





Abstract
Central precocious puberty (CPP) can have a familial form in approximately one-quarter of the children. The recognition of this inherited condition increased after the identification of autosomal dominant CPP with paternal transmission caused by mutations in the MKRN3 and DLK1 genes.
We aimed to characterize the inheritance and estimate the prevalence of familial CPP in a large multiethnic cohort; to compare clinical and hormonal features, as well as treatment response to GnRH analogs (GnRHa), in children with distinct modes of transmission; and to investigate the genetic basis of familial CPP.
We retrospectively studied 586 children with a diagnosis of CPP. Patients with familial CPP (n = 276) were selected for clinical and genetic analysis. Data from previous studies were grouped, encompassing sequencing of MKRN3 and DLK1 genes in 204 patients. Large-scale parallel sequencing was performed in 48 individuals from 34 families.
The prevalence of familial CPP was estimated at 22%, with a similar frequency of maternal and paternal transmission. Pedigree analyses of families with maternal transmission suggested an autosomal dominant inheritance. Clinical and hormonal features, as well as treatment response to GnRHa, were similar among patients with different forms of transmission of familial CPP. MKRN3 loss-of-function mutations were the most prevalent cause of familial CPP, followed by DLK1 loss-of-function mutations, affecting, respectively, 22% and 4% of the studied families; both affected exclusively families with paternal transmission. Rare variants of uncertain significance were identified in CPP families with maternal transmission.
We demonstrated a similar prevalence of familial CPP with maternal and paternal transmission. MKRN3 and DLK1 loss-of-function mutations were the major causes of familial CPP with paternal transmission.
Central precocious puberty (CPP) results from the premature reactivation of the hypothalamic-pituitary-gonadal axis, clinically manifested as the development of secondary sexual characteristics before the age of 8 years in girls and 9 years in boys (1). Familial CPP can be defined by diagnosis or clinical history of early sexual development in one or more first-, second-, or third-degree family member of a confirmed CPP case (2).
The first description of familial CPP dates back to 1981, when 3 related individuals, including 2 siblings and their father, were diagnosed with CPP (3). Subsequently, in 1986, an American brief report described 3 familial cases in a series of 58 CPP girls, estimating a frequency of familial CPP of 5.2% (4). In 2004, de Vries et al (2) demonstrated a notorious higher prevalence of familial CPP of 27.5% in a cohort of 156 Israeli children (42 girls and 1 boy affected) (2). Besides having studied a larger cohort, a more active investigation of the familial pubertal history may have contributed to the higher prevalence reported in the Israeli study. While the American study reported only females of first- and second-degree (mother, sister, and maternal grandmother) as potentially affected relatives, the Israeli study included male and female relatives of first-, second-, and third-degree of both maternal and paternal sides. In fact, a recent French study that evaluated 332 children with non-syndromic CPP found a similar prevalence of familial cases of 25% (5). Interestingly, the inheritance of familial CPP was suggested by de Vries et al (2) and corroborated by more recent studies (5, 6) as autosomal dominant with incomplete, sex-dependent penetrance, with higher penetrance in girls and a high prevalence of maternal transmission.
The clinical recognition of familial CPP has increased significantly after the identification of genetic causes, including loss-of-function mutations in two maternally imprinted genes (Makorin ring finger protein 3 [MKRN3] and Delta-like noncanonical Notch ligand 1 [DLK1]) in families with CPP, providing evidence of the importance of monogenic causes in the familial form of CPP (7, 8). Inactivating defects in both genes cause non-syndromic autosomal dominant CPP with exclusive paternal transmission. Despite the large number of families with maternally transmitted CPP described in previous studies, no genetic etiology has been elucidated in this form of transmission (2, 5, 6).
In the current study, we evaluated a large and multiethnic cohort of familial CPP. We estimated the prevalence of familial CPP and characterized the modes of inheritance and transmission. We also compared clinical and hormonal features, as well as treatment response to gonadotropin-releasing hormone (GnRH) analogs (GnRHa), among children with distinct forms of transmission of familial CPP. In addition, we characterized the genetic basis of a subset of patients submitted to genetic studies.
Methods
Study Design and Participants
In total, 586 children with confirmed diagnosis of CPP without central nervous system lesions were retrospectively evaluated, including Brazilian children (n = 242), Spanish children (n = 179), and a subgroup of children from multiple countries (n = 165) (Fig. 1). The retrospective evaluation was based on the children's medical files from 1988 to 2021. The Brazilian cohort was evaluated at a single tertiary center (Hospital das Clínicas, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brasil). The Spanish cohort was constituted by the Spanish PUBERE Registry, relying on 55 centers throughout the country and supported by the Spanish Society for Pediatric Endocrinology (Sociedad Española de Endocrinología Pediátrica). The cohort of 165 children from multiple countries comprised 10 different ethnic backgrounds: 93 Brazilians (from different health centers), 42 French, 6 Belgians, 4 Greeks, 5 Turkish, 5 Americans, 5 Argentinean, 2 British, 2 Israelis, and 1 Australian. Notably, all of them had familial CPP.
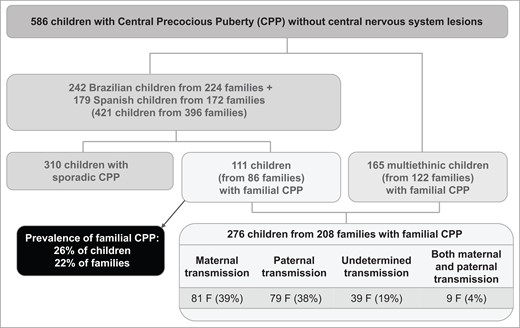
Flow chart of the study. F: Number of families.
The diagnosis of CPP was defined by the development of progressive pubertal signs before 8 years of age in girls and 9 years in boys and confirmed either by basal or GnRH-stimulated luteinizing hormone (LH) levels. Brain magnetic resonance imaging was performed in all CPP cases to rule out organic causes (1).
The familial form was defined by the presence of at least one first-, second-, or third-degree relative with a history of early sexual development. For female relatives, a report of clinically documented CPP or menarche ≤ 9 years old was considered (9). For male relatives, the considered parameters were either a report of clinically documented CPP or a self-report of pubertal signs (such as facial shaving, voice breaking, and early timing of growth spurt or growth completion) having occurred prior to their peers (10). First-degree relatives were defined as mother, father, brother(s) and sister(s); second-degree relatives as grandparents, aunt(s), and uncle(s); and third-degree relatives as cousins. For additional analysis, we considered families in which the menarche of female relatives occurred between 9 and 10 years old (corresponding to the interval between −2 and −1 SD score (SDS) of mean age of menarche in the general population) (11).
The mode of transmission of familial CPP included 4 groups according to the sex of the individual who transmitted the trait to the proband: 1) maternal: mother and or other maternal relatives affected, 2) paternal: father and or other paternal relatives affected, 3) undetermined: only siblings affected, 4) both maternal and paternal: at least one maternal and one paternal relative affected. The study protocol was approved by all Ethics Committees. Written informed consent was obtained from all patients and/or their legal guardians.
Clinical and Hormonal Data Collection
Available routine clinical and hormonal data were collected from medical files from children with familial CPP, including chronological age (CA) at first pubertal signs (breast development in girls and testicular enlargement in boys), CA at pubarche, CA and bone age (BA, Greulich and Pyle method) at first evaluation, bone age advancement (BA − CA), height SDS and body mass index (BMI) SDS at first evaluation. Hormonal data included basal and GnRH-stimulated LH and follicle-stimulating hormone (FSH) levels, measured either by electrochemiluminescence assays (ECLIAs; Roche Diagnostics; Cat# 11732234, RRID:AB_2800498 for LH and Cat# 11775863, RRID:AB_2800499 for FSH) after 2012, or immunofluorometric assays (IFMAs; Perkin Elmer; Cat# B031-101, RRID:AB_2783737 for LH and Cat# B017-201, RRID:AB_2783738 for FSH) before 2012; and basal estradiol or testosterone levels, measured either by ECLIA (Roche Diagnostics; Cat# 03000079, RRID:AB_2893079 for estradiol and Roche Cat# 05200067, RRID:AB_2783736 for testosterone) after 2012, or fluoroimmunoassays (FIA; Perkin Elmer; Cat# B056-101, RRID:AB_2927557 for estradiol and Cat# B050-201, RRID:AB_2783739 for testosterone) before 2012. Basal LH cutoff was considered in the pubertal range if >0.3 IU/L (ECLIA) or >0.6 IU/L (IFMA), and the GnRH-stimulated LH peak cutoff was >5 IU/L using ECLIA or >6.9 IU/L for girls and >9.6 IU/L for boys using IFMA (1). For those CPP patients treated with GnRHa, the CA (years) at the onset of treatment and CA (years), BA (years), height (SDS), and BMI (SDS) at the interruption of treatment were documented. Clinical and hormonal parameters during treatment included: regression or stabilization of secondary sexual characteristics, adequate growth velocity for CA, and suppressed sexual steroids levels with basal and/or GnRH-stimulated LH levels in the prepubertal range (basal LH <0.6 IU/L for IFMA and <0.3 IU/L for ECLIA; LH 120 minutes after GnRHa <4 IU/L) (12). Whenever available, CA at menarche (years), adult height (SDS) and target height (calculated using mid-parental height minus 6.5 cm for girls or plus 6.5 cm for boys; SDS) were also obtained. Target height range was established as target height ± 8.5 cm (13, 14). Additionally, birth weight and birth length were obtained, and children were classified as small, adequate, or large for gestational age.
