. 2).
Subjects With Transient Congenital Hypothyroidism
In the 15 patients with transient CH, one was considered probably solved. This patient had compound heterozygous variants in SLC26A4 (1 likely pathogenic and 1 of uncertain significance) and a homozygous TPO deletion c.2722_2736del, causing the loss of 5 amino acids in the intracellular domain without reading frame modification, of unknown effect (ID 45).
Discussion
We report 103 cases of CH-GIS referred to our center for genetic testing. In 73 out of 103 patients, genetic testing found 95 variants, including 38 variants classified as “pathogenic,” 14 as “likely pathogenic,” and 28 of “uncertain significance.” Targeted NGS found a genetic explanation with solved or probably solved cases in 42% of the patients (n = 43/103), mainly in the TG and TPO genes. The molecular yield increased to 70% when TSH at neonatal screening was ≥ 80 mIU/L or FT4 at CH diagnosis was < 5 pmol/L.
Numerous studies have described genetic variants in patients with CH-GIS. Some studied only 1 gene (38–41); others reported the frequency of the variants found in patients with CH-GIS but with no individual analysis of genetic resolution (42–48). We identified 15 studies that analyzed the rate of genetic resolution by screening at least 2 genes in CH-GIS (Table 2) (15–29). The resolution varied widely between studies, ranging from 10% to 93%. Only 3 studies included 30 patients or more with CH-GIS and analyzed at least 8 different genes, with a genetic resolution between 41% and 60% (20, 23, 25). The molecular yield in these studies was consistent with the 42% yield in the present study.
Literature review of studies that analyzed at least 2 genes in patients having CH-GIS, with the percentage of genetic resolution
| Publication | Year of publication | Country(ies) | Type of CH | Number of patients with GIS | Presence of consanguineous patients | Presence of familial cases | Number of genes analyzed | Percentage of genetic resolution in GIS | More frequently involved gene(s) |
|---|---|---|---|---|---|---|---|---|---|
| Narumi et al. | 2011 | Japan | GIS | 14 | NA | Yes | 7 | 93% | DUOX2 and TPO |
| Wang et al. | 2014 | China | GIS with goiter | 67 | No | No | 3 | 13% | DUOX2 |
| Jiang et al. | 2016 | China | GIS | 12 | No | No | 12 | 92% | DUOX2 |
| Lof et al. | 2016 | Finland | GIS and TD | 21 | NA | Yes | 13 | 55% in familial cases, 20% in sporadic cases | TPO |
| Matsuo et al. | 2016 | Japan | GIS | 48 | NA | NA | 3 | 23% | DUOX2 |
| Nicholas et al. | 2016 | United Kingdom, Oman, Saudi Arabia, United Arab Emirates, Turkey | GIS | 49 | Yes | Yes | 8 | 59% | TG and TPO |
| Park et al. | 2016 | Korea | GIS and TD | NA (among 170) | NA | NA | 6 | 31% | DUOX2 |
| Sun et al. | 2018 | China | GIS and TD | NA (among 110) | No | Yes | 21 | 52% | DUOX2 |
| Zou et al. | 2018 | Saudi Arabia | GIS and TD | 30 | NA | Yes | WES | 60% | TPO |
| Santos-Silva et al. | 2019 | Portugal | GIS | 9 | Yes | Yes | 28 | 67% | TG and TPO |
