





Abstract
Kenny–Caffey syndrome (KCS) is a rare hereditary disorder characterized by short stature, hypoparathyroidism, and electrolyte disturbances. KCS1 and KCS2 are caused by pathogenic variants in TBCE and FAM111A, respectively. Clinically the phenotypes are difficult to distinguish.
The objective was to determine and expand the phenotypic spectrum of KCS1 and KCS2 in order to anticipate complications that may arise in these disorders.
We clinically and genetically analyzed 10 KCS2 patients from 7 families. Because we found unusual phenotypes in our cohort, we performed a systematic review of genetically confirmed KCS cases using PubMed and Scopus. Evaluation by 3 researchers led to the inclusion of 26 papers for KCS1 and 16 for KCS2, totaling 205 patients. Data were extracted following the Cochrane guidelines and assessed by 2 independent researchers.
Several patients in our KCS2 cohort presented with intellectual disability (3/10) and chronic kidney disease (6/10), which are not considered common findings in KCS2. Systematic review of all reported KCS cases showed that the phenotypes of KCS1 and KCS2 overlap for postnatal growth retardation (KCS1: 52/52, KCS2: 23/23), low parathyroid hormone levels (121/121, 16/20), electrolyte disturbances (139/139, 24/27), dental abnormalities (47/50, 15/16), ocular abnormalities (57/60, 22/23), and seizures/spasms (103/115, 13/16). Symptoms more prevalent in KCS1 included intellectual disability (74/80, 5/24), whereas in KCS2 bone cortical thickening (1/18, 16/20) and medullary stenosis (7/46, 27/28) were more common.
Our case series established chronic kidney disease as a new feature of KCS2. In the literature, we found substantial overlap in the phenotypic spectra of KCS1 and KCS2, but identified intellectual disability and the abnormal bone phenotype as the most distinguishing features.
Kenny–Caffey syndrome (KCS) is an inherited disorder that is characterized by short stature, hypoparathyroidism, and skeletal defects that include cortical thickening and medullary stenosis of the tubular bones (1, 2). The molecular basis for KCS has been identified as pathogenic variants in the tubulin-specific chaperone E gene (TBCE) (KCS1, OMIM #244460) or the family with sequence similarity 111 member A gene (FAM111A) (KCS2, OMIM #127000) (3, 4) (Fig. 1A). TBCE is involved in microtubule dynamics (5-7), whereas FAM111A plays a role in DNA replication (8, 9) and viral defense (10-12). KCS1 is an autosomal recessive disorder similar to Sanjad–Sakati syndrome (SSS) (also termed hypoparathyroidism–retardation–dysmorphism syndrome) (OMIM #241410), a disorder that is present almost exclusively in Middle Eastern populations (13). Although KCS1 and SSS have been distinguished in the literature based on the skeletal phenotype and propensity to bacterial infections, the 2 disease entities are caused by the same deletion mutation and are thus likely to represent variable manifestations of the same disorder (14, 15). Missense mutations in TBCE are also associated with the phenotypically different disorder progressive encephalopathy with amyotrophy and optic atrophy (OMIM #617207), and possibly with familial isolated hyperparathyroidism (OMIM #145000) (16, 17). Inheritance of KCS2 is autosomal dominant, although many pathogenic variants occur de novo (1, 18-20). The most commonly reported pathogenic variant is c.1706G>A, which causes a p.Arg569His substitution (20). Phenotypic diversity also exists for KCS2, as heterozygous missense FAM111A mutations have also been found in patients with gracile bone dysplasia (GCLEB; OMIM #602361). The GCLEB pathogenic variants reported until now have been different from those in KCS2, but have occurred in the same protein domain. The clinical presentation of GCLEB is similar to that of KCS but is more severe and typically leads to perinatal death (4).
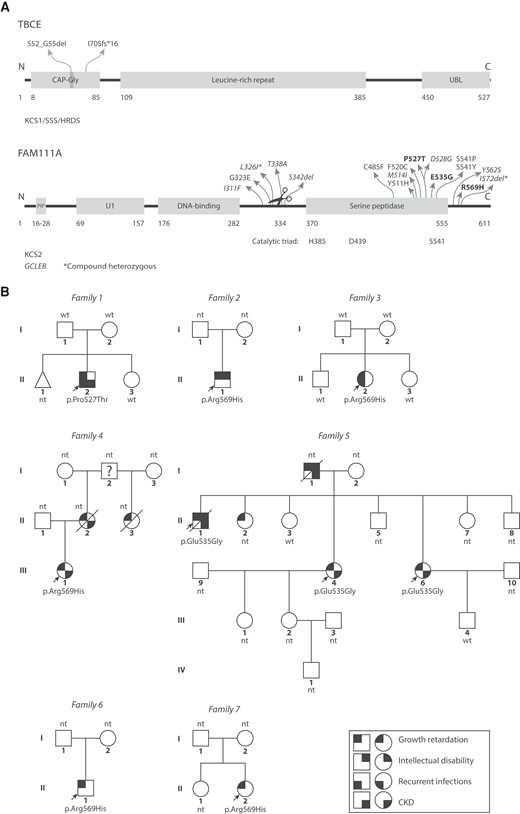
Variants Kenny–Caffey syndrome patients. (A) Top: TBCE is involved in αβ-tubulin dimer formation and dissociation (5-7). KCS1/SSS patient pathogenic variants are located to the CAP-Gly domain that binds the tubulin α-subunit. The vast majority of patients carry the homozygous c.155-166del12 (p.Ser52_Gly55del) pathogenic variant. Bottom: FAM111A is involved in DNA replication (8, 9) and anti-viral defense (10-12). KCS2 and GCLEB (italics) variants in FAM111A show clustering in and around the serine peptidase domain and autocleavage site (scissors). (Likely) pathogenic variants from our case series are shown in bold text. CAP-Gly, cytoskeleton-associated protein-glycine-rich, PIP, PCNA-interacting protein, U1, domain of unknown significance, UBL, ubiquitin-like. *Compound heterozygous variants that were found together in 1 GCLEB patient. (B) Pedigrees of KCS2 patients, indicating patients with symbols partially filled in with black based on their phenotypes. The question mark indicates the person is likely affected but the phenotype is unknown. Probands are indicated with arrows. CKD, chronic kidney disease; nt, not genetically tested; wt, wild type.
Diagnosing KCS can be challenging for several reasons: the heterogeneity of the clinical presentation, the similarity to other syndromes, and the absence of clear KCS hallmarks. KCS1 is classically associated with microcephaly, susceptibility to infections, and intellectual disability, whereas KCS2 is characterized by macrocephaly and cortical thickening and medullary stenosis of the long bones (Table S1 (21)). However, recent publications and our own observations challenge these conventional distinctions. For instance, in several KCS2 patients, cortical thickening or medullary stenosis of the long bones was not observed (19, 22, 23). Moreover, cortical thickening and medullary stenosis of the long bones was reported in a KCS1 patient as well (14). Likewise, intellectual disability was noted in 2 genetically confirmed KCS2 patients (23, 24). Therefore, it is important to establish a comprehensive overview of symptoms in KCS1 and KCS2 to gain a better understanding of the presentation of these disorders.
In this case series, we report 10 patients from 7 families with heterozygous FAM111A pathogenic variants and further extend the clinical spectrum of KCS2 (Fig. 1B). To obtain more insights into the heterogeneity of the KCS phenotype, we subsequently performed a systematic review of all genetically confirmed KCS1 and KCS2 cases from the literature in order to compare the phenotypic spectra of the 2 forms of KCS.
Materials and Methods
Patient Collection
Patients were diagnosed based on genetic testing, clinical data, and laboratory measurements. We only included patients with symptoms that were previously reported in KCS and that had a (likely) pathogenic variant in FAM111A. Informed consent for publication of the findings was obtained from all patients.
Genetic Investigations
Patient F1-II-2 was initially tested for DiGeorge syndrome using multiplex ligation–dependent probe amplification. Targeted sequencing was performed for pathogenic variants in calcium sensing receptor (CASR), claudin 16 (CLDN16), FXYD domain containing ion transport regulator 2 (FXYD2), transient receptor potential cation channel subfamily M member 6 (TRPM6), glial cells missing transcription factor 2 (GCM2), and epidermal growth factor (EGF). Whole exome sequencing eventually identified a pathogenic variant in FAM111A. DNA from patient F2-II-1 was sequenced using Sanger sequencing. For patient F3-II-2, exome capture was performed using Agilent SureSelectXT Human All Exon v4 enrichment kit. Sequencing was performed on Illumina HiSeq using the 2× 100 bp paired-end module. For patient F4-III-1, DNA sequencing of coding exons and flanking splice signal sequences was performed using a skeletal dysplasia gene panel as part of the 100 000 genomes project. In family 5, initially targeted sequencing was performed for CASR variants in the father (F5-I-1), FXYD2, potassium voltage-gated channel subfamily A member 1 (KCNA1), Barttin CLCNK type accessory subunit beta (BSND), and chloride voltage-gated channel Kb (CLCNKB) variants for 1 daughter (F5-II-4), and potassium inwardly rectifying channel subfamily J member 1 (KCNJ1) variants for the son. Subsequent whole exome sequencing of 3 affected and 2 healthy family members identified a novel variant in the FAM111A gene in the patients. A more elaborate description of the sequencing methods of family 5 can be found elsewhere (21). DNA of patient F6-II-1 was sequenced for FAM111A variants by Sanger sequencing for 2 coding exons using polymerase chain reaction amplicons with primer sets 5′-CTAAGGTACCATCTCCAGGGA-3′ and 5′-ATGCCAATACTATGCCATGTT-3′, 5′-TTGGATCCCATGTCAAACAC-3′ and 5′-TAATTCATCAACTGGCTGGGT-3′, 5′-CAATTGGAATTGGGAAGTGTAA-3′ and 5′-TTCCATAGGTACTTGTTGTCCA-3′, and 5′-TAGATAGCATTGTGGGAGACG-3′ and 5′-AATGCCTGGCAGATAGGAAAT-3′. For patient F7-II-2, targeted genetic testing of FAM111A and TBCE was performed by GeneDx. Sequencing was performed on an Illumina sequencing system with 100 bp or greater paired-end reads.