Statistical Analysis
For descriptive statistics, categorical variables were expressed as percentages and continuous variables as medians and interquartile ranges (IQR) due to non-Gaussian distribution (tested by Kolmogorov-Smirnov normality test). Comparisons of categorical variables between CPP groups with different modes of transmission were performed using the chi-square test. The Kruskal-Wallis test was applied to compare continuous variables among groups. The statistical analysis was performed on the R × 64 platform (v 4.1.0, 2021, Vienna, Austria). Statistical significance was set at P < 0.05.
Genetic Analysis
The Developmental Endocrinology Unit at Hospital das Clínicas, São Paulo, has been a pioneer in studying genetic causes of idiopathic CPP (15). Prior to 2013, the genetic study was based on the sequencing of candidate genes by the Sanger method in selected patients. The use of whole-exome sequencing (WES) allowed the identification of loss-of-function mutations in MKRN3 associated with familial CPP in 2013 (7). Since then, all consecutive patients with CPP and normal magnetic resonance imaging were systematically invited to participate in the genetic studies. Therefore, we were able to study 204 patients with familial CPP who had available genomic leukocyte DNA. The coding region of MKRN3 was sequenced in all 204 patients by the Sanger method, a method previously described (16). Since 2017, when loss-of-function mutations in DLK1 were associated with familial CPP (8), the coding regions of this gene were also sequenced in all patients.
The probands’ relatives who were affected by CPP were also invited to participate in the genetic studies, as well as some of the unaffected relatives, for the purpose of genetic segregation. Families in which DNA from more than one family member were available for study were selected for WES (n = 34).
An additional 14 patients with familial CPP without pathogenic mutations in MKRN3 or DLK1 were submitted to targeted gene panel sequencing. These children were selected either for having presented an earlier age of thelarche (≤6 years) or for having the diagnosis of syndromic CPP; this last criterion was part of a previous published study on the genetic characterization of CPP associated with multiple anomalies (17).
The genomic positions and the raw data of all experiments were aligned using the GRCh37/hg19 assembly of the human genome reference.
Large-scale parallel sequencing
WES was performed in 34 patients with familial CPP along with one or more close relatives, according to previously published protocols (18). The libraries were constructed with the SureSelect Target Enrichment system (Agilent Technologies, CA, USA) and the sequences were generated in the Illumina HiSeq 2500 and NovaSeq 6000 platforms running on paired-end mode. In-house bioinformatic analysis was performed. Furthermore, an additional 14 patients with familial CPP were sequenced by a customized panel of targeted genes (746 genes known or candidates to be associated with developmental, endocrine, or metabolic disorders) using Agilent SureSelect assay (Agilent Technologies, CA, USA). Sequencing was performed on an Illumina NextSeq 500 platform in paired-end mode and in-house bioinformatic analysis was performed.
Data analysis of large-scale parallel sequencing
Large-scale parallel sequencing data were primarily filtered for loss-of-function and/or nonsynonymous variants present in the proband. Additionally, WES data were also filtered based on the family pedigree, selecting variants present in affected or possibly asymptomatic carriers’ family members and absent in unaffected family members. Next, sequencing data were screened for rare variants (minor allele frequency ≤0.01% in public and in-house databases) located in exonic regions and consensus splice site sequences. Splice site sequence analyses were performed using dbscSNV and Human Splice Finder. Public database included gnomAD (Genome Aggregation Database) and ABraOM (Online Archive of Brazilian Mutations) (19, 20); while the in-house exome database (SELA Laboratory) included a local cohort of 523 unrelated individuals with distinct medical disorders with the same ethnic background (21). Subsequently, the variant filtration prioritized variants based on their potential to be pathogenic: loss-of-function variants and missense variants predicted to be pathogenic by multiple in silico programs among SIFT, PolyPhen2, Mutation Taster, Mutation Assessor, Likelihood Ratio Test (LRT), Functional Analysis through Hidden Markov Models (FATHMM and FATHMM-MKL), Protein Variation Effect Analyzer (PROVEAN), MetaSVM, MetaLR, Mendelian Clinically Applicable Pathogenicity Score (M-CAP), Rare Exome Variant Ensemble Learner (REVEL), and Combined Annotation Dependent Depletion (CADD). The sequencing reads carrying candidate variants were visually inspected using the Integrative Genomics Viewer (IGV; Broad Institute, Cambridge, MA, USA) (18). Possible candidate variants were classified according to the American College of Medical Genetics and Genomics (ACMG) standards with 5 categories of pathogenicity: pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign (22). Variants classified as pathogenic, likely pathogenic, or VUS were then prioritized according to previous reports linking them—or the genes in which they are located—to CPP or other pubertal phenotypes (ClinVar, PubMed, Online Mendelian Inheritance in Man [OMIM]), gene function and pathway, gene expression in the hypothalamus (Genotype-Tissue Expression [GTEx] Portal), and association with pubertal milestones in Genome Wide Association Studies (GWAS).
Results
Prevalence of Familial CPP Among Brazilian and Spanish Patients
For patients followed at both the Division of Endocrinology of the University of São Paulo and the Spanish PUBERE registry, we had access to the medical registries of the total number of patients with CPP (421 children—402 girls and 19 boys—from 396 families). We estimated the prevalence of familial CPP in this cohort and identified 111 patients from 86 families (considering menarche ≤9 years old), corresponding to 26% of the cases and 22% of the families. An increased prevalence of familial CPP was observed when age of menarche was considered ≤10 years old: 131 patients from 106 families, corresponding to 31% of cases and 27% of the families.
Classification of the Transmission Mode of Familial CPP and Pedigree Analysis Among All Families Evaluated
Classification of the transmission mode was performed in all 208 families with familial CPP (Fig. 1). Maternal transmission was identified in 81 families (39%), paternal transmission in 79 families (38%), undetermined transmission in 39 families (19%), and both paternal and maternal transmission in the remaining 9 families (4%). No consanguinity was recognized among the evaluated families.
Pedigree analysis was possible in 157 out of the 208 (75.5%) families with complete information about family members’ pubertal onset on medical files (Table 1). The proportion of females was markedly predominant among affected individuals in all modes of transmission of familial CPP (369 of 409 individuals [90%]; Table 1). Among the 71 families with maternal transmission of CPP, most of them (n = 53 [75%]) had 2 generations known to be affected, while from the 59 families with the paternal transmission of CPP, almost half of them (n = 29 [49%]) had 2 generations known to be affected (Table 1). Notably, in 66% of the probands with maternally transmitted CPP the mother was affected, while in 22% of the probands with paternally transmitted CPP the father was known to be affected.
Number of affected generations in familial CPP according to the mode of transmission
| Mode of transmission | No. of families | Affected members (F/M) | 1 affected generation | 2 affected generations | ≥3 affected generations |
|---|---|---|---|---|---|
| Maternal | 71 | 183 (172/11) | 11 (15%) | 53 (75%) | 7 (10%) |
| Paternal | 59 | 154 (130/24) | 25 (42%) | 29 (49%) | 5 (9%) |
| Both maternal and paternal | 7 | 31 (27/4) | 0 | 3 (43%) | 4 (57%) |
| Undetermined | 20 | 41 (40/1) | 20 (100%) | 0 | 0 |
| Total | 157 | 409 (369/40) | 56 (36%) | 85 (54%) | 16 (10%) |
| Mode of transmission | No. of families | Affected members (F/M) | 1 affected generation | 2 affected generations | ≥3 affected generations |
|---|---|---|---|---|---|
| Maternal | 71 | 183 (172/11) | 11 (15%) | 53 (75%) | 7 (10%) |
| Paternal | 59 | 154 (130/24) | 25 (42%) | 29 (49%) | 5 (9%) |
| Both maternal and paternal | 7 | 31 (27/4) | 0 | 3 (43%) | 4 (57%) |
| Undetermined | 20 | 41 (40/1) | 20 (100%) | 0 | 0 |
| Total | 157 | 409 (369/40) | 56 (36%) | 85 (54%) | 16 (10%) |
Abbreviations: F, Female; M, Male.
