





Abstract
Immune checkpoint inhibitors (ICI) are cancer therapies that are approved for use in at least 19 different cancers. They function by stimulating immune cell responses against cancer, and their toxicities comprise a host of autoinflammatory syndromes that may impact any organ system. Endocrine toxicities occur in as high as 25% to 50% of ICI recipients, depending on the treatment regimen used. These toxicities vary in severity from mild, asymptomatic cases of subclinical hypothyroidism to severe, fatal cases of adrenal crisis, thyroid dysfunction, or diabetic ketoacidosis. Thus, timely recognition and treatment is critical. Herein, we present clinical cases of ICI-induced thyroid dysfunction, hypophysitis, and insulin-dependent diabetes mellitus. We use these cases to discuss the screening, diagnosis, and management of ICI-associated endocrine dysfunction.
Systemic cancer therapies have historically been dominated by cytotoxic chemotherapies, agents that (through various mechanisms) target and kill rapidly dividing cells in a somewhat indiscriminate fashion. Since 2011 (1), however, a novel class of cancer therapy, known as immune checkpoint inhibitors (ICI) has increasingly emerged. These agents function not by targeting proliferating cells, but by blocking key negative regulators of immune activation, thereby enlisting tumor-specific T cells to attack tumor cells. These agents are now approved for use in at least 19 different cancer types (comprising nearly half of all patients with cancer) (2), and they produce durable or even curative responses in many cases.
Four different classes of ICI are FDA approved. The first, consisting of only ipilimumab, blocks cytotoxic T-lymphocyte antigen 4 (CTLA-4) and is approved in only melanoma, where it produces approximately 20% long-term survival (up from approximately 5% historically), but has minimal activity elsewhere (3). The second and third classes, inhibitors of programmed death-1 (PD-1: nivolumab, pembrolizumab, cemiplimab, dostarlimab) and its ligand (PD-L1; atezolizumab, avelumab, durvalumab) are more widely approved, and produce response rates ranging from 10% to 80% depending on the cancer type (4). PD-1 and PD-L1 inhibitors are also combined with a wide range of agents, including with anti-CTLA-4, chemotherapy, and targeted therapy (5). The fourth class, consisting of the anti-lymphocyte activating-3 (Lag-3; relatlimab) is only approved in combination with nivolumab for advanced melanoma (6, 7).
These agents, broadly speaking, trigger antitumor immunity by removing key regulators that inhibit T cell function. As might be expected, based on their mechanism of action, they also may cause autoimmune-like reactions. These side effects are known as immune-related adverse events (irAEs) and may impact any organ system. Autoinflammatory in nature, they have clinical and histopathologic resemblance to their organ specific counterparts (eg, colitis irAE has similar presentation to Crohn's disease). The most common organs impacted are those with frequent environmental interfaces (skin, lungs, gastrointestinal tract) or frequent smoldering autoimmunity (joints, thyroid) (8). However, other organs such as the liver, heart, brain, pancreas, pituitary, and bone marrow may also be impacted. A high index of suspicion and multidisciplinary care are therefore needed given the range of presentations (9). The majority of ICI-treated patients experience at least one irAE (10-13), the rate and severity of which vary with different agents (14). Severe irAEs (grade 3-4 according to the National Cancer Institute's Common Terminology Criteria for Adverse Events) are most frequent in patients treated with combined anti-PD-1 and anti-CTLA-4 (up to 50%) and less frequent in anti-PD-1/PD-L1 monotherapy (10%-15%) (15, 16). Treatment for adverse events can include the following 3 cornerstones: (1) supportive management (which may be sufficient for mild toxicities and for endocrinopathies as discussed below); 2) high-dose steroids; and 3) ICI cessation; the latter 2 primarily for more severe events, but generally not for endocrine irAEs. Although generally reversible, irAEs can uncommonly present in fulminant and fatal fashion, or evolve into a chronic phenotype (17, 18).