Literature Screening
Details about our approach to systematic review of the literature have been uploaded to PROSPERO prior to the start of screening (registration number CRD42022291565). In order to get a comprehensive overview of symptoms in confirmed cases of KCS1 and KCS2, a search was performed of the PubMed and Scopus databases (Fig. 2). The results were last checked on December 9, 2021. The inclusion criteria were (1) patient report and (2) diagnosis confirmed by pathogenic variants in the TBCE or FAM111A gene regardless of the reported clinical definition. The exclusion criteria were (1) diagnosis other than KCS1, SSS or hypoparathyroidism–retardation–dysmorphism syndrome for KCS1, and KCS2 or GCLEB/OCS for KCS2, (2) reviews of previously reported cases, unless they provide new patient information, (3) reports that do not provide any relevant clinical information, and (4) inaccessible papers that cannot be obtained upon request. Patients were not excluded based on demographics, and manuscripts were not excluded based on their publication status. The search strategy for KCS1 was to search literature from the year 2002 onwards and for KCS2 from the year 2013 onwards, as these were the years in which the causative genetic variants were discovered. The search terms are detailed elsewhere (21). Screening was performed by 2 people. Cases that could not immediately be excluded on the basis of the title and abstract were analyzed through full texts. A third person was responsible for settling disagreements.
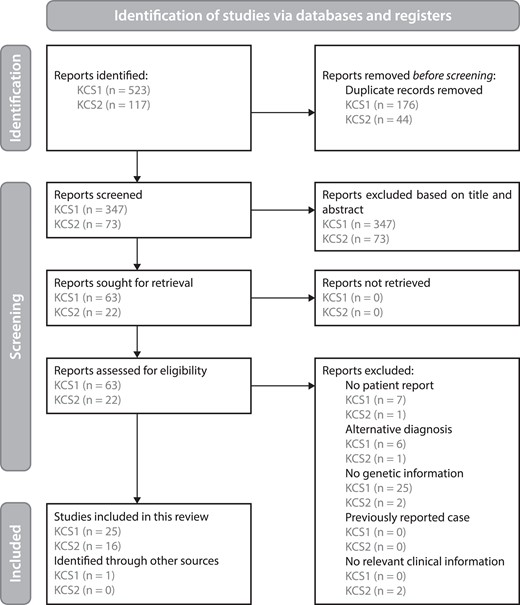
Screening for KCS1 and KCS2 case reports. Flow scheme detailing the screening methods used according to the PRISMA 2020 guidelines (25).
Extraction of Information
The first author's name, year of publication, basic patient information, genetic results, laboratory measurements, and symptoms were extracted from each paper. In the case of missing data, authors were contacted. Consanguinity was defined based on parents who were first-, second-, or third-degree relatives, or on the basis of the definition of consanguinity given by the authors if the relation was not specified. Symptoms were considered present whenever the authors stated this and/or based on a reference range stated in the paper. In the absence of such statements, the following definitions were used. Intrauterine growth retardation was defined as low birth weight of ≤−2 SD (or ≤3rd percentile) (26). In cases where only birth weight was given without SD/percentile, a reference range relevant to the ethnicity (if given) was used and again ≤−2 SD (or ≤3rd percentile) was the cut-off (27-29). Postnatal growth retardation was defined as short height of ≤−2 SD (or ≤3rd percentile). Growth retardation was considered proportional if limbs were in proportion or when the upper to lower segment ratio deviated less than 2 SD. For patients of ≥18 years, ratios between 0.80 and 1.07 were considered proportionate. Parathyroid hormone (PTH) levels were considered low when serum levels were below the lower limit of the normal reference range as described by MedScape (intact [whole] PTH: 1.06 pmol/L). Hypocalcemia was defined as serum total calcium values below the lower limit of the normal reference range for the particular age group as described by MedScape (<10 days: 1.9 mmol/L, umbilical: 2.25 mmol/L, 10 days-2 years: 2.3 mmol/L, child [2 years-18 years]: 2.2 mmol/L, adult [≥18 years]: 2.25 mmol/L), assuming normal serum albumin levels unless otherwise specified. If the age was unknown, the following values were used: a total serum calcium concentration <2.20 mmol/L in the presence of normal plasma protein concentrations or a serum ionized calcium concentration <1.17 mmol/L. Hypomagnesemia was assumed when serum levels were below the lower limit of the normal reference range: 0.7 mmol/L. Hyperphosphatemia was defined as phosphorus serum levels above the normal reference range as described by MedScape: adult and elderly (≥18 years): 1.45 mmol/L, child (2 months-18 years): 2.1 mmol/L, newborn (<2 months): 3 mmol/L. Growth hormone (GH) deficiency was defined as a GH stimulation test using arginine, glucagon, or insulin peak values of <10 ng/mL. Insulin-like growth factor 1 (IGF-1) deficiency was assumed when values were ≤ −2 SD (or ≤3rd percentile). Hypothyroidism was defined as free thyroxine (FT4) <0.8 ng/dL (10 pmol/L) and/or as thyroid stimulating hormone (TSH) >5 mIU/L. Reduced spleen function also entailed asplenia. Sleep apnea was solely based on the author's description. Hypoglycemia was defined as a glucose serum level of <2.775 mmol/L. Anemia was solely based on the author's description and no distinction was made based on the type of anemia. Calcifications in the kidneys were grouped under nephrocalcinosis. Calcifications in other tissues than the kidneys were grouped as calcifications. Nephrolithiasis (kidney stones) was grouped separately. Delayed closure of the anterior fontanelle was defined as a still patent anterior fontanelle from 2.5 years of age, based on information from MedScape. Osteosclerosis and bone cortical thickening and medullary stenosis were defined solely based on the author's statements. Delayed bone age was defined as a bone age of ≤ −2 SD. High alkaline phosphatase (ALP) levels were defined as >90th percentile for age and sex (30). If >18 years old, >120 IU/L was considered high ALP, and if age was not known >300 IU/L. Intellectual disability was defined as any sort of developmental delay, including language delay, fine motor/adaptive delay, cognitive delay, social delay, gross motor developmental delay, and/or behavioral disturbances, based on the definition from MedScape. Under seizures/spasms we grouped seizures, spasms, convulsions, and twitching. Infections were recorded independently of the frequency, and the following cases were included where a noninfectious cause could not be excluded but was unlikely: keratitis and otitis media. Dental abnormalities were defined to include tooth agenesis, edentulism, delayed dentition, premature shedding of permanent dentition, primary tooth retention, severe caries, early decay, taurodontism, and crown and enamel hypoplasia, or when the authors stated there were “dental abnormalities” without specifying. Eye problems were defined to include hyperopia, myopia, (pseudo)papilledema, cataracts, glaucoma, corneal opacity, astigmatism, strabismus, maculopathy, retinopathy, keratopathy, amblyopia, microphthalmia, optic nerve anomalies, and tortuous retinal vessels, or when the authors stated there were “eye abnormalities” without specifying. Male genital abnormalities were defined as any abnormalities to the male reproductive tract, either internal or external. Chronic kidney disease (CKD) was defined based on the diagnosis described by the authors, or based on the definition from UpToDate: an estimated glomerular filtration rate (eGFR) value of <60 mL/min per 1.73 m2 or albuminuria of >30 mg/day for a period of >3 months, urinary sediment abnormalities (detected by urinalysis); anatomic abnormalities were determined by imaging studies, and pathologic abnormalities were discovered upon biopsy and/or a history of kidney transplantation. Factors such as age and race were not taken into account. CKD was assumed to be absent in patients who were reported to have “normal kidney function.” Hypercalciuria was defined as a ratio of urine calcium to urine creatinine above the 95th percentile or below the 5th percentile for age (31). Renal magnesium wasting was defined as a fractional excretion >4% or a urinary magnesium excretion of >1 mmol/day, combined with low serum magnesium. Exact laboratory values were extracted as well. In the case of electrolyte values, only those measurements were used that were taken before the start of treatment with supplementals or GH. In addition, the age at the time of measurement was extracted. When multiple values were available for a patient, the most extreme values were included.
Data Analysis Systematic Literature Review
PTH, calcium, magnesium, phosphorus, GH, IGF-1, TSH, FT4, glucose, and ALP values that were reported in different units than the ones presented in this review were converted using the online tool www.unitslab.com.
Percentages were calculated for the occurrence of features in KCS. To determine whether features were specific to the KCS type, the percentage difference (PD) was calculated by dividing the absolute difference of the percentages by the average of the percentages, multiplied by 100. If the PD was >100% the feature was considered to be specific to either KCS type 1 or 2. Likewise, if the PD was <100%, the feature was considered to be overlapping both KCS types.
Laboratory measurement data are expressed as mean. Statistical analysis was performed using Graphpad Prism v8 (San Diego, CA, USA). For statistical analysis of 2 groups, the assumption of equal variances was tested using an F test. If assumptions were not met, data were first logarithmically transformed to achieve equal normality. Then, an unpaired t-test was used to test for differences in the data. If equal variance was still not achieved, the Mann–Whitney U test was performed. Differences with P < .05 were considered to be statistically significant.
Results
Clinical and Genetic Case Presentation
We identified 7 families with pathogenic variants in FAM111A. The main clinical presentations and genetic findings of 10 patients from these families are summarized in Table 1 and the pedigrees are indicated in Fig. 1B, with further clinical description below and in more detail elsewhere (21).