Number of affected generations in familial CPP according to the mode of transmission
| Mode of transmission | No. of families | Affected members (F/M) | 1 affected generation | 2 affected generations | ≥3 affected generations |
|---|---|---|---|---|---|
| Maternal | 71 | 183 (172/11) | 11 (15%) | 53 (75%) | 7 (10%) |
| Paternal | 59 | 154 (130/24) | 25 (42%) | 29 (49%) | 5 (9%) |
| Both maternal and paternal | 7 | 31 (27/4) | 0 | 3 (43%) | 4 (57%) |
| Undetermined | 20 | 41 (40/1) | 20 (100%) | 0 | 0 |
| Total | 157 | 409 (369/40) | 56 (36%) | 85 (54%) | 16 (10%) |
| Mode of transmission | No. of families | Affected members (F/M) | 1 affected generation | 2 affected generations | ≥3 affected generations |
|---|---|---|---|---|---|
| Maternal | 71 | 183 (172/11) | 11 (15%) | 53 (75%) | 7 (10%) |
| Paternal | 59 | 154 (130/24) | 25 (42%) | 29 (49%) | 5 (9%) |
| Both maternal and paternal | 7 | 31 (27/4) | 0 | 3 (43%) | 4 (57%) |
| Undetermined | 20 | 41 (40/1) | 20 (100%) | 0 | 0 |
| Total | 157 | 409 (369/40) | 56 (36%) | 85 (54%) | 16 (10%) |
Abbreviations: F, Female; M, Male.
Clinical and Hormonal Features of Patients With Familial CPP
Clinical and hormonal data were available in 145 girls and 9 boys from 127 families with familial CPP. Due to the small number of male patients, boys were excluded from the comparative analysis. Five girls who had CPP with both maternal and paternal transmission were also excluded from the comparative analysis. Therefore, comparison was made among 56 girls with maternal transmission, 56 girls with paternal transmission, and 28 girls with undetermined transmission of CPP (Table 2).
Clinical and hormonal features of patients with familial CPP according to the mode of transmission
| Clinical and hormonal features | Maternal transmission (n = 56) Median/IQR | Paternal transmission (n = 56) Median/IQR | Undetermined transmission (n = 28) Median/IQR | Pa |
|---|---|---|---|---|
| Thelarche CA, y | 6.5 (6, 7.1) | 6.2 (5.6, 7) | 7.1 (6.1, 7.4) | 0.029 |
| Pubarche CA, y | 7 (6.5, 8) | 7 (6, 7.5) | 7.4 (6.2, 7.7) | 0.485 |
| First evaluation | ||||
| CA, y | 7.6 (6.9, 8.3) | 7.7 (6.4, 8.3) | 7.8 (7.5, 8.5) | 0.339 |
| BA, y | 10 (9, 11) | 10 (8.3, 11) | 11 (8.9, 11.4) | 0.541 |
| BA advancement, y | 2.3 (1.5, 3.1) | 2.1 (0.9, 2.9) | 1.5 (1.1, 2.6) | 0.152 |
| Height SDS | 1.7 (1.1, 2.3) | 1.6 (0.5, 2.2) | 1.0 (0.4, 1.8) | 0.104 |
| BMI SDS | 0.8 (0.1, 1.6) | 0.7 (0.4, 1.4) | 0.6 (0.04, 1.4) | 0.789 |
| Basal LH,bIU/L | 0.8 (0.1, 1.2) | 1.4 (0.3, 4.2) | 1.1 (0.2, 1.8) | 0.126 |
| Basal FSH,bIU/L | 3.4 (2.0, 4.7) | 4.2 (2.4, 6.0) | 4.4 (3.0, 5.9) | 0.508 |
| Basal LH within pubertal rangec | 33/51 (64.7%) | 31/50 (62%) | 16/26 (61.5%) | 0.947d |
| Estradiol, pg/mL | 18.2 (12.0, 33.2) | 20.1 (14.9, 36.3) | 17 (13.0, 29) | 0.398 |
| GnRH-stimulated peak LH,bIU/L | 13.0 (9.8, 17.1) | 9.7 (5.7, 18.5) | 15.5 (9.3, 19.1) | 0.652 |
| GnRH-stimulated peak FSH,bIU/L | 13.9 (10.3, 15.7) | 14.0 (10.3, 24.2) | 15.5 (14.3, 16.6) | 0.375 |
| GnRH-stimulated LH peak in pubertal rangec | 30/32 (93.7%) | 20/21 (95.2%) | 10/11 (90.9%) | 0.891e |
| Clinical and hormonal features | Maternal transmission (n = 56) Median/IQR | Paternal transmission (n = 56) Median/IQR | Undetermined transmission (n = 28) Median/IQR | Pa |
|---|---|---|---|---|
| Thelarche CA, y | 6.5 (6, 7.1) | 6.2 (5.6, 7) | 7.1 (6.1, 7.4) | 0.029 |
| Pubarche CA, y | 7 (6.5, 8) | 7 (6, 7.5) | 7.4 (6.2, 7.7) | 0.485 |
| First evaluation | ||||
| CA, y | 7.6 (6.9, 8.3) | 7.7 (6.4, 8.3) | 7.8 (7.5, 8.5) | 0.339 |
| BA, y | 10 (9, 11) | 10 (8.3, 11) | 11 (8.9, 11.4) | 0.541 |
| BA advancement, y | 2.3 (1.5, 3.1) | 2.1 (0.9, 2.9) | 1.5 (1.1, 2.6) | 0.152 |
| Height SDS | 1.7 (1.1, 2.3) | 1.6 (0.5, 2.2) | 1.0 (0.4, 1.8) | 0.104 |
| BMI SDS | 0.8 (0.1, 1.6) | 0.7 (0.4, 1.4) | 0.6 (0.04, 1.4) | 0.789 |
| Basal LH,bIU/L | 0.8 (0.1, 1.2) | 1.4 (0.3, 4.2) | 1.1 (0.2, 1.8) | 0.126 |
| Basal FSH,bIU/L | 3.4 (2.0, 4.7) | 4.2 (2.4, 6.0) | 4.4 (3.0, 5.9) | 0.508 |
| Basal LH within pubertal rangec | 33/51 (64.7%) | 31/50 (62%) | 16/26 (61.5%) | 0.947d |
| Estradiol, pg/mL | 18.2 (12.0, 33.2) | 20.1 (14.9, 36.3) | 17 (13.0, 29) | 0.398 |
| GnRH-stimulated peak LH,bIU/L | 13.0 (9.8, 17.1) | 9.7 (5.7, 18.5) | 15.5 (9.3, 19.1) | 0.652 |
| GnRH-stimulated peak FSH,bIU/L | 13.9 (10.3, 15.7) | 14.0 (10.3, 24.2) | 15.5 (14.3, 16.6) | 0.375 |
| GnRH-stimulated LH peak in pubertal rangec | 30/32 (93.7%) | 20/21 (95.2%) | 10/11 (90.9%) | 0.891e |
Abbreviations: BA, bone age; BMI, body mass index; CA, chronological age; CPP, central precocious puberty; FSH, follicle-stimulating hormone; GnRH, gonadotropin-releasing hormone; IQR, interquartile range; LH, luteinizing hormone; SDS, standard deviation score.
P value was calculated for comparison among the 3 groups simultaneously using nonparametric Kruskal-Wallis test for continuous variables or chi-square test for categorical variables. Statistically significant P values were marked in bold.
For statistical purposes, only hormones measured by electrochemiluminescence assay (ECLIA) were included.
Hormones measured either by ECLIA or immunofluorometric assay (IFMA) were considered.
X-squared = 0.109.
X-squared = 0.231.