Endocrine toxicities from ICI may range from the common and routine (eg, subclinical hypothyroidism) to life-threatening (diabetic ketoacidosis, thyroid crisis, and adrenal crisis). Management of the endocrine toxicities differs from that of most other systems in several fundamental ways (19-22). First, high-dose steroids do not appear to decrease the severity or duration of the event (with the possible exception of minimizing neuro-inflammatory symptoms of hypophysitis). Thus, high-dose steroids generally have little to no role in ICI-endocrinopathies. Second, endocrine toxicities do not necessitate discontinuation of therapy, allowing patients to continue to receive their anticancer treatment. Third, institution of hormone therapy to correct the underlying deficiency is the major cornerstone of treatment. Finally, in contrast to other reversible irAEs, hormone deficiencies tend to be lifelong, and rarely resolve. It is speculated that the initial immune response triggered by the irAE results in sufficient damage or elimination of hormone-producing cells that precludes functional recovery, but the exact mechanism has not been established, is likely multifactorial, and requires further research.
Clinical Case 1: Thyroid Dysfunction
A 69-year-old man was diagnosed with early-stage melanoma on the left temple and underwent resection. Four years later, he developed a regional recurrence and underwent left parotidectomy and cervical lymph node dissection. Following resection, he was treated with nivolumab (anti-PD-1) for a planned total of 1 year. Regular screening with thyrotropin (TSH) and free thyroxine (FT4) levels showed normal thyroid function for the first 11 months of ICI therapy. Eleven months into treatment, he developed increasing malaise and tremors. His laboratory values demonstrated thyrotoxicosis, with suppressed TSH and elevated FT4 (Fig. 1) and total triiodothyronine (T3). He was treated symptomatically with propranolol. After 5 weeks, his symptoms had largely resolved and his TSH, FT4, and total T3 had normalized. One month later, he presented complaining of increasing fatigue. His TSH was elevated to 6.0 mcU/mL, and his FT4 was at the lower limit of normal, so he was started on levothyroxine 50 µg daily. His symptoms improved, and his dose was eventually increased to 75 µg daily, which he continues currently.
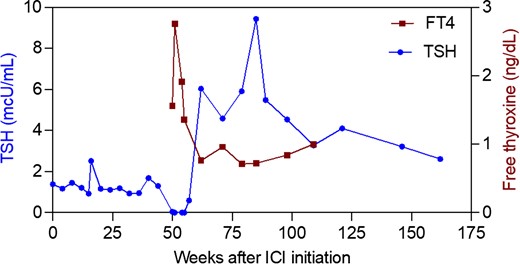
Longitudinal monitoring of thyroid stimulating hormone (TSH) and free thyroxine (FT4) in a patient with ICI-thyrotoxicosis. Suppressed TSH and elevated FT4 were noted 50 weeks after ICI initiation. Propanolol (Pr.) was given until thyroid function tests normalized. Levothyroxine (LT4) was started at a dose of 50 µg and titrated up to 75 µg.
Clinical Presentation
The most common endocrine irAE, thyroid dysfunction (including subclinical disease) occurs in as many as 25% of people treated with CTLA-4 monotherapy, 38% of those treated with PD-1/PD-L1 monotherapy, and 56% of those treated with dual CTLA-4 and PD-1/PD-L1 dual therapy (23-28). Although data are limited on the recently approved Lag-3 inhibitors, Lag-3/PD-1 combination therapy appears to modestly increase occurrence of thyroid toxicity compared with PD-1 monotherapy (7).
Patients with ICI-induced thyroid dysfunction commonly present with clinical or biochemical evidence of thyrotoxicosis, as occurred in our case, but initial presentation with signs and symptoms of hypothyroidism or with asymptomatic disease is not uncommon. Most cases develop within 6 weeks of ICI initiation, although some have been reported even up to years into therapy (29). The large majority of ICI-induced thyrotoxicosis is caused by thyroiditis, an acute immune-mediated destruction of thyroid follicular cells resulting in unregulated spilling of preformed thyroid hormone into systemic circulation. ICI-induced thyroiditis is generally self-limited, evolving into hypothyroidism or, less commonly, resolving into a euthyroid state within 6 weeks. Cases of ICI-induced Graves’ disease have been reported (30-35) but are very rare. Other non-autoimmune causes of thyrotoxicosis, including autonomous nodules and multinodular goiter, may be coincident with cancer treatment, but are not thought to be affected by ICI therapy.