Phenotypic, clinical, and genetic characteristics of the patients in our cohort
| Demographics | F1-II-2 | F2-II-1 | F3-II-2 | F4-III-1 | F5-I-1 | F5-II-1 | F5-II-4 | F5-II-6 | F6-II-1 | F7-II-2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Female | Male | Male | Female | Female | Male | Female |
| Consanguinity | No | No | No | No | No | No | No | No | No | No |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Black | Caucasian |
| Genetics | ||||||||||
| FAM111A variant | c.1579C>A p.Pro527Thr | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His | ND | c.1604A>G p.Glu535Gly | c.1604A>G p.Glu535Gly | c.1604A>G p.Glu535Gly | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His |
| Zygosity | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | |
| Inheritance | de novo | de novo? | de novo | Autosomal dominant? | de novo? | Autosomal dominant? | Autosomal dominant? | Autosomal dominant? | de novo? | de novo? |
| ACMG score | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Uncertain significance | Uncertain significance | Uncertain significance | Pathogenic | Pathogenic | |
| Laboratory measurements | ||||||||||
| Hypoparathyroidism | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypocalcemia | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypomagnesemia | Yes | Yes | Yes | Yes | No | Yes | Yes | No | Yes | No |
| Hyperphosphatemia | Yes | Yes | No | No | No | No | No | No | Yes | Yes |
| Hypokalemia | ND | ND | Yes | Yes | ND | Yes | Yes | No | ND | ND |
| Clinical features | ||||||||||
| IUGR | No | No | Yes | No | No | |||||
| PNGR | Yes | Yes | Yes | Yes | Yes | Yes | ||||
| Proportionate short stature | No | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | |
| Calcifications | None evident | No | Cerebral | Cerebral | Hip | No | ||||
| Seizures/spasms | Yes | Yes | Yes (spasms only) | No | Yes | Yes | Yes | No | Yes | Yes |
| Del. Clos. AF | Yes | Yes | Yes | Yes | ||||||
| Cort. Thick. | Yes | No | Yes | Yes | Yes | |||||
| Med. Sten. | Yes | No | Yes | Yes | Yes | |||||
| ID | No | Mild | No | No | Yes | Yes | No | No | No | |
| Infections | Meningo-encephalitis with severe liver involvement, human herpesvirus 6 infection with acute hepatic failure | Hepatitis of unknown origin | Recurrent respiratory infections | No | No | No | Urinary tract infections | No | No | No |
| Dental problems | Oligodontia | Severe hypodontia, enamel hypoplasia | Delayed dental eruption, extensive dental caries | Loose dentition, hypodontia | ||||||
| Ocular defects | Microphthalmia, hypermetropia, astigmatism, anisometropia and macular scarring | No | High hypermetropia, ametropic amblyopia | Manifest hypermetropia, dot opacities off axis in the left lens, hyperopic discs, slight anterior protrusion and surrounding drusen in the right eye | Anterior ischemic optic neuropathy in the left eye | Yes | No | Papilledema, bilateral amblyopia, high hyperopia | ||
| Hearing problems | No | No | Deaf | No | ||||||
| Repr. organ abnormalities | One ovary missing, mild primary ovarian failure | Labial fusion | ||||||||
| Chronic kidney disease | G2A1 | No | No | G3bA1 | Kidney failure | G3a | G3a | Kidney transplant | No | |
| Nephrocalcinosis | No | |||||||||
| Other renal abnormalities | Kidney volumes at lower limit | No | Small kidneys | Small kidneys |
| Demographics | F1-II-2 | F2-II-1 | F3-II-2 | F4-III-1 | F5-I-1 | F5-II-1 | F5-II-4 | F5-II-6 | F6-II-1 | F7-II-2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Female | Male | Male | Female | Female | Male | Female |
| Consanguinity | No | No | No | No | No | No | No | No | No | No |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Black | Caucasian |
| Genetics | ||||||||||
| FAM111A variant | c.1579C>A p.Pro527Thr | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His | ND | c.1604A>G p.Glu535Gly | c.1604A>G p.Glu535Gly | c.1604A>G p.Glu535Gly | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His |
| Zygosity | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | |
| Inheritance | de novo | de novo? | de novo | Autosomal dominant? | de novo? | Autosomal dominant? | Autosomal dominant? | Autosomal dominant? | de novo? | de novo? |
| ACMG score | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Uncertain significance | Uncertain significance | Uncertain significance | Pathogenic | Pathogenic | |
| Laboratory measurements | ||||||||||
| Hypoparathyroidism | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypocalcemia | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypomagnesemia | Yes | Yes | Yes | Yes | No | Yes | Yes | No | Yes | No |
| Hyperphosphatemia | Yes | Yes | No | No | No | No | No | No | Yes | Yes |
| Hypokalemia | ND | ND | Yes | Yes | ND | Yes | Yes | No | ND | ND |
| Clinical features | ||||||||||
| IUGR | No | No | Yes | No | No | |||||
| PNGR | Yes | Yes | Yes | Yes | Yes | Yes | ||||
| Proportionate short stature | No | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | |
| Calcifications | None evident | No | Cerebral | Cerebral | Hip | No | ||||
| Seizures/spasms | Yes | Yes | Yes (spasms only) | No | Yes | Yes | Yes | No | Yes | Yes |
| Del. Clos. AF | Yes | Yes | Yes | Yes | ||||||
| Cort. Thick. | Yes | No | Yes | Yes | Yes | |||||
| Med. Sten. | Yes | No | Yes | Yes | Yes | |||||
| ID | No | Mild | No | No | Yes | Yes | No | No | No | |
| Infections | Meningo-encephalitis with severe liver involvement, human herpesvirus 6 infection with acute hepatic failure | Hepatitis of unknown origin | Recurrent respiratory infections | No | No | No | Urinary tract infections | No | No | No |
| Dental problems | Oligodontia | Severe hypodontia, enamel hypoplasia | Delayed dental eruption, extensive dental caries | Loose dentition, hypodontia | ||||||
| Ocular defects | Microphthalmia, hypermetropia, astigmatism, anisometropia and macular scarring | No | High hypermetropia, ametropic amblyopia | Manifest hypermetropia, dot opacities off axis in the left lens, hyperopic discs, slight anterior protrusion and surrounding drusen in the right eye | Anterior ischemic optic neuropathy in the left eye | Yes | No | Papilledema, bilateral amblyopia, high hyperopia | ||
| Hearing problems | No | No | Deaf | No | ||||||
| Repr. organ abnormalities | One ovary missing, mild primary ovarian failure | Labial fusion | ||||||||
| Chronic kidney disease | G2A1 | No | No | G3bA1 | Kidney failure | G3a | G3a | Kidney transplant | No | |
| Nephrocalcinosis | No | |||||||||
| Other renal abnormalities | Kidney volumes at lower limit | No | Small kidneys | Small kidneys |
For the inheritance, question marks are used when the parents were not genetically tested and so the mode of inheritance was assumed based on parental phenotype only.
Abbreviations: ACMG, American College of Medical Genetics; Cort. Thick., cortical thickening; Del. Clos. AF, delayed closure anterior fontanelle; ID, intellectual disability; IUGR, intrauterine growth retardation; Med. Sten.:, medullary stenosis; ND, not determined; PNGR, postnatal growth retardation; Repr. organ abnormalities, reproductive organ abnormalities.
Phenotypic, clinical, and genetic characteristics of the patients in our cohort
| Demographics | F1-II-2 | F2-II-1 | F3-II-2 | F4-III-1 | F5-I-1 | F5-II-1 | F5-II-4 | F5-II-6 | F6-II-1 | F7-II-2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Female | Male | Male | Female | Female | Male | Female |
| Consanguinity | No | No | No | No | No | No | No | No | No | No |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Black | Caucasian |
| Genetics | ||||||||||
| FAM111A variant | c.1579C>A p.Pro527Thr | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His | ND | c.1604A>G p.Glu535Gly | c.1604A>G p.Glu535Gly | c.1604A>G p.Glu535Gly | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His |
| Zygosity | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | |
| Inheritance | de novo | de novo? | de novo | Autosomal dominant? | de novo? | Autosomal dominant? | Autosomal dominant? | Autosomal dominant? | de novo? | de novo? |
| ACMG score | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Uncertain significance | Uncertain significance | Uncertain significance | Pathogenic | Pathogenic | |
| Laboratory measurements | ||||||||||
| Hypoparathyroidism | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypocalcemia | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypomagnesemia | Yes | Yes | Yes | Yes | No | Yes | Yes | No | Yes | No |
| Hyperphosphatemia | Yes | Yes | No | No | No | No | No | No | Yes | Yes |
| Hypokalemia | ND | ND | Yes | Yes | ND | Yes | Yes | No | ND | ND |
| Clinical features | ||||||||||
| IUGR | No | No | Yes | No | No | |||||
| PNGR | Yes | Yes | Yes | Yes | Yes | Yes | ||||
| Proportionate short stature | No | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | |
| Calcifications | None evident | No | Cerebral | Cerebral | Hip | No | ||||
| Seizures/spasms | Yes | Yes | Yes (spasms only) | No | Yes | Yes | Yes | No | Yes | Yes |
| Del. Clos. AF | Yes | Yes | Yes | Yes | ||||||
| Cort. Thick. | Yes | No | Yes | Yes | Yes | |||||
| Med. Sten. | Yes | No | Yes | Yes | Yes | |||||
| ID | No | Mild | No | No | Yes | Yes | No | No | No | |
| Infections | Meningo-encephalitis with severe liver involvement, human herpesvirus 6 infection with acute hepatic failure | Hepatitis of unknown origin | Recurrent respiratory infections | No | No | No | Urinary tract infections | No | No | No |
| Dental problems | Oligodontia | Severe hypodontia, enamel hypoplasia | Delayed dental eruption, extensive dental caries | Loose dentition, hypodontia | ||||||
| Ocular defects | Microphthalmia, hypermetropia, astigmatism, anisometropia and macular scarring | No | High hypermetropia, ametropic amblyopia | Manifest hypermetropia, dot opacities off axis in the left lens, hyperopic discs, slight anterior protrusion and surrounding drusen in the right eye | Anterior ischemic optic neuropathy in the left eye | Yes | No | Papilledema, bilateral amblyopia, high hyperopia | ||
| Hearing problems | No | No | Deaf | No | ||||||
| Repr. organ abnormalities | One ovary missing, mild primary ovarian failure | Labial fusion | ||||||||
| Chronic kidney disease | G2A1 | No | No | G3bA1 | Kidney failure | G3a | G3a | Kidney transplant | No | |
| Nephrocalcinosis | No | |||||||||
| Other renal abnormalities | Kidney volumes at lower limit | No | Small kidneys | Small kidneys |