Clinical and hormonal features of patients with familial CPP according to the mode of transmission
| Clinical and hormonal features | Maternal transmission (n = 56) Median/IQR | Paternal transmission (n = 56) Median/IQR | Undetermined transmission (n = 28) Median/IQR | Pa |
|---|---|---|---|---|
| Thelarche CA, y | 6.5 (6, 7.1) | 6.2 (5.6, 7) | 7.1 (6.1, 7.4) | 0.029 |
| Pubarche CA, y | 7 (6.5, 8) | 7 (6, 7.5) | 7.4 (6.2, 7.7) | 0.485 |
| First evaluation | ||||
| CA, y | 7.6 (6.9, 8.3) | 7.7 (6.4, 8.3) | 7.8 (7.5, 8.5) | 0.339 |
| BA, y | 10 (9, 11) | 10 (8.3, 11) | 11 (8.9, 11.4) | 0.541 |
| BA advancement, y | 2.3 (1.5, 3.1) | 2.1 (0.9, 2.9) | 1.5 (1.1, 2.6) | 0.152 |
| Height SDS | 1.7 (1.1, 2.3) | 1.6 (0.5, 2.2) | 1.0 (0.4, 1.8) | 0.104 |
| BMI SDS | 0.8 (0.1, 1.6) | 0.7 (0.4, 1.4) | 0.6 (0.04, 1.4) | 0.789 |
| Basal LH,bIU/L | 0.8 (0.1, 1.2) | 1.4 (0.3, 4.2) | 1.1 (0.2, 1.8) | 0.126 |
| Basal FSH,bIU/L | 3.4 (2.0, 4.7) | 4.2 (2.4, 6.0) | 4.4 (3.0, 5.9) | 0.508 |
| Basal LH within pubertal rangec | 33/51 (64.7%) | 31/50 (62%) | 16/26 (61.5%) | 0.947d |
| Estradiol, pg/mL | 18.2 (12.0, 33.2) | 20.1 (14.9, 36.3) | 17 (13.0, 29) | 0.398 |
| GnRH-stimulated peak LH,bIU/L | 13.0 (9.8, 17.1) | 9.7 (5.7, 18.5) | 15.5 (9.3, 19.1) | 0.652 |
| GnRH-stimulated peak FSH,bIU/L | 13.9 (10.3, 15.7) | 14.0 (10.3, 24.2) | 15.5 (14.3, 16.6) | 0.375 |
| GnRH-stimulated LH peak in pubertal rangec | 30/32 (93.7%) | 20/21 (95.2%) | 10/11 (90.9%) | 0.891e |
| Clinical and hormonal features | Maternal transmission (n = 56) Median/IQR | Paternal transmission (n = 56) Median/IQR | Undetermined transmission (n = 28) Median/IQR | Pa |
|---|---|---|---|---|
| Thelarche CA, y | 6.5 (6, 7.1) | 6.2 (5.6, 7) | 7.1 (6.1, 7.4) | 0.029 |
| Pubarche CA, y | 7 (6.5, 8) | 7 (6, 7.5) | 7.4 (6.2, 7.7) | 0.485 |
| First evaluation | ||||
| CA, y | 7.6 (6.9, 8.3) | 7.7 (6.4, 8.3) | 7.8 (7.5, 8.5) | 0.339 |
| BA, y | 10 (9, 11) | 10 (8.3, 11) | 11 (8.9, 11.4) | 0.541 |
| BA advancement, y | 2.3 (1.5, 3.1) | 2.1 (0.9, 2.9) | 1.5 (1.1, 2.6) | 0.152 |
| Height SDS | 1.7 (1.1, 2.3) | 1.6 (0.5, 2.2) | 1.0 (0.4, 1.8) | 0.104 |
| BMI SDS | 0.8 (0.1, 1.6) | 0.7 (0.4, 1.4) | 0.6 (0.04, 1.4) | 0.789 |
| Basal LH,bIU/L | 0.8 (0.1, 1.2) | 1.4 (0.3, 4.2) | 1.1 (0.2, 1.8) | 0.126 |
| Basal FSH,bIU/L | 3.4 (2.0, 4.7) | 4.2 (2.4, 6.0) | 4.4 (3.0, 5.9) | 0.508 |
| Basal LH within pubertal rangec | 33/51 (64.7%) | 31/50 (62%) | 16/26 (61.5%) | 0.947d |
| Estradiol, pg/mL | 18.2 (12.0, 33.2) | 20.1 (14.9, 36.3) | 17 (13.0, 29) | 0.398 |
| GnRH-stimulated peak LH,bIU/L | 13.0 (9.8, 17.1) | 9.7 (5.7, 18.5) | 15.5 (9.3, 19.1) | 0.652 |
| GnRH-stimulated peak FSH,bIU/L | 13.9 (10.3, 15.7) | 14.0 (10.3, 24.2) | 15.5 (14.3, 16.6) | 0.375 |
| GnRH-stimulated LH peak in pubertal rangec | 30/32 (93.7%) | 20/21 (95.2%) | 10/11 (90.9%) | 0.891e |
Abbreviations: BA, bone age; BMI, body mass index; CA, chronological age; CPP, central precocious puberty; FSH, follicle-stimulating hormone; GnRH, gonadotropin-releasing hormone; IQR, interquartile range; LH, luteinizing hormone; SDS, standard deviation score.
P value was calculated for comparison among the 3 groups simultaneously using nonparametric Kruskal-Wallis test for continuous variables or chi-square test for categorical variables. Statistically significant P values were marked in bold.
For statistical purposes, only hormones measured by electrochemiluminescence assay (ECLIA) were included.
Hormones measured either by ECLIA or immunofluorometric assay (IFMA) were considered.
X-squared = 0.109.
X-squared = 0.231.
Thelarche occurred at a median CA of 6.5 years (IQR 6, 7.1) in girls with maternally transmitted CPP, 6.2 years (IQR 5.6, 7) in girls with paternally transmitted CPP, and 7.1 years (IQR 6.1, 7.4) in girls with undetermined transmission of CPP (P = 0.029). Post hoc analysis demonstrated that the difference in thelarche CA between girls with paternal vs undetermined transmission of CPP was statically significant (P = 0.037, vs P = 0.101 for maternal vs undetermined transmission and P = 0.269 for maternal vs paternal transmission). At first visit, girls with maternally transmitted CPP had a median BA advancement of 2.3 years (IQR 1.5, 3.1) and median height SDS of 1.7 (IQR 1.1, 2.3), while girls with paternally transmitted CPP had median BA advancement of 2.1 years (IQR 0.9, 2.9) and median height SDS of 1.6 (IQR 0.5, 2.2), and girls with undetermined transmission of CPP had median BA advancement of 1.5 years (IQR 1.1, 2.6) and median height SDS of 1.0 (IQR 0.4, 1.8) (P > 0.05). The median BMI SDS was in the normal range (> −1.0 and <1.0) for all 3 groups (P = 0.789).
Thirty-three of the 51 girls (64.7%) with maternally transmitted CPP who had available data for basal LH levels had values in the pubertal range, while 30 out of the 32 (93.7%) who had GnRH-stimulated LH peak measured reached a pubertal response. Thirty-one of the 50 girls (62%) with paternally transmitted CPP who had available data for basal LH levels had values in the pubertal range, while 20 out of 21 (95.2%) who had GnRH-stimulated LH peak measured reached a pubertal response. Sixteen of the 26 girls (61.5%) with undetermined transmission of CPP who had available data for basal LH levels had values in the pubertal range, while 10 out of 11 (90.9%) who had GnRH-stimulated LH peak measured reached a pubertal response. Hormone levels did not differ between groups (P > 0.05; Table 2).
Treatment of CPP with long-acting GnRHa
Among children treated with GnRHa, data were available in 31 girls with maternally transmitted CPP, 24 girls with paternally transmitted CPP, and 13 girls with the undetermined transmission of CPP (Table 3). The median treatment duration was 2.2 years (IQR 2.0, 2.9) in girls with maternally transmitted CPP, 2.6 years (IQR 2.0, 3.5) in girls with paternally transmitted CPP, and 2.0 years (IQR 1.6, 2.2) in girls with the undetermined transmission of CPP (P = 0.229; Table 3). All children reached an adequate clinical and hormone control levels, according to the criteria mentioned above in this manuscript (data not shown). In girls who achieved adult height before the conclusion of this study, it was within the target height range in 87.5% of those with maternally transmitted CPP, 81.8% of those with paternally transmitted CPP, and 75% of those with an undetermined transmission of CPP (P = 0.915); while it was below the target height range in the remaining girls, despite treatment with GnRHa.