| Wang et al. | 2020 | China | GIS and TD | 32 | No | Yes | 29 | 41% | DUOX2 |
| Zdraveska et al. | 2020 | Macedonia | GIS and hypoplasia | 34 | No | Yes | 2 to 9 | 10% | NA |
| Li et al. | 2021 | China | GIS and TD | 468 | NA | Yes | 5 | 4% | PAX8 |
| Shin et al. | 2021 | South Korea | GIS | 20 | NA | NA | 8 | 80% | DUOX2 and TSHR |
| Stoupa et al. | 2021 | France, USA, Turkey | GIS | 19 | Yes | Yes | 78 | 53% | TG |
| Present study | France | GIS | 103 | Yes | Yes | 48 | 42% | TG and TPO |
| Publication | Year of publication | Country(ies) | Type of CH | Number of patients with GIS | Presence of consanguineous patients | Presence of familial cases | Number of genes analyzed | Percentage of genetic resolution in GIS | More frequently involved gene(s) |
|---|---|---|---|---|---|---|---|---|---|
| Narumi et al. | 2011 | Japan | GIS | 14 | NA | Yes | 7 | 93% | DUOX2 and TPO |
| Wang et al. | 2014 | China | GIS with goiter | 67 | No | No | 3 | 13% | DUOX2 |
| Jiang et al. | 2016 | China | GIS | 12 | No | No | 12 | 92% | DUOX2 |
| Lof et al. | 2016 | Finland | GIS and TD | 21 | NA | Yes | 13 | 55% in familial cases, 20% in sporadic cases | TPO |
| Matsuo et al. | 2016 | Japan | GIS | 48 | NA | NA | 3 | 23% | DUOX2 |
| Nicholas et al. | 2016 | United Kingdom, Oman, Saudi Arabia, United Arab Emirates, Turkey | GIS | 49 | Yes | Yes | 8 | 59% | TG and TPO |
| Park et al. | 2016 | Korea | GIS and TD | NA (among 170) | NA | NA | 6 | 31% | DUOX2 |
| Sun et al. | 2018 | China | GIS and TD | NA (among 110) | No | Yes | 21 | 52% | DUOX2 |
| Zou et al. | 2018 | Saudi Arabia | GIS and TD | 30 | NA | Yes | WES | 60% | TPO |
| Santos-Silva et al. | 2019 | Portugal | GIS | 9 | Yes | Yes | 28 | 67% | TG and TPO |
| Wang et al. | 2020 | China | GIS and TD | 32 | No | Yes | 29 | 41% | DUOX2 |
| Zdraveska et al. | 2020 | Macedonia | GIS and hypoplasia | 34 | No | Yes | 2 to 9 | 10% | NA |
| Li et al. | 2021 | China | GIS and TD | 468 | NA | Yes | 5 | 4% | PAX8 |
| Shin et al. | 2021 | South Korea | GIS | 20 | NA | NA | 8 | 80% | DUOX2 and TSHR |
| Stoupa et al. | 2021 | France, USA, Turkey | GIS | 19 | Yes | Yes | 78 | 53% | TG |
| Present study | France | GIS | 103 | Yes | Yes | 48 | 42% | TG and TPO |
Abbreviations: CH, congenital hypothyroidism; GIS, gland-in-situ; NA, not available; TD, thyroid dysgenesis.
Literature review of studies that analyzed at least 2 genes in patients having CH-GIS, with the percentage of genetic resolution
| Publication | Year of publication | Country(ies) | Type of CH | Number of patients with GIS | Presence of consanguineous patients | Presence of familial cases | Number of genes analyzed | Percentage of genetic resolution in GIS | More frequently involved gene(s) |
|---|---|---|---|---|---|---|---|---|---|
| Narumi et al. | 2011 | Japan | GIS | 14 | NA | Yes | 7 | 93% | DUOX2 and TPO |
| Wang et al. | 2014 | China | GIS with goiter | 67 | No | No | 3 | 13% | DUOX2 |
| Jiang et al. | 2016 | China | GIS | 12 | No | No | 12 | 92% | DUOX2 |