| Demographics | F1-II-2 | F2-II-1 | F3-II-2 | F4-III-1 | F5-I-1 | F5-II-1 | F5-II-4 | F5-II-6 | F6-II-1 | F7-II-2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Female | Male | Male | Female | Female | Male | Female |
| Consanguinity | No | No | No | No | No | No | No | No | No | No |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | Black | Caucasian |
| Genetics | ||||||||||
| FAM111A variant | c.1579C>A p.Pro527Thr | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His | ND | c.1604A>G p.Glu535Gly | c.1604A>G p.Glu535Gly | c.1604A>G p.Glu535Gly | c.1706G>A p.Arg569His | c.1706G>A p.Arg569His |
| Zygosity | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | |
| Inheritance | de novo | de novo? | de novo | Autosomal dominant? | de novo? | Autosomal dominant? | Autosomal dominant? | Autosomal dominant? | de novo? | de novo? |
| ACMG score | Pathogenic | Pathogenic | Pathogenic | Pathogenic | Uncertain significance | Uncertain significance | Uncertain significance | Pathogenic | Pathogenic | |
| Laboratory measurements | ||||||||||
| Hypoparathyroidism | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypocalcemia | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypomagnesemia | Yes | Yes | Yes | Yes | No | Yes | Yes | No | Yes | No |
| Hyperphosphatemia | Yes | Yes | No | No | No | No | No | No | Yes | Yes |
| Hypokalemia | ND | ND | Yes | Yes | ND | Yes | Yes | No | ND | ND |
| Clinical features | ||||||||||
| IUGR | No | No | Yes | No | No | |||||
| PNGR | Yes | Yes | Yes | Yes | Yes | Yes | ||||
| Proportionate short stature | No | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | |
| Calcifications | None evident | No | Cerebral | Cerebral | Hip | No | ||||
| Seizures/spasms | Yes | Yes | Yes (spasms only) | No | Yes | Yes | Yes | No | Yes | Yes |
| Del. Clos. AF | Yes | Yes | Yes | Yes | ||||||
| Cort. Thick. | Yes | No | Yes | Yes | Yes | |||||
| Med. Sten. | Yes | No | Yes | Yes | Yes | |||||
| ID | No | Mild | No | No | Yes | Yes | No | No | No | |
| Infections | Meningo-encephalitis with severe liver involvement, human herpesvirus 6 infection with acute hepatic failure | Hepatitis of unknown origin | Recurrent respiratory infections | No | No | No | Urinary tract infections | No | No | No |
| Dental problems | Oligodontia | Severe hypodontia, enamel hypoplasia | Delayed dental eruption, extensive dental caries | Loose dentition, hypodontia | ||||||
| Ocular defects | Microphthalmia, hypermetropia, astigmatism, anisometropia and macular scarring | No | High hypermetropia, ametropic amblyopia | Manifest hypermetropia, dot opacities off axis in the left lens, hyperopic discs, slight anterior protrusion and surrounding drusen in the right eye | Anterior ischemic optic neuropathy in the left eye | Yes | No | Papilledema, bilateral amblyopia, high hyperopia | ||
| Hearing problems | No | No | Deaf | No | ||||||
| Repr. organ abnormalities | One ovary missing, mild primary ovarian failure | Labial fusion | ||||||||
| Chronic kidney disease | G2A1 | No | No | G3bA1 | Kidney failure | G3a | G3a | Kidney transplant | No | |
| Nephrocalcinosis | No | |||||||||
| Other renal abnormalities | Kidney volumes at lower limit | No | Small kidneys | Small kidneys |
For the inheritance, question marks are used when the parents were not genetically tested and so the mode of inheritance was assumed based on parental phenotype only.
Abbreviations: ACMG, American College of Medical Genetics; Cort. Thick., cortical thickening; Del. Clos. AF, delayed closure anterior fontanelle; ID, intellectual disability; IUGR, intrauterine growth retardation; Med. Sten.:, medullary stenosis; ND, not determined; PNGR, postnatal growth retardation; Repr. organ abnormalities, reproductive organ abnormalities.
Family 1
The proband (patient F1-II-2) is Caucasian and was born with a weight of 2960 g and a length of 49 cm. At 8 weeks of age, he presented with seizures in the setting of hypocalcemia, hypomagnesemia, hyperphosphatemia, and inappropriately low PTH. He developed growth retardation and closure of the anterior fontanelle was delayed. An ophthalmic examination revealed microphthalmia with hypermetropia, astigmatism, anisometropia, and pigmented areas in the right macula, which was suspected to be due to macular scarring. At 4 years old, cortical thickening and medullary stenosis of the long bones could be demonstrated. CKD stage G2A1 (eGFR 76 mL/min/1.73 m2, urinary albumin/creatinine ratio 7 mg/g) in the absence of nephrocalcinosis was present at 9 years of age. The patient has also suffered recurrent severe infections. Whole exome sequencing revealed a heterozygous c.1579C>A variant in FAM111A, predicted to cause a p.Pro527Thr substitution. This FAM111A variant was not detected in the parents or the sister of the proband.
Family 2
Patient F2-II-1 is a Caucasian boy with a weight of 3250 g at birth, a length of 52 centimeters, and a head circumference of 32 cm. Growth retardation, determined by low weight and height, began postnatally and was proportionate. GH levels were not tested and the IGF-1 level was normal at 91 ng/mL at the age of 8 years. He has had persistent hypomagnesemia since birth, while low PTH levels, hypocalcemia, and hyperphosphatemia were only detected during the first month of life, at which time the patient experienced seizures. Renal function was normal. Bone age was delayed (6 years and 3 months at 8 years and 9 months). No ophthalmic abnormalities were noted. Notably, the patient had mild intellectual disability, although this was not formally assessed. At the age of 9 months, the patient had hepatitis of unknown origin. Genetic testing revealed the recurrent heterozygous FAM111A variant c.1706G>A (p.Arg569His).
Family 3
Patient F3-II-2 is a Caucasian woman with a disproportionate short stature (−1.3 SD difference between subischial leg length and sitting height), due to intrauterine and postnatal growth retardation (previously reported in (32)). She had small, puffy hands and feet. Her long bones had a gracile appearance without cortical thickening or medullary stenosis. She also had delayed closure of the anterior fontanelle, oligodontia, and a slight widening of the pubic symphysis. At 18 years of age, she presented with symptomatic hypocalcemia, hypomagnesemia, and low PTH, consistent with hypoparathyroidism. She suffered from recurrent respiratory infections, most likely viral in origin. In addition, only 1 ovary was detected radiologically, which showed mild primary ovarian insufficiency. Since her initial description (32), whole exome sequencing revealed a heterozygous de novo FAM111A variant c.1706G>A (p.Arg569His), confirming the diagnosis of KCS2.
Family 4
Patient F4-III-1 is a Caucasian woman of short stature, weighing 60.3 kg and measuring 130 cm in height at the age of 25. Skeletal X-rays demonstrated disproportional shortening of the femora, with diaphyseal medullary stenosis affecting the femora, fibula, tibia, and humeri. However, cortical thickening was not observed. She had hypocalcemia, normal phosphate levels, hypomagnesemia, and a low PTH level. The patient was diagnosed with CKD stage 3b A1 at an eGFRcreat-cysC of 44 to 65 mL/min/1.73 m2 at age 20. She carries a heterozygous variant (c.1706G>A, p.Arg569His) in FAM111A as tested by whole genome sequencing. Her family members were not genetically tested. However, the mother was diagnosed with KCS based on her clinical features and passed away after having been on renal replacement therapy for 18 months. The mother's half-sister was also suspected to have KCS based on classic skull and facial characteristics. She passed away at the age of 47 from heart failure.
Family 5
Patient F5-I-1 was a Caucasian male with a proportionate short stature (weight 50 kg, length 146 cm). He was diagnosed with hypocalcemia due to hypoparathyroidism. He also had intellectual disability, epilepsy, and calcifications in the basal ganglia. He had a transient ischemic attack at the age of 65. At the age of 66, he was diagnosed with CKD stage G3b, which progressed to kidney failure for which no kidney replacement therapy was started and of which he died at the age of 77 years (Fig. 3A). He had 8 children, of whom 4 had short stature. Three of these 4 children were medically evaluated, including a son (F5-II-1) and 2 daughters (F5-II-4 and F5-II-6). Similar to their father, these 3 patients also had hypoparathyroidism with hypocalcemia and developed CKD. CKD was of variable severity with slow progression in patient F5-II-4 (current eGFR 45 mL/min/1.73 m2, eGFR decline ∼1 mL/min/1.73 m2/year) (Fig. 3B), and more rapid CKD progression in patient F5-II-6 requiring kidney transplantation at the age of 59 years. In patient F5-II-1, CKD stage G3a (eGFR 64 mL/min/1.73 m2) was observed at age 57; he died a year later. In all 4 patients no cause for CKD was found other than possibly KCS2 or the related treatment, including vitamin D and calcium supplements, although no nephrocalcinosis or hypercalciuria were detected. Other notable findings were hypokalemia and hypomagnesemia, both due to renal losses in patient F5-II-4, and intellectual disability and cerebral calcifications in patient F5-II-1. The 3 patients who were investigated with whole exome sequencing (F5-II-1, F5-II-4, and F5-II-6) carried a heterozygous missense variant c.1604A>G in FAM111A, which was not previously reported and is predicted to result in the amino acid substitution p.Glu535Gly. This variant was not found in 2 unaffected family members (F5-II-3 and F5-III-4).
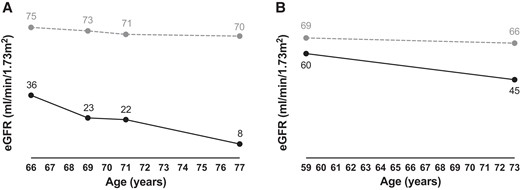
CKD progression. Progression of CKD shown as eGFR for age for patient (A) F5-I-1 and (B) F5-II-4. The dashed line represents the healthy (A) male and (B) female population (33).
Family 6
Patient F6-II-1, a Black male, was previously reported in the literature (34). Briefly, his phenotype consisted of postnatal growth retardation, thin calvaria with absent diploic space, and cortical thickening with medullary stenosis of the long bones. Additionally, biochemical analysis demonstrated low PTH levels with hypomagnesemia, hypocalcemia, and borderline hyperphosphatemia. No eye abnormalities were present and he had extensive dental caries at the age of 10 years. At the time of previous publication, the diagnosis of KCS was proposed on the basis of his clinical and biochemical features, but genetic testing was not available. We later confirmed this diagnosis by Sanger sequencing, which identified the heterozygous recurrent FAM111A variant (c.1706G>A, p.Arg569His).