Clinical features of patients with familial CPP before and after treatment with GnRHa according to the mode of transmission
| Treatment data | Maternal transmission (n = 31) Median/IQR | Paternal transmission (n = 24) Median/IQR | Undetermined transmission (n = 13) Median/IQR | Pa |
|---|---|---|---|---|
| Initial CA, y | 7.9 (7.0, 8.4) | 8.0 (7.3, 8.4) | 8.1 (7.9, 9.0) | 0.206 |
| Final CA, | 10.5 (9.9, 10.7) | 10.6 (10.0, 11.0) | 10.5 (10, 11.0) | 0.632 |
| Treatment duration, y | 2.2 (2.0, 2.9) | 2.6 (2.0, 3.5) | 2.0 (1.6, 2.2) | 0.229 |
| First evaluation after treatment | ||||
| BA, y | 12 (11.5, 12) | 12 (12, 13) | 12 (11.6, 12) | 0.256 |
| Height SDS | 1.6 (1.0, 1.9) | 1.3 (0.9, 2.1) | 0.6 (0.2, 1.3) | 0.152 |
| BMI SDS | 0.7 (0.2, 1.4) | 0.8 (0.3, 1.4) | 1.0 (0.6, 1.7) | 0.269 |
| Age at menarche, y | n= 27 11.2 (10.6, 12) | n= 21 12 (11.0, 12.0) | n= 8 11.8 (11.4, 12.1) | 0.289 |
| Time between interruption of treatment and menarche, y | 1.0 (0.6, 1.1) | 1.0 (0.5, 1.5) | 1.1 (1.0, 1.4) | 0.382 |
| AH SDS | n= 8 0.35 (0.0, 0.8) | n= 13 −0.45 (−0.8, 0.4) | n= 4 −1.25 (−2.1, −0.6) | 0.086 |
| TH SDS | −0.4 (−1.0, 0.1) | −0.5 (−1.0, 0.5) | −1.0 (−1.4, 0.5) | 0.612 |
| AH − TH SDS | 0.7 (0.4, 1.0) | 0.2 (−1.0, 1.2) | 0.2 (−0.9, 1.1) | 0.695 |
| AH within the TH range | 7/8 (87.5%) | 9/11 (81.8%) | 3/4 (75%) | 0.915b |
| Treatment data | Maternal transmission (n = 31) Median/IQR | Paternal transmission (n = 24) Median/IQR | Undetermined transmission (n = 13) Median/IQR | Pa |
|---|---|---|---|---|
| Initial CA, y | 7.9 (7.0, 8.4) | 8.0 (7.3, 8.4) | 8.1 (7.9, 9.0) | 0.206 |
| Final CA, | 10.5 (9.9, 10.7) | 10.6 (10.0, 11.0) | 10.5 (10, 11.0) | 0.632 |
| Treatment duration, y | 2.2 (2.0, 2.9) | 2.6 (2.0, 3.5) | 2.0 (1.6, 2.2) | 0.229 |
| First evaluation after treatment | ||||
| BA, y | 12 (11.5, 12) | 12 (12, 13) | 12 (11.6, 12) | 0.256 |
| Height SDS | 1.6 (1.0, 1.9) | 1.3 (0.9, 2.1) | 0.6 (0.2, 1.3) | 0.152 |
| BMI SDS | 0.7 (0.2, 1.4) | 0.8 (0.3, 1.4) | 1.0 (0.6, 1.7) | 0.269 |
| Age at menarche, y | n= 27 11.2 (10.6, 12) | n= 21 12 (11.0, 12.0) | n= 8 11.8 (11.4, 12.1) | 0.289 |
| Time between interruption of treatment and menarche, y | 1.0 (0.6, 1.1) | 1.0 (0.5, 1.5) | 1.1 (1.0, 1.4) | 0.382 |
| AH SDS | n= 8 0.35 (0.0, 0.8) | n= 13 −0.45 (−0.8, 0.4) | n= 4 −1.25 (−2.1, −0.6) | 0.086 |
| TH SDS | −0.4 (−1.0, 0.1) | −0.5 (−1.0, 0.5) | −1.0 (−1.4, 0.5) | 0.612 |
| AH − TH SDS | 0.7 (0.4, 1.0) | 0.2 (−1.0, 1.2) | 0.2 (−0.9, 1.1) | 0.695 |
| AH within the TH range | 7/8 (87.5%) | 9/11 (81.8%) | 3/4 (75%) | 0.915b |
Abbreviations: CPP, central precocious puberty; GnRHa, gonadotropin-releasing hormone analog; IQR, interquartile range; CA, chronological age; BA, bone age; SDS, standard deviation score; BMI, body mass index; AH, adult height; TH, target height.
P value was calculated for comparison among the 3 groups simultaneously using nonparametric Kruskal-Wallis test for continuous variables or chi-square test for categorical variables.
X-squared = 0.964.
Clinical features of patients with familial CPP before and after treatment with GnRHa according to the mode of transmission
| Treatment data | Maternal transmission (n = 31) Median/IQR | Paternal transmission (n = 24) Median/IQR | Undetermined transmission (n = 13) Median/IQR | Pa |
|---|---|---|---|---|
| Initial CA, y | 7.9 (7.0, 8.4) | 8.0 (7.3, 8.4) | 8.1 (7.9, 9.0) | 0.206 |
| Final CA, | 10.5 (9.9, 10.7) | 10.6 (10.0, 11.0) | 10.5 (10, 11.0) | 0.632 |
| Treatment duration, y | 2.2 (2.0, 2.9) | 2.6 (2.0, 3.5) | 2.0 (1.6, 2.2) | 0.229 |
| First evaluation after treatment | ||||
| BA, y | 12 (11.5, 12) | 12 (12, 13) | 12 (11.6, 12) | 0.256 |
| Height SDS | 1.6 (1.0, 1.9) | 1.3 (0.9, 2.1) | 0.6 (0.2, 1.3) | 0.152 |
| BMI SDS | 0.7 (0.2, 1.4) | 0.8 (0.3, 1.4) | 1.0 (0.6, 1.7) | 0.269 |
| Age at menarche, y | n= 27 11.2 (10.6, 12) | n= 21 12 (11.0, 12.0) | n= 8 11.8 (11.4, 12.1) | 0.289 |
| Time between interruption of treatment and menarche, y | 1.0 (0.6, 1.1) | 1.0 (0.5, 1.5) | 1.1 (1.0, 1.4) | 0.382 |
| AH SDS | n= 8 0.35 (0.0, 0.8) | n= 13 −0.45 (−0.8, 0.4) | n= 4 −1.25 (−2.1, −0.6) | 0.086 |
| TH SDS | −0.4 (−1.0, 0.1) | −0.5 (−1.0, 0.5) | −1.0 (−1.4, 0.5) | 0.612 |
| AH − TH SDS | 0.7 (0.4, 1.0) | 0.2 (−1.0, 1.2) | 0.2 (−0.9, 1.1) | 0.695 |
| AH within the TH range | 7/8 (87.5%) | 9/11 (81.8%) | 3/4 (75%) | 0.915b |
| Treatment data | Maternal transmission (n = 31) Median/IQR | Paternal transmission (n = 24) Median/IQR | Undetermined transmission (n = 13) Median/IQR | Pa |
|---|---|---|---|---|
| Initial CA, y | 7.9 (7.0, 8.4) | 8.0 (7.3, 8.4) | 8.1 (7.9, 9.0) | 0.206 |
| Final CA, | 10.5 (9.9, 10.7) | 10.6 (10.0, 11.0) | 10.5 (10, 11.0) | 0.632 |
| Treatment duration, y | 2.2 (2.0, 2.9) | 2.6 (2.0, 3.5) | 2.0 (1.6, 2.2) | 0.229 |
| First evaluation after treatment | ||||
| BA, y | 12 (11.5, 12) | 12 (12, 13) | 12 (11.6, 12) | 0.256 |
| Height SDS | 1.6 (1.0, 1.9) | 1.3 (0.9, 2.1) | 0.6 (0.2, 1.3) | 0.152 |
| BMI SDS | 0.7 (0.2, 1.4) | 0.8 (0.3, 1.4) | 1.0 (0.6, 1.7) | 0.269 |
| Age at menarche, y | n= 27 11.2 (10.6, 12) | n= 21 12 (11.0, 12.0) | n= 8 11.8 (11.4, 12.1) | 0.289 |
| Time between interruption of treatment and menarche, y | 1.0 (0.6, 1.1) | 1.0 (0.5, 1.5) | 1.1 (1.0, 1.4) | 0.382 |
| AH SDS | n= 8 0.35 (0.0, 0.8) | n= 13 −0.45 (−0.8, 0.4) | n= 4 −1.25 (−2.1, −0.6) | 0.086 |
| TH SDS | −0.4 (−1.0, 0.1) | −0.5 (−1.0, 0.5) | −1.0 (−1.4, 0.5) | 0.612 |
| AH − TH SDS | 0.7 (0.4, 1.0) | 0.2 (−1.0, 1.2) | 0.2 (−0.9, 1.1) | 0.695 |
| AH within the TH range | 7/8 (87.5%) | 9/11 (81.8%) | 3/4 (75%) | 0.915b |
Abbreviations: CPP, central precocious puberty; GnRHa, gonadotropin-releasing hormone analog; IQR, interquartile range; CA, chronological age; BA, bone age; SDS, standard deviation score; BMI, body mass index; AH, adult height; TH, target height.
P value was calculated for comparison among the 3 groups simultaneously using nonparametric Kruskal-Wallis test for continuous variables or chi-square test for categorical variables.
X-squared = 0.964.
Most children (78 of 97 with birth data available [80.4%]) were born appropriate for gestational age, while 17 of them were born small for gestational age and 2 were born large for gestational age. There was no statistically significant difference in the proportion of children born small, appropriate, or large for gestational age among these 3 groups (P = 0.428).
Genetic Analysis
DNA sequencing of MKRN3 and DLK1 was performed in 204 patients from 165 families. WES was performed in 101 individuals from 34 families. Additionally, 14 probands from distinct families underwent targeted gene panel sequencing. The pedigrees of families submitted to WES and targeted gene panel are shown in Fig. 2.

Pedigrees of families with familial central precocious puberty (CPP) submitted to whole-exome sequencing (WES) (A, B, C) and targeted panel sequencing (D). Squares indicate male family members, circles female family members, symbols with a slash deceased family members and bifurcated vertical lines dizygotic twins. Black symbols are affected subjects, white symbols are unaffected subjects, gray symbols denote early puberty (thelarche between 8 and 9 years of age and/or menarche between 9 and 10 years for females, and testicular enlargement between 9 and 10 years for males) and symbols with black circles mark asymptomatic carriers. Family members whose phenotype is unknown are marked with a question mark and brothers younger than 9 years and sisters younger than 8 years are labeled with an asterisk. Arrows indicate the proband in each family. A, Families with maternal transmission of CPP submitted to WES. B, Families with paternal transmission of CPP submitted to WES. Families 20-24 harbor MKRN3 loss-of-function mutations and family 25 DLK1 loss-of-function mutation. C, Families with both maternal and paternal (29 and 30) and undetermined (31-34) transmission of CPP submitted to WES. D, Families with maternal (35-39), paternal (40-43), both maternal and paternal (44-45), and undetermined (46-48) transmission of CPP submitted to targeted panel sequencing.