| Lof et al. | 2016 | Finland | GIS and TD | 21 | NA | Yes | 13 | 55% in familial cases, 20% in sporadic cases | TPO |
| Matsuo et al. | 2016 | Japan | GIS | 48 | NA | NA | 3 | 23% | DUOX2 |
| Nicholas et al. | 2016 | United Kingdom, Oman, Saudi Arabia, United Arab Emirates, Turkey | GIS | 49 | Yes | Yes | 8 | 59% | TG and TPO |
| Park et al. | 2016 | Korea | GIS and TD | NA (among 170) | NA | NA | 6 | 31% | DUOX2 |
| Sun et al. | 2018 | China | GIS and TD | NA (among 110) | No | Yes | 21 | 52% | DUOX2 |
| Zou et al. | 2018 | Saudi Arabia | GIS and TD | 30 | NA | Yes | WES | 60% | TPO |
| Santos-Silva et al. | 2019 | Portugal | GIS | 9 | Yes | Yes | 28 | 67% | TG and TPO |
| Wang et al. | 2020 | China | GIS and TD | 32 | No | Yes | 29 | 41% | DUOX2 |
| Zdraveska et al. | 2020 | Macedonia | GIS and hypoplasia | 34 | No | Yes | 2 to 9 | 10% | NA |
| Li et al. | 2021 | China | GIS and TD | 468 | NA | Yes | 5 | 4% | PAX8 |
| Shin et al. | 2021 | South Korea | GIS | 20 | NA | NA | 8 | 80% | DUOX2 and TSHR |
| Stoupa et al. | 2021 | France, USA, Turkey | GIS | 19 | Yes | Yes | 78 | 53% | TG |
| Present study | France | GIS | 103 | Yes | Yes | 48 | 42% | TG and TPO |
| Publication | Year of publication | Country(ies) | Type of CH | Number of patients with GIS | Presence of consanguineous patients | Presence of familial cases | Number of genes analyzed | Percentage of genetic resolution in GIS | More frequently involved gene(s) |
|---|---|---|---|---|---|---|---|---|---|
| Narumi et al. | 2011 | Japan | GIS | 14 | NA | Yes | 7 | 93% | DUOX2 and TPO |
| Wang et al. | 2014 | China | GIS with goiter | 67 | No | No | 3 | 13% | DUOX2 |
| Jiang et al. | 2016 | China | GIS | 12 | No | No | 12 | 92% | DUOX2 |
| Lof et al. | 2016 | Finland | GIS and TD | 21 | NA | Yes | 13 | 55% in familial cases, 20% in sporadic cases | TPO |
| Matsuo et al. | 2016 | Japan | GIS | 48 | NA | NA | 3 | 23% | DUOX2 |
| Nicholas et al. | 2016 | United Kingdom, Oman, Saudi Arabia, United Arab Emirates, Turkey | GIS | 49 | Yes | Yes | 8 | 59% | TG and TPO |
| Park et al. | 2016 | Korea | GIS and TD | NA (among 170) | NA | NA | 6 | 31% | DUOX2 |
| Sun et al. | 2018 | China | GIS and TD | NA (among 110) | No | Yes | 21 | 52% | DUOX2 |
| Zou et al. | 2018 | Saudi Arabia | GIS and TD | 30 | NA | Yes | WES | 60% | TPO |
| Santos-Silva et al. | 2019 | Portugal | GIS | 9 | Yes | Yes | 28 | 67% | TG and TPO |
| Wang et al. | 2020 | China | GIS and TD | 32 | No | Yes | 29 | 41% | DUOX2 |
| Zdraveska et al. | 2020 | Macedonia | GIS and hypoplasia | 34 | No | Yes | 2 to 9 | 10% | NA |
| Li et al. | 2021 | China | GIS and TD | 468 | NA | Yes | 5 | 4% | PAX8 |
| Shin et al. | 2021 | South Korea | GIS | 20 | NA | NA | 8 | 80% | DUOX2 and TSHR |
| Stoupa et al. | 2021 | France, USA, Turkey | GIS | 19 | Yes | Yes | 78 | 53% | TG |
| Present study | France | GIS | 103 | Yes | Yes | 48 | 42% | TG and TPO |
Abbreviations: CH, congenital hypothyroidism; GIS, gland-in-situ; NA, not available; TD, thyroid dysgenesis.