Family 7
Patient F7-II-2 is a Caucasian female who was diagnosed with postnatal growth retardation: by 1 month of age, growth retardation was noted with a length of 47 cm (<0.01 percentile for age/sex). In addition to growth concerns, she was noted to have dysmorphic facial features with frontal bossing and delayed closure of her anterior fontanelle. She was found to have hypoparathyroidism around 2 weeks of life when she presented with seizures due to hypocalcemia. At that time, she had hyperphosphatemia. At the age of 4, she had decreased visual acuity (20-80 bilaterally), as well as bilateral amblyopia and high hyperopia, and she was noted to have bilateral optic disk swelling, requiring corrective lenses. Further work-up included a skeletal survey that showed calvarial hyperostosis, cortical thickening with narrowing of the medullary canal involving all of the long bones. She also has abnormal dentition and labial fusion. Targeted gene testing was performed for the patient (parents were not tested at the time, but both parents are of average height) and revealed the FAM111A c.1706G>A hotspot variant (p.Arg569His).
Patient Characteristics of our Cohort
Collectively, the KCS2 patients (n = 10 from 7 families) showed symptoms such as hypoparathyroidism, growth retardation, and skeletal dysplasia commonly described in KCS2 in the literature. However, we also demonstrated novel features including CKD and intellectual disability for some patients. To determine whether these findings are common in KCS, we performed a systematic review of all genetically confirmed KCS1 and KCS2 cases reported in the literature.
Systematic Literature Review
Using the search terms for our systematic literature review (strategy described in Methods, search strings (21)) we initially obtained a total of 1168 results for KCS1 and 305 for KCS2. After screening, we selected 26 papers with 175 cases for KCS1 and 16 papers with 30 cases for KCS2 (full list of included papers in Table S2 (21)). Demographical data and reported features were extracted from these papers and are presented in Table 2 (a similar table including our patient cohort can be found elsewhere (Table S3 (21)). The majority of KCS1 patients (≥90%) presented with intrauterine and postnatal growth retardation, delayed bone age, seizures, intellectual disability, and ocular and dental abnormalities. Biochemical investigations demonstrated hypocalcemia, hyperphosphatemia, and low PTH, consistent with hypoparathyroidism. KCS2 patients were generally (≥90%) reported to have postnatal growth retardation, delayed closure of the anterior fontanelle, medullary stenosis of the long bones and male genital, and ocular and dental abnormalities.
Comparison of KCS1 and 2 characteristics from the literature
| KCS1 (n = 175) | KCS2 (n = 30) | |||
|---|---|---|---|---|
| Number (total) | Valid % | Number (total) | Valid % | |
| Demographics | ||||
| Male/Female | 68/66 (134) | 18/11 (29) | ||
| Consanguinity | 87 (92) | 95% | 1 (16) | 6% |
| Laboratory measurements | ||||
| Low PTH | 121 (121) | 100% | 16 (20) | 80% |
| Hypocalcemia | 139 (139) | 100% | 24 (27) | 89% |
| Hypomagnesemia | 23 (30) | 77% | 10 (13) | 77% |
| Hyperphosphatemia | 69 (69) | 100% | 12 (18) | 67% |
| Hypercalciuria | 23 (27) | 85% | 1 (11) | 9% |
| Hypermagnesuria | 0 (0) | 0 (3) | ||
| GH deficiency | 9 (12) | 4 (11) | 36% | |
| Low IGF-1 | 12 (13) | 3 (8) | 38% | |
| Hypothyroidism | 12 (24) | 33% | 3 (4) | 75% |
| Hypoglycemia | 11 (16) | 0 (5) | 0% | |
| High ALP | 29 (61) | 48% | 2 (8) | 25% |
| Clinical features | ||||
| Growth retardation | 136 (136) | 100% | 29 (29) | 100% |
| IUGR | 127 (131) | 97% | 5 (16) | 31% |
| PNGR | 52 (52) | 100% | 23 (23) | 100% |
| Proportionate | 4 (7) | 10 (13) | 77% | |
| Delayed closure anterior fontanelle | 2 (5) | 14 (15) | 93% | |
| Cortical thickening | 1 (18) | 6% | 16 (20) | 80% |
| Medullary stenosis | 7 (46) | 15% | 27 (28) | 96% |
| Delayed bone age | 27 (30) | 90% | 3 (6) | 50% |
| Seizures/spasms | 103 (115) | 90% | 13 (16) | 81% |
| Sleep apnea | 20 (42) | 48% | 0 (7) | 0% |
| Intellectual disability | 74 (80) | 93% | 5 (24) | 21% |
| Infections | 43 (54) | 80% | 6 (10) | 60% |
| Sepsis | 8 | 15%a | 2 | 20%a |
| Otitis media | 20 | 37%a | 3 | 30%a |
| Respiratory infections | 20 | 37%a | 1 | 10%a |
| Viral infections | 2 | 4%a | 0 | 0%a |
| Bacterial infections | 2 | 4%a | 1 | 10%a |
| Reduced spleen function | 10 (16) | 1 (7) | 14% | |
| Anemia | 7 (28) | 25% | 5 (10) | 50% |
| Dental abnormalities | 47 (50) | 94% | 15 (16) | 94% |
| Delayed dentition | 41 | 82%a | 3 | 19%a |
| Tooth agenesis | 4 | 8%a | 8 | 50%a |
| Early decay/Extensive caries | 39 | 78%a | 4 | 25%a |
| Taurodontism | 3 | 6%a | 0 | 0%a |
| Ocular abnormalities | 57 (60) | 95% | 22 (23) | 96% |
| Hypermetropia | 6 | 10%a | 15 | 65%a |
| Microphthalmia | 27 | 45%a | 4 | 17%a |
| (Pseudo)papilledema | 5 | 8%a | 3 | 13%a |
| Corneal opacity | 10 | 17%a | 0 | 0%a |
| Cataracts | 6 | 10%a | 1 | 4%a |
| Strabismus | 25 | 42%a | 0 | 0%a |
| Maculopathy | 0 | 0%a | 2 | 9%a |
| Retinal vascular tortuosity | 21 | 35%a | 1 | 4%a |
| Male genital abnormalities | 9 (44) | 21% | 9 (10) | 90% |
| Micropenis | 7 | 16%a | 7 | 70%a |
| Microorchidism | 2 | 5%a | 5 | 50%a |
| Cryptorchidism | 5 | 11%a | 1 | 10%a |
| Nonrenal calcifications | 17 (33) | 52% | 8 (12) | 67% |
| Nephrocalcinosis | 30 (53) | 57% | 4 (10) | 40% |
| Nephrolithiasis | 4 (18) | 22% | 0 (6) | 0% |
| Chronic kidney disease | 0 (31) | 0% | 1 (7) | 14% |
| KCS1 (n = 175) | KCS2 (n = 30) | |||
|---|---|---|---|---|
| Number (total) | Valid % | Number (total) | Valid % | |
| Demographics | ||||
| Male/Female | 68/66 (134) | 18/11 (29) | ||
| Consanguinity | 87 (92) | 95% | 1 (16) | 6% |
| Laboratory measurements | ||||
| Low PTH | 121 (121) | 100% | 16 (20) | 80% |
| Hypocalcemia | 139 (139) | 100% | 24 (27) | 89% |
| Hypomagnesemia | 23 (30) | 77% | 10 (13) | 77% |
| Hyperphosphatemia | 69 (69) | 100% | 12 (18) | 67% |
| Hypercalciuria | 23 (27) | 85% | 1 (11) | 9% |
| Hypermagnesuria | 0 (0) | 0 (3) | ||
| GH deficiency | 9 (12) | 4 (11) | 36% | |
| Low IGF-1 | 12 (13) | 3 (8) | 38% | |
| Hypothyroidism | 12 (24) | 33% | 3 (4) | 75% |
| Hypoglycemia | 11 (16) | 0 (5) | 0% | |
| High ALP | 29 (61) | 48% | 2 (8) | 25% |
| Clinical features | ||||
| Growth retardation | 136 (136) | 100% | 29 (29) | 100% |
| IUGR | 127 (131) | 97% | 5 (16) | 31% |
| PNGR | 52 (52) | 100% | 23 (23) | 100% |
| Proportionate | 4 (7) | 10 (13) | 77% | |
| Delayed closure anterior fontanelle | 2 (5) | 14 (15) | 93% | |
| Cortical thickening | 1 (18) | 6% | 16 (20) | 80% |
| Medullary stenosis | 7 (46) | 15% | 27 (28) | 96% |
| Delayed bone age | 27 (30) | 90% | 3 (6) | 50% |
| Seizures/spasms | 103 (115) | 90% | 13 (16) | 81% |
| Sleep apnea | 20 (42) | 48% | 0 (7) | 0% |
| Intellectual disability | 74 (80) | 93% | 5 (24) | 21% |
| Infections | 43 (54) | 80% | 6 (10) | 60% |
| Sepsis | 8 | 15%a | 2 | 20%a |
| Otitis media | 20 | 37%a | 3 | 30%a |
| Respiratory infections | 20 | 37%a | 1 | 10%a |
| Viral infections | 2 | 4%a | 0 | 0%a |
| Bacterial infections | 2 | 4%a | 1 | 10%a |
| Reduced spleen function | 10 (16) | 1 (7) | 14% | |
| Anemia | 7 (28) | 25% | 5 (10) | 50% |
| Dental abnormalities | 47 (50) | 94% | 15 (16) | 94% |
| Delayed dentition | 41 | 82%a | 3 | 19%a |
| Tooth agenesis | 4 | 8%a | 8 | 50%a |
| Early decay/Extensive caries | 39 | 78%a | 4 | 25%a |
| Taurodontism | 3 | 6%a | 0 | 0%a |
| Ocular abnormalities | 57 (60) | 95% | 22 (23) | 96% |
| Hypermetropia | 6 | 10%a | 15 | 65%a |
| Microphthalmia | 27 | 45%a | 4 | 17%a |
| (Pseudo)papilledema | 5 | 8%a | 3 | 13%a |
| Corneal opacity | 10 | 17%a | 0 | 0%a |
| Cataracts | 6 | 10%a | 1 | 4%a |
| Strabismus | 25 | 42%a | 0 | 0%a |
| Maculopathy | 0 | 0%a | 2 | 9%a |
| Retinal vascular tortuosity | 21 | 35%a | 1 | 4%a |
| Male genital abnormalities | 9 (44) | 21% | 9 (10) | 90% |
| Micropenis | 7 | 16%a | 7 | 70%a |
| Microorchidism | 2 | 5%a | 5 | 50%a |
| Cryptorchidism | 5 | 11%a | 1 | 10%a |
| Nonrenal calcifications | 17 (33) | 52% | 8 (12) | 67% |
| Nephrocalcinosis | 30 (53) | 57% | 4 (10) | 40% |
| Nephrolithiasis | 4 (18) | 22% | 0 (6) | 0% |
| Chronic kidney disease | 0 (31) | 0% | 1 (7) | 14% |
Numbers represent the number of patients reported as having the feature out of the total number of patients for whom the feature was measured/described (thus omitting missing data) and from this a percentage was calculated (valid %) if the feature was described in more than 10% of the reported patients (KCS1: 17 patients, KCS2: 3 patients). The total number of patients we reviewed is n = 175 for KCS1 and n = 30 for KCS2.