Symptoms of ICI-induced thyrotoxicosis are similar to those seen in other forms of thyrotoxicosis, including anxiety, emotional lability, palpitations, tremor, hyperdefecation, heat intolerance, and increased perspiration, although the majority of patients are asymptomatic (24). Significant weight loss is less common due to the time-limited course of ICI-induced thyrotoxicosis. Signs of thyrotoxicosis may include rapid speech, hyperactivity, tachycardia, new-onset atrial fibrillation, fine tremor, hyper-reflexia, and warm, moist skin. Individuals may have eye lid retraction or lid lag, which are signs of sympathetic hyperactivity. Other ocular signs such as exophthalmos, periorbital edema, and limitation of eye movement are not generally seen in ICI-induced thyroiditis, and if present should prompt investigation into possible Graves’ disease.
Symptoms of hypothyroidism tend to be more subtle than symptoms of thyrotoxicosis. They include fatigue, depressed mood, slowed speech, skin dryness, cold sensitivity, and constipation. Physical exam findings include bradycardia and delayed relaxation of deep tendon reflexes. Weight gain, hair loss, and other findings associated with longstanding hypothyroidism, such as coarsening of hair and skin, enlargement of the tongue, and hoarseness may be absent since these patients tend to be diagnosed early due to frequent monitoring and interaction with their healthcare providers.
Asymptomatic Screening
Given the relative frequency of ICI-induced thyroid dysfunction, patients should have routine screening with TSH and free thyroxine (T4) at least every 4 to 6 weeks while receiving ICI, in accordance with recommendations from the American Society of Clinical Oncology (ASCO) (21), the National Comprehensive Cancer Network (NCCN) (22), and the Society for Immunotherapy of Cancer (SITC) (20). Frequency of screening can be adjusted based on an individual's baseline thyroid function while receiving ICI (22). Since thyroid dysfunction can occur any time after ICI initiation, SITC recommends ongoing surveillance every 6 to 12 months after ICI cessation (20). Some controversy still exists regarding the appropriate upper limit of normal (ULN) for TSH values and the need for age-adjusted normal ranges (36). Most labs report an ULN between 3.5 and 4.5 mIU/L. The American Society of Clinical Oncology bases its guidelines on an ULN of 4.5, while the National Comprehensive Cancer Network uses an ULN of 4.0. The American Thyroid Association and American Association of Clinical Endocrinologists (ATA/AACE) guidelines acknowledge the controversy while recommending use of assay-specific normal ranges or an upper limit of 4.12 mIU/L in the absence of assay-specific reference ranges (36). Increasing evidence has shown that anti-thyroid antibodies (anti-thyroglobulin or anti-thyroid peroxidase) at ICI initiation predicts ICI-induced thyroid disease (26, 37, 38), suggesting that measurement of anti-thyroid antibodies may help to guide screening in at-risk individuals. Since the rate of ICI-induced thyroid dysfunction remains relatively high when antibodies are negative (39), particularly in those treated with combination therapies (26), we have not yet incorporated thyroid antibody measurement into our routine clinical practice. To reliably identify all patients with thyroid disease, we screen thyroid function every 4 to 6 weeks in ICI patients, so the presence of anti-thyroid antibodies would not change our management. However, the use of these tests may evolve over time and can be the source of reasonable debate. See Fig. 2 for graphical summary of management of ICI-induced thyroid dysfunction.
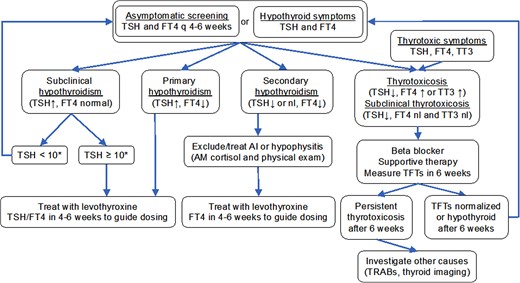
Algorithm for management of thyroid dysfunction in patients receiving ICI. *Consider lower threshold to treat subclinical hypothyroidism in younger patients, symptomatic patients, or in those with positive TPO or Tg antibodies. **Starting dose 1.6 µg/kg/day for severe symptoms or marked TSH elevation; 1.45 µg/kg/day for hypothyroidism following ICI-thyroiditis; 0.8 to 1.2 µg/kg/day for mild symptoms or TSH <10 mIU/L. Reduce doses for those at risk for complications (elderly or those with coronary artery disease). ***Starting dose 1.6 µg/kg/day with reduction for high-risk individuals. Abbreviation: TRAb, thyroid receptor antibody.