Genetic findings previously reported
The most prevalent monogenic cause of familial CPP was MKRN3 loss-of-function mutations (71 cases from 36 families), followed by DLK1 loss-of-function mutations (12 cases from 6 families). These pathogenic genetic defects were previously described, as referenced in Table 4.
Loss-of-function mutations in MKRN3 and DLK1 genes among patients with familial CPP
| Genetic diagnosis | No. of affected cases and families | Proportion among tested familial CPP cases | Proportion among tested paternally transmitted CPP cases | Inheritance and transmission | Sequencing methods | References |
|---|---|---|---|---|---|---|
| MKRN3mutations | 71 cases from 36 families | 35% of cases/22% of families | 77% of cases/57% of families | AD with paternal transmission | WES, Sanger | (7, 23, 24, 25, 26) |
| DLK1mutations | 12 cases from 6 families | 6% of cases/4% of families | 13% of cases/10% of families | AD with paternal transmission | Whole-genome sequencing, WES, Sanger | (8, 27) |
| Genetic diagnosis | No. of affected cases and families | Proportion among tested familial CPP cases | Proportion among tested paternally transmitted CPP cases | Inheritance and transmission | Sequencing methods | References |
|---|---|---|---|---|---|---|
| MKRN3mutations | 71 cases from 36 families | 35% of cases/22% of families | 77% of cases/57% of families | AD with paternal transmission | WES, Sanger | (7, 23, 24, 25, 26) |
| DLK1mutations | 12 cases from 6 families | 6% of cases/4% of families | 13% of cases/10% of families | AD with paternal transmission | Whole-genome sequencing, WES, Sanger | (8, 27) |
Abbreviations: AD, autosomal dominant; CPP, central precocious puberty; WES, whole-exome sequencing.
Loss-of-function mutations in MKRN3 and DLK1 genes among patients with familial CPP
| Genetic diagnosis | No. of affected cases and families | Proportion among tested familial CPP cases | Proportion among tested paternally transmitted CPP cases | Inheritance and transmission | Sequencing methods | References |
|---|---|---|---|---|---|---|
| MKRN3mutations | 71 cases from 36 families | 35% of cases/22% of families | 77% of cases/57% of families | AD with paternal transmission | WES, Sanger | (7, 23, 24, 25, 26) |
| DLK1mutations | 12 cases from 6 families | 6% of cases/4% of families | 13% of cases/10% of families | AD with paternal transmission | Whole-genome sequencing, WES, Sanger | (8, 27) |
| Genetic diagnosis | No. of affected cases and families | Proportion among tested familial CPP cases | Proportion among tested paternally transmitted CPP cases | Inheritance and transmission | Sequencing methods | References |
|---|---|---|---|---|---|---|
| MKRN3mutations | 71 cases from 36 families | 35% of cases/22% of families | 77% of cases/57% of families | AD with paternal transmission | WES, Sanger | (7, 23, 24, 25, 26) |
| DLK1mutations | 12 cases from 6 families | 6% of cases/4% of families | 13% of cases/10% of families | AD with paternal transmission | Whole-genome sequencing, WES, Sanger | (8, 27) |
Abbreviations: AD, autosomal dominant; CPP, central precocious puberty; WES, whole-exome sequencing.
In a family with maternal transmission of CPP, the male proband had CPP associated with other anomalies (autism spectrum disorder, tall stature, and dysmorphic features) (Family 11; Fig. 2A). The boy and his relatives (3 affected and 2 unaffected relatives) were submitted to WES, and 2 rare candidate variants segregating with the early onset of puberty were identified: 1) a missense substitution (p.Pro267Leu) in the Uridine diphosphate glycosyltransferase 2 family member 4 (UGT2B4) gene, classified as a VUS; and 2) a frameshift deletion (p.Phe144Leufs*14) in the McKusick-Kaufman syndrome (MKKS) gene. These genetic defects were previously described (17).
New possible candidate genes
A rare heterozygous missense variant (p.His650Arg) in the Tet methylcytosine dioxygenase 2 (TET2) was identified in a Spanish girl with maternally transmitted CPP (Family 4; Fig. 2A). This girl had thelarche at 7.5 years and her mother, who had a history of menarche at 9 years, presented the same variant (p.His650Arg), whereas her unaffected father had the wild-type sequence. Although absent in public databases, the missense variant (p.His650Arg) had a benign prediction in most in silico programs (Table 5). The TET2 gene was previously associated with age of menarche onset in a GWAS analysis (28).
Rare heterozygous variants of uncertain significance identified in patients with familial CPP
| Gene | Position | Variant | dbSNP ID | Allele frequency | In silico analysis | ACMG | Variant inheritance | Clinical and laboratory data of affected individuals | |
|---|---|---|---|---|---|---|---|---|---|
| gnomAD | ABraOM | ||||||||
| TET2 | chr4: 106157048 | NM_001127208: c.1949A > G: p.His650Arg | — | 0 | 0 | 4P/9B | VUS | Maternally inherited (Family 4; Fig. 2A) | Proband: Thelarche at CA 7.5 yr. First visit at CA 7.9 yr, BA 11 yr, B2PH1, H = 1.3 SDS, BMI = −0.05 SDS, basal LH = 1 IU/L (ECLIA). Mother: Menarche at CA 9 yr. |
| BRWD1 | chr21: 40568453 | NM_018963: c.6542C > T: p.Thr2181Met | rs148068634 | 0.0001138 | 0 | 0P/13B | VUS | Maternally inherited (Family 36; Fig. 2D) | Proband: Thelarche at CA 6 yr. First visit at CA 8.2 yr, BA 11.5 yr, B4PH3, H = 2.3 SDS, BMI = 1.4 SDS, basal LH = 1.5 IU/L (IFMA). Mother: Menarche at CA 10 yr. |
| chr21: 40574398 | NM_018963: c.4438A > G: p.Thr1480Ala | rs775782176 | 0.0000159 | 0 | 2P/11B | VUS | Sporadic CPP | Proband: Thelarche at CA 7.8 yr. First visit at CA 20 yr, B5PH5, H = −0.95 SDS, BMI = 1.3 SDS. | |
| chr21: 40585477 | NM_018963: c.3788A > G: p.Asn1263Ser | rs752901254 | 0.0000041 | 0 | 2P/11B | VUS | Sporadic CPP | Proband: Thelarche at CA 6 yr. First visit at CA 6.5 yr, BA 8.9 yr, B2PH2, H = 0.7 SDS, BMI = −0.3 SDS, basal LH = 0.6 IU/L (IFMA), GnRH-stimulated LH peak 9.5 IU/L (IFMA). | |
| UGT2B4 | chr4: 70350967 | NM_021139.3: c.1266_1269del: p.Ser422Argfs*7 | rs765274693 | 0.0000040 | 0 | — | VUS | Paternally inherited (Family 30; Fig. 2C) | Proband: Thelarche at CA 7.8 yr. First visit at CA 8 yr, BA 10.5 yr, B2PH1, H = 2.5 SDS, BMI = −0.2 SDS, basal LH = 0.6 IU/L (ECLIA). Father: Early puberty. |
| Gene | Position | Variant | dbSNP ID | Allele frequency | In silico analysis | ACMG | Variant inheritance | Clinical and laboratory data of affected individuals | |
|---|---|---|---|---|---|---|---|---|---|
| gnomAD | ABraOM | ||||||||
| TET2 | chr4: 106157048 | NM_001127208: c.1949A > G: p.His650Arg | — | 0 | 0 | 4P/9B | VUS | Maternally inherited (Family 4; Fig. 2A) | Proband: Thelarche at CA 7.5 yr. First visit at CA 7.9 yr, BA 11 yr, B2PH1, H = 1.3 SDS, BMI = −0.05 SDS, basal LH = 1 IU/L (ECLIA). Mother: Menarche at CA 9 yr. |
| BRWD1 | chr21: 40568453 | NM_018963: c.6542C > T: p.Thr2181Met | rs148068634 | 0.0001138 | 0 | 0P/13B | VUS | Maternally inherited (Family 36; Fig. 2D) | Proband: Thelarche at CA 6 yr. First visit at CA 8.2 yr, BA 11.5 yr, B4PH3, H = 2.3 SDS, BMI = 1.4 SDS, basal LH = 1.5 IU/L (IFMA). Mother: Menarche at CA 10 yr. |
| chr21: 40574398 | NM_018963: c.4438A > G: p.Thr1480Ala | rs775782176 | 0.0000159 | 0 | 2P/11B | VUS | Sporadic CPP | Proband: Thelarche at CA 7.8 yr. First visit at CA 20 yr, B5PH5, H = −0.95 SDS, BMI = 1.3 SDS. | |
| chr21: 40585477 | NM_018963: c.3788A > G: p.Asn1263Ser | rs752901254 | 0.0000041 | 0 | 2P/11B | VUS | Sporadic CPP | Proband: Thelarche at CA 6 yr. First visit at CA 6.5 yr, BA 8.9 yr, B2PH2, H = 0.7 SDS, BMI = −0.3 SDS, basal LH = 0.6 IU/L (IFMA), GnRH-stimulated LH peak 9.5 IU/L (IFMA). | |
| UGT2B4 | chr4: 70350967 | NM_021139.3: c.1266_1269del: p.Ser422Argfs*7 | rs765274693 | 0.0000040 | 0 | — | VUS | Paternally inherited (Family 30; Fig. 2C) | Proband: Thelarche at CA 7.8 yr. First visit at CA 8 yr, BA 10.5 yr, B2PH1, H = 2.5 SDS, BMI = −0.2 SDS, basal LH = 0.6 IU/L (ECLIA). Father: Early puberty. |
Abbreviations: ABraOM, Online Archive of Brazilian Mutations; ACMG, American College of Medical Genetics and Genomics; B, benign; BA, bone age; BMI, body mass index; B-PH, Breast and pubic hair stages according to Tanner and Marshal (12); CA, chronological age; chr, chromosome; CPP, central precocious puberty; dbSNP ID, Single Nucleotide Polymorphism Database Identifier; ECLIA, electrochemiluminescence assay; gnomAD, Genome Aggregation Database; GnRH, gonadotropin-releasing hormone; H, height; IFMA, immunofluorometric assay; LH, luteinizing hormone; P, pathogenic; SDS, standard deviation score; VUS, Variant of Uncertain Significance.