Definitions for the variables can be found in “Materials and Methods.”
Abbreviations: IUGR, intrauterine growth retardation; PNGR, postnatal growth retardation; PTH, parathyroid hormone.
Percentages calculated using the total number of patients for whom infections or dental/ocular/genital defects were described.
Comparison of KCS1 and 2 characteristics from the literature
| KCS1 (n = 175) | KCS2 (n = 30) | |||
|---|---|---|---|---|
| Number (total) | Valid % | Number (total) | Valid % | |
| Demographics | ||||
| Male/Female | 68/66 (134) | 18/11 (29) | ||
| Consanguinity | 87 (92) | 95% | 1 (16) | 6% |
| Laboratory measurements | ||||
| Low PTH | 121 (121) | 100% | 16 (20) | 80% |
| Hypocalcemia | 139 (139) | 100% | 24 (27) | 89% |
| Hypomagnesemia | 23 (30) | 77% | 10 (13) | 77% |
| Hyperphosphatemia | 69 (69) | 100% | 12 (18) | 67% |
| Hypercalciuria | 23 (27) | 85% | 1 (11) | 9% |
| Hypermagnesuria | 0 (0) | 0 (3) | ||
| GH deficiency | 9 (12) | 4 (11) | 36% | |
| Low IGF-1 | 12 (13) | 3 (8) | 38% | |
| Hypothyroidism | 12 (24) | 33% | 3 (4) | 75% |
| Hypoglycemia | 11 (16) | 0 (5) | 0% | |
| High ALP | 29 (61) | 48% | 2 (8) | 25% |
| Clinical features | ||||
| Growth retardation | 136 (136) | 100% | 29 (29) | 100% |
| IUGR | 127 (131) | 97% | 5 (16) | 31% |
| PNGR | 52 (52) | 100% | 23 (23) | 100% |
| Proportionate | 4 (7) | 10 (13) | 77% | |
| Delayed closure anterior fontanelle | 2 (5) | 14 (15) | 93% | |
| Cortical thickening | 1 (18) | 6% | 16 (20) | 80% |
| Medullary stenosis | 7 (46) | 15% | 27 (28) | 96% |
| Delayed bone age | 27 (30) | 90% | 3 (6) | 50% |
| Seizures/spasms | 103 (115) | 90% | 13 (16) | 81% |
| Sleep apnea | 20 (42) | 48% | 0 (7) | 0% |
| Intellectual disability | 74 (80) | 93% | 5 (24) | 21% |
| Infections | 43 (54) | 80% | 6 (10) | 60% |
| Sepsis | 8 | 15%a | 2 | 20%a |
| Otitis media | 20 | 37%a | 3 | 30%a |
| Respiratory infections | 20 | 37%a | 1 | 10%a |
| Viral infections | 2 | 4%a | 0 | 0%a |
| Bacterial infections | 2 | 4%a | 1 | 10%a |
| Reduced spleen function | 10 (16) | 1 (7) | 14% | |
| Anemia | 7 (28) | 25% | 5 (10) | 50% |
| Dental abnormalities | 47 (50) | 94% | 15 (16) | 94% |
| Delayed dentition | 41 | 82%a | 3 | 19%a |
| Tooth agenesis | 4 | 8%a | 8 | 50%a |
| Early decay/Extensive caries | 39 | 78%a | 4 | 25%a |
| Taurodontism | 3 | 6%a | 0 | 0%a |
| Ocular abnormalities | 57 (60) | 95% | 22 (23) | 96% |
| Hypermetropia | 6 | 10%a | 15 | 65%a |
| Microphthalmia | 27 | 45%a | 4 | 17%a |
| (Pseudo)papilledema | 5 | 8%a | 3 | 13%a |
| Corneal opacity | 10 | 17%a | 0 | 0%a |
| Cataracts | 6 | 10%a | 1 | 4%a |
| Strabismus | 25 | 42%a | 0 | 0%a |
| Maculopathy | 0 | 0%a | 2 | 9%a |
| Retinal vascular tortuosity | 21 | 35%a | 1 | 4%a |
| Male genital abnormalities | 9 (44) | 21% | 9 (10) | 90% |
| Micropenis | 7 | 16%a | 7 | 70%a |
| Microorchidism | 2 | 5%a | 5 | 50%a |
| Cryptorchidism | 5 | 11%a | 1 | 10%a |
| Nonrenal calcifications | 17 (33) | 52% | 8 (12) | 67% |
| Nephrocalcinosis | 30 (53) | 57% | 4 (10) | 40% |
| Nephrolithiasis | 4 (18) | 22% | 0 (6) | 0% |
| Chronic kidney disease | 0 (31) | 0% | 1 (7) | 14% |
| KCS1 (n = 175) | KCS2 (n = 30) | |||
|---|---|---|---|---|
| Number (total) | Valid % | Number (total) | Valid % | |
| Demographics | ||||
| Male/Female | 68/66 (134) | 18/11 (29) | ||
| Consanguinity | 87 (92) | 95% | 1 (16) | 6% |
| Laboratory measurements | ||||
| Low PTH | 121 (121) | 100% | 16 (20) | 80% |
| Hypocalcemia | 139 (139) | 100% | 24 (27) | 89% |
| Hypomagnesemia | 23 (30) | 77% | 10 (13) | 77% |
| Hyperphosphatemia | 69 (69) | 100% | 12 (18) | 67% |
| Hypercalciuria | 23 (27) | 85% | 1 (11) | 9% |
| Hypermagnesuria | 0 (0) | 0 (3) | ||
| GH deficiency | 9 (12) | 4 (11) | 36% | |
| Low IGF-1 | 12 (13) | 3 (8) | 38% | |
| Hypothyroidism | 12 (24) | 33% | 3 (4) | 75% |
| Hypoglycemia | 11 (16) | 0 (5) | 0% | |
| High ALP | 29 (61) | 48% | 2 (8) | 25% |
| Clinical features | ||||
| Growth retardation | 136 (136) | 100% | 29 (29) | 100% |
| IUGR | 127 (131) | 97% | 5 (16) | 31% |
| PNGR | 52 (52) | 100% | 23 (23) | 100% |
| Proportionate | 4 (7) | 10 (13) | 77% | |
| Delayed closure anterior fontanelle | 2 (5) | 14 (15) | 93% | |
| Cortical thickening | 1 (18) | 6% | 16 (20) | 80% |
| Medullary stenosis | 7 (46) | 15% | 27 (28) | 96% |
| Delayed bone age | 27 (30) | 90% | 3 (6) | 50% |
| Seizures/spasms | 103 (115) | 90% | 13 (16) | 81% |
| Sleep apnea | 20 (42) | 48% | 0 (7) | 0% |
| Intellectual disability | 74 (80) | 93% | 5 (24) | 21% |
| Infections | 43 (54) | 80% | 6 (10) | 60% |
| Sepsis | 8 | 15%a | 2 | 20%a |
| Otitis media | 20 | 37%a | 3 | 30%a |
| Respiratory infections | 20 | 37%a | 1 | 10%a |
| Viral infections | 2 | 4%a | 0 | 0%a |
| Bacterial infections | 2 | 4%a | 1 | 10%a |
| Reduced spleen function | 10 (16) | 1 (7) | 14% | |
| Anemia | 7 (28) | 25% | 5 (10) | 50% |
| Dental abnormalities | 47 (50) | 94% | 15 (16) | 94% |
| Delayed dentition | 41 | 82%a | 3 | 19%a |
| Tooth agenesis | 4 | 8%a | 8 | 50%a |
| Early decay/Extensive caries | 39 | 78%a | 4 | 25%a |
| Taurodontism | 3 | 6%a | 0 | 0%a |
| Ocular abnormalities | 57 (60) | 95% | 22 (23) | 96% |
| Hypermetropia | 6 | 10%a | 15 | 65%a |
| Microphthalmia | 27 | 45%a | 4 | 17%a |
| (Pseudo)papilledema | 5 | 8%a | 3 | 13%a |
| Corneal opacity | 10 | 17%a | 0 | 0%a |
| Cataracts | 6 | 10%a | 1 | 4%a |
| Strabismus | 25 | 42%a | 0 | 0%a |
| Maculopathy | 0 | 0%a | 2 | 9%a |
| Retinal vascular tortuosity | 21 | 35%a | 1 | 4%a |
| Male genital abnormalities | 9 (44) | 21% | 9 (10) | 90% |
| Micropenis | 7 | 16%a | 7 | 70%a |
| Microorchidism | 2 | 5%a | 5 | 50%a |
| Cryptorchidism | 5 | 11%a | 1 | 10%a |
| Nonrenal calcifications | 17 (33) | 52% | 8 (12) | 67% |
| Nephrocalcinosis | 30 (53) | 57% | 4 (10) | 40% |
| Nephrolithiasis | 4 (18) | 22% | 0 (6) | 0% |
| Chronic kidney disease | 0 (31) | 0% | 1 (7) | 14% |
Numbers represent the number of patients reported as having the feature out of the total number of patients for whom the feature was measured/described (thus omitting missing data) and from this a percentage was calculated (valid %) if the feature was described in more than 10% of the reported patients (KCS1: 17 patients, KCS2: 3 patients). The total number of patients we reviewed is n = 175 for KCS1 and n = 30 for KCS2.