Symptomatic Testing and Diagnosis
The most common symptoms of early thyroid dysfunction are nonspecific, often overlapping with symptoms that are common in cancer patients and in other ICI-mediated adverse events. Thus, the clinician should have a low threshold to order thyroid function tests in symptomatic patients. Individuals with signs and symptoms suggestive of hypothyroidism should have TSH and free T4 measured. Those with suspected thyrotoxicosis should also have total T3 measured, due to the possibility of elevated T3 with relatively normal free T4. Individuals with mild or absent symptoms and mildly abnormal labs should have thyroid function tests (TFTs) repeated after approximately 4 weeks to confirm the diagnosis, while those with signs, symptoms, and laboratory values that strongly support the diagnosis can begin treatment immediately, as described below.
Management
Subclinical hypothyroidism
Subclinical hypothyroidism (SCH) is defined as elevated TSH with normal free T4 levels. Our practice in SCH is consistent with guidelines from the American Thyroid Association and the European Thyroid Association, which recommend initiation of thyroid hormone replacement in SCH if TSH is greater than 10 mIU/L (36, 40, 41). These guidelines are supported by recent studies demonstrating a lack of symptomatic benefit when TSH is less than 10 mIU/L in older adults (42, 43). Individuals with SCH and TSH between 7 and 10 mIU/L have increased long-term cardiovascular risk (44), so treatment starting at a lower TSH threshold may be of benefit in younger patients. The presence of anti-thyroid antibodies increases the risk of progression to overt hypothyroidism in spontaneous SCH (45, 46) and possibly in ICI-related SCH (24), so the threshold to initiate thyroid hormone replacement may be lower in individuals with positive antibodies. The decision to treat should also be tailored to the individual patient situation, based on age, symptoms, comorbidities, and TSH value. SCH often requires a lower starting dose than overt hypothyroidism (see below), and doses of 25 to 50 micrograms are often sufficient. After starting levothyroxine, TSH should be remeasured in 4 to 6 weeks to adjust the dose. Thereafter, TSH should be measured every 6 to 12 months or 4 to 6 weeks after any dose change (47). ICI treatment can continue without interruption in SCH.
Overt primary hypothyroidism
Primary hypothyroidism is characterized by elevated TSH and low FT4. In individuals with mildly elevated TSH (< 10) and mildly depressed FT4, or with mild or no symptoms, repeating TFT measurement in 4 weeks may be appropriate to confirm the diagnosis. In those with frankly elevated TSH or with convincing symptoms, initiation of thyroid hormone replacement is appropriate. Starting thyroid hormone dose varies depending on degree of hypothyroidism and other patient characteristics. Individuals with severe symptoms or with markedly elevated TSH (> 10-20 mIU/L) will likely require a full weight-based starting dose of 1.6 µg/kg/day (21, 22). Those with hypothyroidism after ICI-thyroiditis generally require 1.45 µg/kg/day (48), and those with mild symptoms or TSH < 10 can be started on doses of 0.8 to 1.2 µg/kg/day. In those who may be at risk of complications from iatrogenic thyrotoxicosis (the elderly, those with coronary artery disease, etc.), the starting dose should be reduced. TSH should be measured 4 to 6 weeks after starting levothyroxine to guide dosing adjustments, targeting a normal TSH level. Since TSH normalization may lag behind T4, particularly in patients recovering from thyrotoxicosis, some clinicians also measure free T4 until TSH target is reached. Ongoing dose adjustments should be based on TSH levels. ICI treatment should generally be continued during and after diagnosis with hypothyroidism, though in rare severe cases that require hospitalization (Grade 3-4), ICI should be temporarily held until clinical stabilization and then may be resumed (21).