Rare heterozygous variants of uncertain significance identified in patients with familial CPP
| Gene | Position | Variant | dbSNP ID | Allele frequency | In silico analysis | ACMG | Variant inheritance | Clinical and laboratory data of affected individuals | |
|---|---|---|---|---|---|---|---|---|---|
| gnomAD | ABraOM | ||||||||
| TET2 | chr4: 106157048 | NM_001127208: c.1949A > G: p.His650Arg | — | 0 | 0 | 4P/9B | VUS | Maternally inherited (Family 4; Fig. 2A) | Proband: Thelarche at CA 7.5 yr. First visit at CA 7.9 yr, BA 11 yr, B2PH1, H = 1.3 SDS, BMI = −0.05 SDS, basal LH = 1 IU/L (ECLIA). Mother: Menarche at CA 9 yr. |
| BRWD1 | chr21: 40568453 | NM_018963: c.6542C > T: p.Thr2181Met | rs148068634 | 0.0001138 | 0 | 0P/13B | VUS | Maternally inherited (Family 36; Fig. 2D) | Proband: Thelarche at CA 6 yr. First visit at CA 8.2 yr, BA 11.5 yr, B4PH3, H = 2.3 SDS, BMI = 1.4 SDS, basal LH = 1.5 IU/L (IFMA). Mother: Menarche at CA 10 yr. |
| chr21: 40574398 | NM_018963: c.4438A > G: p.Thr1480Ala | rs775782176 | 0.0000159 | 0 | 2P/11B | VUS | Sporadic CPP | Proband: Thelarche at CA 7.8 yr. First visit at CA 20 yr, B5PH5, H = −0.95 SDS, BMI = 1.3 SDS. | |
| chr21: 40585477 | NM_018963: c.3788A > G: p.Asn1263Ser | rs752901254 | 0.0000041 | 0 | 2P/11B | VUS | Sporadic CPP | Proband: Thelarche at CA 6 yr. First visit at CA 6.5 yr, BA 8.9 yr, B2PH2, H = 0.7 SDS, BMI = −0.3 SDS, basal LH = 0.6 IU/L (IFMA), GnRH-stimulated LH peak 9.5 IU/L (IFMA). | |
| UGT2B4 | chr4: 70350967 | NM_021139.3: c.1266_1269del: p.Ser422Argfs*7 | rs765274693 | 0.0000040 | 0 | — | VUS | Paternally inherited (Family 30; Fig. 2C) | Proband: Thelarche at CA 7.8 yr. First visit at CA 8 yr, BA 10.5 yr, B2PH1, H = 2.5 SDS, BMI = −0.2 SDS, basal LH = 0.6 IU/L (ECLIA). Father: Early puberty. |
| Gene | Position | Variant | dbSNP ID | Allele frequency | In silico analysis | ACMG | Variant inheritance | Clinical and laboratory data of affected individuals | |
|---|---|---|---|---|---|---|---|---|---|
| gnomAD | ABraOM | ||||||||
| TET2 | chr4: 106157048 | NM_001127208: c.1949A > G: p.His650Arg | — | 0 | 0 | 4P/9B | VUS | Maternally inherited (Family 4; Fig. 2A) | Proband: Thelarche at CA 7.5 yr. First visit at CA 7.9 yr, BA 11 yr, B2PH1, H = 1.3 SDS, BMI = −0.05 SDS, basal LH = 1 IU/L (ECLIA). Mother: Menarche at CA 9 yr. |
| BRWD1 | chr21: 40568453 | NM_018963: c.6542C > T: p.Thr2181Met | rs148068634 | 0.0001138 | 0 | 0P/13B | VUS | Maternally inherited (Family 36; Fig. 2D) | Proband: Thelarche at CA 6 yr. First visit at CA 8.2 yr, BA 11.5 yr, B4PH3, H = 2.3 SDS, BMI = 1.4 SDS, basal LH = 1.5 IU/L (IFMA). Mother: Menarche at CA 10 yr. |
| chr21: 40574398 | NM_018963: c.4438A > G: p.Thr1480Ala | rs775782176 | 0.0000159 | 0 | 2P/11B | VUS | Sporadic CPP | Proband: Thelarche at CA 7.8 yr. First visit at CA 20 yr, B5PH5, H = −0.95 SDS, BMI = 1.3 SDS. | |
| chr21: 40585477 | NM_018963: c.3788A > G: p.Asn1263Ser | rs752901254 | 0.0000041 | 0 | 2P/11B | VUS | Sporadic CPP | Proband: Thelarche at CA 6 yr. First visit at CA 6.5 yr, BA 8.9 yr, B2PH2, H = 0.7 SDS, BMI = −0.3 SDS, basal LH = 0.6 IU/L (IFMA), GnRH-stimulated LH peak 9.5 IU/L (IFMA). | |
| UGT2B4 | chr4: 70350967 | NM_021139.3: c.1266_1269del: p.Ser422Argfs*7 | rs765274693 | 0.0000040 | 0 | — | VUS | Paternally inherited (Family 30; Fig. 2C) | Proband: Thelarche at CA 7.8 yr. First visit at CA 8 yr, BA 10.5 yr, B2PH1, H = 2.5 SDS, BMI = −0.2 SDS, basal LH = 0.6 IU/L (ECLIA). Father: Early puberty. |
Abbreviations: ABraOM, Online Archive of Brazilian Mutations; ACMG, American College of Medical Genetics and Genomics; B, benign; BA, bone age; BMI, body mass index; B-PH, Breast and pubic hair stages according to Tanner and Marshal (12); CA, chronological age; chr, chromosome; CPP, central precocious puberty; dbSNP ID, Single Nucleotide Polymorphism Database Identifier; ECLIA, electrochemiluminescence assay; gnomAD, Genome Aggregation Database; GnRH, gonadotropin-releasing hormone; H, height; IFMA, immunofluorometric assay; LH, luteinizing hormone; P, pathogenic; SDS, standard deviation score; VUS, Variant of Uncertain Significance.
A rare heterozygous missense variant (p.Thr2181Met) in the Bromodomain and WD repeat domain containing 1 (BRWD1) was identified in a Brazilian girl with maternally transmitted CPP submitted to target panel gene sequencing (Family 36; Fig. 2D). This girl had thelarche at 6 years and her mother, who had menarche at 10 years, harbored the same variant (p.Thr2181Met). The proband's brother also had a history of early sexual development, and a maternal first cousin had been treated for CPP with GnRHa. Only the mother's DNA was available for genetic study. Although rare in public databases, the missense variant (p.Thr2181Met) had a benign prediction in all in silico programs analyzed (Table 5). The BRWD1 gene was previously associated with age of menarche in 2 distinct GWAS (28, 29), as well as with male puberty timing (onset voice breaking) (10). Additionally, 2 other rare heterozygous variants of uncertain significance (p.Thr1480Ala and p.Asn1263Ser) were identified in the BRWD1 gene in 2 Brazilian girls with sporadic CPP who had been previously submitted to target panel gene sequencing as part of genetic investigation of CPP at the University of São Paulo laboratory (Table 5).