The factors that would explain this variability may be the number of genes studied, the proportion of familial cases, the severity of CH, and the classification of solved or unsolved cases between studies. First, the molecular yield was 13% to 23% when 3 genes were studied (16, 19), and this increased to 41% to 93% when at least 8 genes were studied (17, 18, 20, 22–25, 28, 29). In addition, familial cases accounted for 6% to 73% of patients in published studies (15, 18, 20, 23–26, 29), compared with 25% in our study. One study assessed genetic resolution in sporadic (20%) and familial cases (55%) (18). This compares with 36% and 58% in sporadic and familial cases in our study, respectively. None of these studies evaluated the severity of CH as a factor associated with genetic resolution, but 1 reported a median screening TSH of 23.3 mIU/L, with a genetic resolution of 41% (25), while another reported a median TSH at diagnosis of 75 mIU/L with 59% genetic resolution (20). In our study, the median TSH at screening and at diagnosis were 60 and 73 mIU/L, respectively, for a genetic resolution of 42%. These values were consistent with a similar association between CH severity and genetic resolution. Regarding the classification of solved or unsolved cases, the criteria vary between studies. We used strict criteria for the classification of solved or probably solved cases, as we classified as unsolved patients with pathogenic or probably pathogenic variants in silico when they were present only at heterozygous state, without in vitro evidence of their pathogenicity at heterozygous state. Also, variants located in the nonessential splice site were not considered as involved. This may underestimate the genetic resolution of our study.
In most European and Middle Eastern studies, the genes most frequently involved in CH-GIS have been the TG and TPO genes, in agreement with the present study (18, 20, 23, 24, 26, 29). Conversely, studies from Asia have found cases that were mainly explained by DUOX2 variants (15–17, 19, 21, 22, 25, 28), whereas DUOX2 mutations explained none of the solved or probably solved cases in the present study. Our results may not superimposable to those from other regions of the world, as the mutations found in the CH-GIS depend on the ethnic background. PAX8 and NKX2-1 mutations, which were first described in thyroid dysgenesis, have been shown to be more frequently associated with GIS in populations of patients suffering from CH (4, 11, 14, 27), in agreement with the finding in our study, which focused solely on GIS.
Our study has several limitations. The NNT (49) and SLC26A7 (9, 10) genes involved in CH-GIS were not studied, nor were some of the genes involved in CH with thyroid dysgenesis, such as GLI-similar zinc finger protein family 3 (GLIS3), cell division cycle associated 8 (CDCA8) coding borealin, jagged1 (JAG1), netrin 1 (NTN1) (4), and TUBB1 (50). Another limitation of this study is the lack of functional studies for all variants. To assess the responsibility of the variants, we used the referent ACMG classification (31), together with familial segregation, in silico prediction software, location in the protein structure, previous published studies of CH with the same variants, and functional studies when available. Additional new tools for predicting protein structure from computational methods, such as the AlphaFold protein structure database, may prove useful in the future to better characterize the consequences of missense variants (51, 52). In France the median TSH at screening and diagnosis for in situ gland were 34 and 91 mIU/L, respectively, in 2016 (unpublished), whereas median TSH were 60 and 73 mIU/L, respectively, in our study: these higher values are in favor of a bias in referral to the most severe cases. However, we believe that the relationship we have shown between CH severity and molecular yield would be true over the entire range of TSH at screening and may contribute to explain the variation in the molecular yield between studies.
In conclusion, we report 103 patients with CH-GIS in France, with a genetic resolution for 42% of them. The percentage of genetic resolution increased to 70% when TSH on neonatal screening was ≥ 80 mIU/L or FT4 at diagnosis was below 5 pmol/L. Even some transient forms had a molecular explanation, and long-term follow-up will be needed, especially when thyroid hormone requirements are high, such as during puberty and later during pregnancy.
Acknowledgments
We warmly thank all the pediatricians who sent a blood sample for genetic analysis in our Reference Center for Rare Diseases of Thyroid and Hormone Receptivity.
Data Availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Disclosures
The authors have no conflicts of interest relevant to this article to disclose.
References

{kind=link}
{kind=link}