Definitions for the variables can be found in “Materials and Methods.”
Abbreviations: IUGR, intrauterine growth retardation; PNGR, postnatal growth retardation; PTH, parathyroid hormone.
Percentages calculated using the total number of patients for whom infections or dental/ocular/genital defects were described.
Our analysis revealed a significant overlap (here defined as PD < 100%, calculated by dividing the absolute difference by the average of the percentages, multiplied by 100) of the phenotypic spectrum of KCS1 and KCS2, including postnatal growth retardation (KCS1: 100%, KCS2: 100% [PD 0%]), low PTH (100%, 80% [PD 22%]), hypocalcemia (100%, 89% [PD 12%]), hypomagnesemia (77%, 77% [PD 0%]), hyperphosphatemia (100%, 67% [PD 40%]), seizures or spasms (90%, 81% [PD 11%]), and nonrenal calcifications (52%, 67% [PD 25%]) that mostly pertained to the brain, especially the basal ganglia. Eye abnormalities were present in almost all KCS patients (95%, 96% [PD 1%]), with hypermetropia and maculopathy being the most typical for KCS2 (10% vs 65% [PD 147%] and 0% vs 9% [PD 200%], respectively), while corneal opacity, strabismus, and retinal vascular tortuosity were more common in KCS1 (17% vs 0% [PD 200%], 42% vs 0% [PD 200%], and 35% vs 4% [PD 159%], respectively). (Pseudo)papilledema, microphthalmia, and cataracts occurred in both KCS1 and KCS2 (8% vs 13% [PD 48%], 45% vs 17% [PD 90%], and 10% vs 4% [PD 86%], respectively). Dental abnormalities were also reported in most patients (94%, 94% [PD 0%]). Delayed dentition and extensive dental caries were reported more in KCS1 patients, although these symptoms also occur relatively frequently in KCS2 (82% vs 19% [PD 125%] and 78% vs 25% [PD 103%], respectively). Taurodontism was only found in KCS1, albeit at a low frequency (6%, 0% [PD 200%]). Tooth agenesis appeared to be more specific for KCS2 (8%, 50% [PD 145%]). Both KCS types showed a high occurrence of infections (80%, 60% [PD 29%]). In contrast, medullary stenosis of the long bones occurred more frequently in KCS2 than KCS1 (15%, 96% [PD 146%]), as well as male genital abnormalities (21%, 90% [PD 124%]). Delayed closure of the anterior fontanelle and cortical thickening of the long bones were also likely more common in KCS2, but were not routinely checked for in KCS1 (2/5 vs 14/15 and 1/18 vs 16/20). Only intrauterine growth retardation and intellectual disability were observed more frequently in KCS1 than in KCS2 (97% vs 31% [PD 103%] and 93% vs 21% [PD 126%], respectively). Sleep apnea and hypoglycemia may also be specific to KCS1, although reporting was low in literature. Combining clinical data of our patient cohort with the data from literature (Table S3 (21)), sleep apnea was reported absent in 9 out of 9 KCS2 patients and hypoglycemia in 11 out of 11 KCS2 patients, supporting that these features may be KCS1 specific (48% vs 0% [PD 200%] and 69% vs 0% [PD 200%], respectively).
To determine whether there was genotype–phenotype correlation within KCS2, all KCS2 cases were separated in 2 groups: hotspot mutation (c.1706G>A) and other pathogenic variants (Table S4 (21)). Only CKD has been significantly more reported in patients with different mutations (6/8 patients) compared with patients carrying the hotspot mutation (1/8 patients). However, it should be noted that 4 out of the 6 affected patients were family members. Interestingly, there was a lot of heterogeneity in symptoms even within the hotspot mutation group, suggesting the genotype and phenotype are not closely related in KCS2.
Laboratory measurements were extracted from the papers when available (Fig. 4; Fig. S1 including our patient cohort (21)). For the electrolytes, only measurements taken before the start of treatment were included. The results showed that calcium and magnesium levels were similarly low in KCS1 and KCS2 patients and that phosphorus was similarly increased (Fig. 4A-4C; Fig. S1A-1C (21)). While the variance in PTH levels was significantly different between the groups (F test, P < .05), a nonparametric test showed no significance (Mann–Whitney U test, P > .05) (Fig. 4D; Fig. S1D (21)). Vitamin D levels were reduced in several KCS patients (Fig. 4E; Fig. S1E (21)). The urine calcium to creatinine ratio was variable in KCS2 patients and increased in the only KCS1 patient for which a value was available (Fig. 4F; Fig. S1F (21)). Peak GH levels and IGF-1 levels were very rarely measured in KCS2 patients (1 and 2 times, respectively), so the groups could not be compared (Fig. 4G and 4H; Fig. S1G and S1H (21)). In KCS1 patients these values tended to be low. Glucose levels were normal in all patients for whom measurements were available, except 1 KCS1 patient who had high glucose levels, in contrast to the multiple reports of hypoglycemia in literature (Fig. 4I; Fig. S1I (21)). ALP levels appeared to be higher in KCS1 patients, which was only significant when patients from our cohort were included (unpaired t-test on logarithmically transformed values, P < .05) (Fig. 4J; Fig. S1J (21)). TSH was increased in KCS1 and significantly higher than in KCS2 patients (unpaired t-test on logarithmically transformed values, P < .05), while free FT4 levels were reduced in most KCS patients, although this was rarely measured in KCS2 (Fig. 4K and 4L; Fig. S1K and 1L (21)). Many KCS1 patients showed increased TSH and many had decreased FT4 levels.

Laboratory values KCS1 and KCS2 patients. Measurements of serum (A) total calcium, (B) phosphorus, (C) magnesium, (D) parathyroid hormone, and (E) 25-hydroxy vitamin D in KCS1 and KCS2 patients reported in literature, before the start of treatment, and (F) measurements of urine calcium:creatinine ratio. Squares represent patients older than 10 days (calcium), 2 months (phosphorus) or 7 years (calcium:creatinine), triangles younger than 10 days (calcium), 2 months (phosphorus) or 7 years (calcium:creatinine), and diamonds exact age unknown. Circles are used when age is not specified. Values related to the growth axis with (G) peak growth hormone and (H) IGF-1 levels (in SD: standard deviation from the mean). In addition, (I) glucose and (J) ALP levels where squares represent individuals with high ALP for age. Measurements related to thyroid functioning with (K) TSH and (L) FT4 levels. The indicated areas represent the reference range (age-dependent for calcium, phosphorus and calcium creatinine). For PTH, values below the detection limit were included as 0 pmol/L. ***P < .001.
Discussion
Here, we present 10 genetically confirmed KCS2 cases from 7 families. Our patient cohort further extends the spectrum of KCS2-associated features, showing the presence of CKD (60%) and intellectual disability (30%) in a subset of patients. Moreover, we performed a systematic review of the literature on KCS1 and KCS2, describing 205 KCS patients. Our systematic approach demonstrates that the phenotypes of KCS1 and KCS2 substantially overlap.
Overlapping symptoms in KCS1 and KCS2 include postnatal growth retardation, cerebral calcifications, seizures or muscle spasms, eye abnormalities such as pseudopapilledema and cataracts, and dental abnormalities including delayed dentition and early tooth decay. In addition, hypocalcemia, hypomagnesemia, low PTH, and hyperphosphatemia occur in both KCS types. This is a unique combination of electrolyte disturbances, otherwise only described in autosomal dominant hypocalcemia, caused by pathogenic variants in the calcium sensing receptor or G protein subunit alpha 11 (35, 36), and in 1 rare case of Gitelman syndrome caused by a pathogenic variant in the Na/Cl cotransporter (37). The fact that KCS1 patients show the electrolyte disturbances more consistently than KCS2 patients may indicate that serum electrolyte levels in KCS2 patients are more variable over time. This is supported by the fact that the range of PTH levels was significantly greater in KCS2 patients. Indeed, while a previous report described the absence of parathyroid glands in a KCS2 patient suggesting this to be the primary defect (38), patients with normal PTH have also been described (23, 39-41). Three of these 4 patients with normal PTH levels also had normal calcium levels, of whom 1 did have low serum magnesium (23, 39-41). Interestingly, hypercalciuria occurred much more frequently in KCS1 than in KCS2 (85% vs 9%), which may point toward a different cause of hypocalcemia in KCS2. We did find urinary magnesium loss in several KCS2 patients (F1-II-2, F3-II-2) with hypomagnesemia. In another previously reported KCS2 patient, magnesium supplementation corrected serum calcium and PTH levels (20), suggesting that hypocalcemia and low PTH levels were secondary to magnesium loss in this patient. We were also able to identify some features that are useful in discriminating between KCS1 and KCS2. Delayed closure of the anterior fontanelle, as well as cortical thickening and medullary stenosis of the long bones, are much more common in KCS2 than KCS1. Hypoparathyroidism typically leads to reduced bone turnover (42). However, as the specific bone phenotype of KCS2 is unique compared with other hypoparathyroidism-related disorders, the KCS2 bone phenotype appears to be caused by something else. Potentially, imbalance of other hormones such as calcitriol, GH, thyroid hormones, or sex hormones could contribute, but values for these hormones have not been reported frequently in KCS. Hypothyroidism has been reported in several KCS patients (12/24 in KCS1, 3/4 in KCS2), occurring both overtly and subclinically (18, 39, 43, 44). In KCS1, both primary and secondary hypothyroidism have been reported. In KCS2, TSH values in hypothyroidism were low, suggestive of secondary or tertiary hypothyroidism. Tertiary hypothyroidism was confirmed in 1 KCS2 patient (39). Other discriminatory features include intellectual disability and intrauterine growth retardation, which are more common in KCS1. In addition, sleep apnea, hypoglycemia, and reduced spleen function sometimes occur in KCS1 and have rarely been reported in KCS2.