Finally, a rare heterozygous frameshift variant (p.Ser422Argfs*7) in the UGT2B4 was identified in a Spanish girl from a family with both maternal and paternal history of CPP (Family 30; Fig. 2C). This girl had thelarche at 7.8 years and her father, who had a history of early puberty, presented the same variant (p.Ser422Argfs*7), while her mother had the wild-type sequence. The UGT2B4 gene was previously associated with age of menarche in specific populations (30).
Discussion
A significant prevalence of familial CPP (25% to 27.5%) has been demonstrated in a few clinical studies in the last decades (2, 6). Interestingly, the current study demonstrated a prevalence of 22% of familial CPP in the largest Brazilian-Spanish cohort using a more restricted criterion of familial history (menarche ≤9 years). Notably, the prevalence of familial CPP increased to 27% if considering the age of menarche ≤10 years old, the same criteria used in previous studies (2, 5).
The predominance of maternal transmission in most previous familial CPP studies is remarkable (2, 5, 6). However, this form of transmission has not been associated with a definitive and recurrent monogenic cause so far. The prevalence of maternally and paternally transmitted CPP was very similar in this cohort of 208 families (39% of maternal transmission and 38% of paternal transmission). Noteworthy, familial history of precocious puberty is more easily recognized in mothers than in fathers, which could create a referral bias for maternally transmitted CPP. However, in this particular study, the proportion of families with the paternal transmission of CPP may also have been overestimated due to the more frequent referral of these families for genetic studies of MKRN3 and DLK1. Families with loss-of-function mutations in these maternally imprinted genes can have nonconsecutive generations affected due to male asymptomatic carriers who inherited the defect from their mothers. This is reflected by the significant proportion (42%) of only one generation known to be affected in CPP families with paternal transmission. In addition, while women often remember their age of menarche, most men do not have a precise remembrance of their pubertal milestones. The lack of this information can underestimate the number of affected fathers (22%) and other male relatives in these families. Oppositely, most families (85%) with maternal transmission of CPP had at least 2 generations known to be affected, suggesting an autosomal dominant inheritance, with direct maternal transmission in 67% of the cases. The great predominance of affected females in the family pedigrees (90%) suggests sex-dependent penetrance.
At diagnosis of CPP, the clinical and hormonal presentations were similar among children with maternal, paternal, or undetermined transmission of CPP, except for the CA of thelarche, which was earlier in girls with paternally transmitted CPP than in girls with undetermined transmission of CPP. In addition, an adequate and similar response to GnRHa treatment among the 3 groups with distinct modes of transmission of familial CPP was consistent with a previous study showing that patients with CPP due to MKRN3 loss-of-function mutations have anthropometric, metabolic, and reproductive outcomes after GnRHa treatment similar to those of idiopathic CPP (31). These findings may suggest that familial CPP from distinct modes of inheritance may involve factors from the same pathways or gene network.
The first description of loss-of-function mutations in the MKRN3 gene associated with familial CPP occurred in 2013 and was followed by many other studies (7, 23-26, 32, 33). In the current study, among the 204 familial CPP cases (from 165 families) submitted to genetic testing, MKRN3 loss-of-function mutations were identified in 35% of the patients and 22% of the families, contrasting with previous prevalence data ranging from 33% to 46% (7, 33). We demonstrated a high prevalence of MKRN3 mutations among paternally transmitted CPP patients (77%) and families (57%) in this study.
DLK1 loss-of-function mutations associated with familial CPP were first identified in a Brazilian family with 5 affected members by Dauber et al (8) in 2017. Subsequently, other distinct inactivating mutations were identified in familial CPP cases with paternal transmission and 1 de novo mutation was identified in a sporadic CPP case (25, 27). DLK1 mutations are especially relevant for their association with metabolic abnormalities manifested mainly in adulthood, which was attributed to the inhibitory effect of DLK1 over adipogenesis (27). The pathways by which DLK1 regulates puberty onset are still unknown. DLK1 loss-of-function mutations were identified in 6% of the patients and 4% of the families in this study. As expected, all patients harboring mutations in this maternally imprinted gene had paternal transmission of CPP. Although DLK1 mutations are currently considered a very rare cause of familial CPP, its prevalence in the paternally transmitted CPP cohort was notable: 13% of the patients and 10% of the families.
Despite the significant number of maternally transmitted CPP cases submitted to large-scale parallel sequencing in this study (62 individuals from 24 families), no definite monogenic cause was identified. We hypothesize that this form of familial CPP might be caused by either copy number variation, single nucleotide variation located in regulatory regions not covered by WES, epigenetic defects, mitochondrial DNA mutations, and/or oligogenic or polygenic inheritance. Interestingly, we have identified a rare heterozygous missense variant of uncertain significance in the TET2 (p.His650Arg) in a family with maternally transmitted CPP. TET2 is part of the family of ten-eleven translocation (tet) enzymes, responsible for DNA demethylation (34). Notably, DNA methylation is one of the best studied epigenetic mechanisms involved in modulating gene activity, and the importance of this process in pubertal onset regulation has been recognized through studies of kisspeptin repressors in animals (35, 36), and, more recently, through a study of the global methylation profile of girls with physiological or central precocious puberty showing a pattern of hypermethylation during the pubertal period compared to the prepubertal period (37). Interestingly, in vitro and mice experiments showed that GnRH transcription and secretion increased with tet2 overexpression, while its ablation led to a significant drop in GnRH expression (34). Moreover, TET2 was shown to be recruited by the Methyl-CPG-Binding Domain Protein 3 (MBD3) to promote DNA demethylase and may be involved in the mechanism by which MKRN3 promotes epigenetic silencing of GNRH1 expression (38). We hypothesized that the TET2 variant (p.His650Arg) found in this family with maternal transmission of CPP could lead to a gain-of-function of TET2, increasing the expression of GNRH1 via demethylation of its promoter, and ultimately leading to premature activation of the hypothalamic-pituitary-gonadal axis. TET2 was also associated with age at menarche in GWAS (28) and the role of this gene in a possible oligogenic or polygenic inheritance of familial CPP must be considered. Likewise, we identified rare heterozygous variants of uncertain significance (p.Thr2181Met, p.Thr1480Ala, and p.Asn1263Ser) in the BRWD1 in 3 different patients (1 with maternally transmitted familial CPP and 2 with sporadic CPP). BRWD1 is a transcriptional regulator whose gene was also associated with age at menarche and male puberty timing in GWAS (10, 28, 29) suggesting that this gene may also be associated with an oligogenic or polygenic inheritance model of CPP. Additionally, we identified a rare heterozygous frameshift variant of uncertain significance (p.Ser422Rfs*7) in the UGT2B4 in a girl with CPP and in her father, who had a history of premature sexual development (Family 30; Fig. 2C). The UGT2B4 gene encodes an enzyme involved in the glucuronidation of androgens and was also associated with age at menarche in specific populations (30, 39). We have previously described a rare heterozygous missense variant in this gene, segregating with maternally transmitted CPP in a Brazilian family submitted to WES (17), and we might consider that this gene could also be associated with an oligogenic or polygenic inheritance model of CPP.
In conclusion, we demonstrated that familial CPP can be prevalent, with a similar frequency of maternal and paternal transmission in a large multiethnic cohort of CPP. Although paternally transmitted CPP has been genetically characterized after the identification of mutations in the imprinted genes MKRN3 and DLK1, the genetic basis of maternally transmitted CPP remains to be elucidated. The current study revealed that CPP with maternal transmission seems to be autosomal dominant, with higher penetrance in females. In addition, clinical and hormonal features at diagnosis and the response to GnRHa treatment were similar among maternal, paternal, or undetermined transmission forms of familial CPP. MKRN3 loss-of-function mutations were the most prevalent cause of familial CPP with paternal transmission, followed by DLK1 loss-of-function mutations. Finally, a family history of CPP should be actively and routinely investigated allowing potential early diagnosis and treatment.
Financial Support
F.R.T. was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq # 140289/2020-8) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP # 2021/12205-2). A.P.M.C was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP # 2022/00719-4). B.B.M. is supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq # 303002/2016-6). V.N.B. and A.C.L are supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP # 2019/27631-7). A.C.L is supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq # 303183/2020-9).
Data Availability
The dataset generated and analyzed during the current study is not publicly available but is available from the corresponding author on reasonable request.
References
Abbreviations
- BA
bone age
- BMI
body mass index
- CA
chronological age
- CPP
central precocious puberty
- ECLIA
electrochemiluminescence assay
- FSH
follicle-stimulating hormone
- GnRH
gonadotropin-releasing hormone
- GnRHa
gonadotropin-releasing hormone analog
- LH
luteinizing hormone
- SDS
standard deviation score
- VUS
variant of uncertain significance
- WES
whole-exome sequencing

{kind=link}
{kind=link}