Within our cohort, 6 patients from 3 different families had CKD at various stages and 1 even required kidney transplantation. In addition, the affected mother (F4-I-1) of 1 of the patients died from kidney failure. Previously, nephrocalcinosis has been reported as the main renal abnormality in KCS described in 30 KCS1 patients (45-49) and in 4 KCS2 patients (24, 50) (Table S5 (21)). In KCS1 there were also reports of nephrolithiasis and hydronephrosis (51). The fact that CKD has not been associated with KCS before may be because patients with KCS are often reported in childhood or adolescence (50), suggesting that KCS patients may have an increased risk of developing CKD with age. However, in 1 previously reported patient about whom we obtained additional information, CKD and nephrocalcinosis were present at a very young age (<2 years of age), challenging this hypothesis. It has been postulated that renal complications and eGFR decline could potentially be caused by supplementation of calcium and active vitamin D in patients with hypoparathyroidism (52, 53). However, in our CKD patients, the absence of hypercalciuria and/or renal calcifications does not support that the CKD resulted from calcium and vitamin D supplementation. Also, KCS1 patients present more frequently with hypercalciuria and nephrocalcinosis, but CKD has not been reported in these patients. While renal magnesium wasting was detected in some patients, calcium in urine was generally within the normal range in KCS2, as was the case in patients F3-II-2 and F5-II-4. However, in the context of low serum calcium, “normocalciuria” can still point toward impaired renal Ca2+ reabsorption. In patient F5-II-4, a thiazide test showed a limited increase in chloride excretion upon inhibition of the sodium chloride cotransporter, suggesting a defect in the distal convoluted tubule, which is an important segment in reabsorption of calcium and magnesium from the primary urine. However, at the RNA level, FAM111A is expressed in all segments of the kidney (54, 55). Further investigations on the role of FAM111A in the kidney are therefore warranted.
Intellectual disability and infections were also reported in several of our KCS2 cases, whereas these symptoms are classically associated with KCS1. Our systematic literature review supports the occurrence of intellectual disability in some patients with KCS2 (5/24), which is significantly less prevalent than in KCS1 (74/80). The literature also supports the occurrence of infections in both disorders (43/54 in KCS1 and 6/10 in KCS2). Although the prevalence is higher in KCS2, the infections appear to be more recurrent in KCS1. In both KCS types, sepsis has been reported that resulted in death (8/43 in KCS1 (56-58) and 2/6 in KCS2 (59, 60)). It needs to be taken into account that some cases in which infections were reported could be unrelated to the disorder. This finding therefore requires further analysis with a larger number of patients to be able to compare occurrence in the general population and occurrence in KCS patients. However, studies have shown that KCS1 patients have functional hyposplenism, high immunoglobulin A and E levels, and abnormal B- and T-cell distributions (57, 61). In a KCS2 patient, neutropenia, eosinophilia, increased immunoglobulin E levels, and a low T lymphocyte helper/suppressor ratio were described (62). These findings could point toward an underlying immune deficiency in KCS (57, 62). Infections may also be underrepresented in our systematic analysis as these were often not reported in case studies. We find in our systematic literature review that both bacterial and viral infections are reported in KCS1 and 2, and that otitis media and respiratory infections are most common. Specifically, Streptococcus pneumoniae infections have been reported a number of times (57, 62, 63). Infections by these bacteria include respiratory infections, meningitis, and otitis media, all of which have been reported in KCS patients. In line with this, it was found that KCS1 patients had low levels of antipneumococcal antibodies (61). However, these data are based on limited reports, and thus prospective cohort studies are required to identify the types of infections that occur in KCS, as this could provide more insight into a potential molecular mechanism.
KCS2 males were believed to be infertile or subfertile because there had been no reports of male KCS2 patients with children and due to reports of genital abnormalities, including microorchidism and micropenis (4, 18, 50, 64). However, here we report the first paternal inheritance of KCS2, demonstrating that not all men with KCS2, if any, are infertile. To date, all reported familial cases of KCS2 demonstrated maternal inheritance (1, 18, 19, 65). We also report on the absence of 1 ovary and mild primary ovarian insufficiency in a female patient (F3-II-2) and labial fusion in another (F7-II-2). Previously, a Couvelaire uterus was reported in KCS2 (1). This suggests that abnormalities of the reproductive system may not be a male-specific feature of KCS.
We also report a novel variant (c.1604A>G; p.Glu535Gly), which is of uncertain significance based on the American College of Medical Genetics criteria PM2, BP4, and PP1 (66-72). Filtering of whole exome sequencing data indicated this variant as the most likely candidate, in line with the phenotype of the patients. In addition, the glutamate at position 535 is conserved in primates and the variant occurs in the same peptidase domain as previously identified pathogenic variants. In another KCS2 patient, we identified the p.Pro527Thr pathogenic variant, which was previously reported in a patient diagnosed with GCLEB who passed away at the age of 8 months (4). As previously mentioned, GCLEB is also caused by missense mutations in FAM111A and represents a more severe phenotype that is perinatally lethal. Until now, all pathogenic variants found in GCLEB were different from the ones in KCS2, albeit in the same region of the protein. The fact that we now see a GCLEB pathogenic variant in a KCS2 patient challenges the close genotype–phenotype correlation that has been assumed between KCS2 and GCLEB (4). This correlation is further challenged by the heterogeneity in symptoms we observed in KCS2 patients who carry the hotspot mutation.
A limitation of our retrospective analysis is missing data. Although we have contacted the corresponding authors of the selected publications, it has not been possible to retrieve all missing demographic and clinical information. Consequently, for some of the less common symptoms in KCS, it remains difficult to determine the exact prevalence. The minimum sample sizes using a confidence interval of 95% and a 5% margin of error were 121 for KCS1 and 28 for KCS2, which was only reached for biological sex and growth retardation, so we could not calculate P values for population proportions to determine statistical differences. In the case of KCS1, the high consanguinity rate may contribute to additional clinical features in these patients that are not directly related to KCS. Additionally, the young age of many reported cases does not exclude the development of clinical features later in life. While the genetic confirmation enables us to make a proper distinction between KCS1 and KCS2, it could also introduce bias as genetic studies can often not be performed in developing countries. In addition, diagnosis often follows symptomatic episodes of hypocalcemia, which could introduce bias toward the reporting of features such as hypocalcemia and seizures. Finally, for the laboratory measurement data, there may an underrepresentation of values within the normal range, as in these cases sometimes no exact values are reported. A strength of our study is the analysis of the large number of genetically confirmed KCS patients, totaling over 250 patients, greater than any cohort to date. As more patients are genetically investigated, we will gain an even better understanding of the overlapping and the defining features of each KCS type, aiding diagnosis. Therefore, we encourage the genetic screening of KCS patients.
In conclusion, the phenotypes of KCS type 1 and 2 most commonly manifest with postnatal growth retardation and hypoparathyroidism. Intellectual disability is more common in KCS1, whereas KCS2 is associated with cortical thickening and medullary stenosis of the long bones, as well as delayed closure of the anterior fontanelle. We propose that renal function be monitored closely in KCS2 patients, even those without hypoparathyroidism, as these patients may have an increased risk of developing CKD with increased age.
Acknowledgments
We would like to acknowledge D. H. H. Viering for his help with the systematic review. We thank Joost Verlouw and the Department of Human Genetics of the Radboudumc for bioinformatics support of whole exome sequencing data. In addition, we would like to acknowledge and thank all authors who were willing to provide additional patient data for the systematic literature review: Dr. C. S. Choong and Dr. M. B. Abraham (73), Dr. S. A. Ajarmeh (74), Dr. O. David (51, 75), Dr. E. P. Buchanan and Dr. H. Streff (76), Dr. E. Eren (59), Dr. E. Lang, Dr. D. Konrad and Dr. C. Gerth-Kahlert (41), Dr. S. M. Nikkel and Dr. K.M. Boycott (18), Dr. A. Touati (49), and Dr. C. Yerawar (77).
Funding
This work was financially supported by the IMAGEN project which is cofunded by the PPP Allowance made available by Health∼Holland, Top Sector Life Sciences & Health, to stimulate public–private partnerships (IMplementation of Advancements in GENetic Kidney Disease, LSHM20009), the Dutch Kidney Foundation (19OP004), the Radboud-Glasgow Collaboration Fund 2020 and the HORIZON EUROPE European Research Council (ERC STG 101040682).
Author Contributions
Conceptualization: H.S., B.P.I., J.H.F.d.B. and J.G.J.H. Investigation: A.J.M.H.V., B.S., C.J.M., C.M.T., C.P., D.L., E.J.H., H.S., K.P.S., L.G., M.C.Z., M.B., M.K., M.A.L., R.M.T., and S.M. Validation: H.S., L.G., B.P.I., and J.H.F.d.B. Data curation: H.S. Writing—Original draft: H.S. and J.H.F.d.B. Writing—Review and editing: A.J.M.H.V., B.C.J.v.d.E., B.S., B.P.I., C.J.M., C.M.T., C.P., D.L., E.J.H., F.J.R., H.S., J.G.J.H., J.H.F.d.B., K.P.S., L.G., M.C.Z., M.B., M.K., M.A.L., R.M.T., and S.M. Visualization: HS. Supervision: J.H.F.d.B., E.J.H., R.M.T., and J.G.J.H.
Disclosures
The authors have nothing to disclose.
Data Availability
Restrictions apply to the availability of some or all data generated or analyzed during this study to preserve patient confidentiality. The corresponding author will on request detail the restrictions and any conditions under which access to some data may be provided.
Consent for Publication
Informed consent was obtained from all participants.
References
Abbreviations
- ALP
alkaline phosphatase
- CKD
chronic kidney disease
- eGFR
estimated glomerular filtration rate
- FAM111A
family with sequence similarity 111 member A
- FT4
free thyroxine
- GCLEB
gracile bone dysplasia
- GH
growth hormone
- IGF
insulin-like growth factor
- KCS
Kenny–Caffey syndrome
- PD
percentage difference
- PTH
parathyroid hormone
- SSS
Sanjad–Sakati syndrome
- TBCE
tubulin-specific chaperone E
- TSH
thyroid-stimulating hormone

{kind=link}
{kind=link}
{kind=link}
{kind=link}