Secondary hypothyroidism
Low FT4 and low or inappropriately normal TSH characterize secondary hypothyroidism (also called central hypothyroidism). Secondary hypothyroidism generally is accompanied by hypophysitis/hypopituitarism and disruption of other pituitary hormones, such as the adrenal or gonadal axes (49). Thus, patients with suspected secondary hypothyroidism should be evaluated for secondary adrenal insufficiency and started on glucocorticoid replacement therapy, if appropriate, prior to thyroid hormone replacement. TSH is not accurate in guiding treatment decisions in secondary hypothyroidism (50). Thus, FT4 levels, rather than TSH, should guide dosing decisions, targeting a level in the upper half of the normal range. It is important to note that thyroid function tests in secondary hypothyroidism can be indistinguishable from those seen in individuals with normal thyroid function during acute systemic illnesses. Hospitalized and critically ill patients often have a transient decrease in TSH, FT4, and total T3. Thyroid hormone replacement in these individuals is not generally beneficial, so the entire clinical situation should be considered before initiating treatment for secondary hypothyroidism. Depending on the clinical scenario and severity of laboratory changes, confirmation of the diagnosis with repeat TFTs in 1 to 2 weeks is often recommended. The decision to temporarily hold ICI should be based on the presence of adrenal insufficiency or hypophysitis, as described below.
Thyrotoxicosis
Suppressed TSH with elevated FT4 or total T3 characterizes thyrotoxicosis. Patients may also present with subclinical thyrotoxicosis (suppressed TSH with normal or high-normal FT4 or total T3). In the absence of clinical evidence of Graves’ disease, such as thyroid eye disease or an audible thyroid bruit, patients with ICI-induced thyroiditis should be treated symptomatically with beta blockers and monitored for 6 weeks for resolution of thyrotoxicosis. If symptoms and laboratory abnormalities persist longer than 6 weeks, alternative causes of thyrotoxicosis should be investigated by measuring thyroid receptor antibodies and obtaining a thyroid ultrasound or thyroid uptake and scan. For severe cases, ICI should be temporarily held (21) and use of additional medical therapies, such as thionamides, steroids, potassium iodide, cholesterol-binding resins, or surgery, can be considered in consultation with an endocrinologist.
Routine screening with free T4 and TSH every 4 to 6 weeks after initiation of ICI therapy in asymptomatic patients is recommended. If secondary hypothyroidism is detected, then evaluation for hypophysitis and secondary adrenal insufficiency is recommended PRIOR to initiation of thyroid hormone therapy.
ICI-thyrotoxicosis is generally self-limited and often evolves into hypothyroidism; thyrotoxicosis that persists > 6 weeks should prompt evaluation of other causes such as Graves’ disease.
ICI-thyroid dysfunction occurs in 20% to 50% of treated patients; treatment consists of weight-based thyroid hormone replacement. High-dose steroids and discontinuation of ICI are NOT indicated, although ICIs may be held in severe cases until the patient is stabilized clinically.
Clinical Case 2: Hypophysitis
A 34-year-old woman with a history of hypothyroidism due to Hashimoto thyroiditis was diagnosed with an early-stage melanoma on the left flank. Two years after initial resection, she developed lymphadenopathy and pelvic pain while pregnant at 33 weeks, and she was found to have inguinal and pelvic lymph nodes involved with melanoma. Labor was induced and she began treatment with ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1). Approximately 2 months later, she noted increasing fatigue and dull headache; morning serum cortisol was low (2.7 µg/dL), and magnetic resonance imaging (MRI) of her brain demonstrated marked enlargement of the pituitary gland and infundibulum, consistent with hypophysitis, that was not present at her baseline brain MRI from 2 months prior (Fig. 3). Due to her symptoms, definitively low cortisol, and classic imaging findings, the diagnosis of hypophysitis was made, and she was started on glucocorticoid therapy before adrenocorticotropin hormone (ACTH) could be obtained. Her levothyroxine dose, TSH, and free T4 levels were unchanged. Insulin-like growth factor 1 (IGF-1), which can be helpful in identifying hypopituitarism in ambiguous cases, was not measured. She briefly received prednisone 60 mg with a plan to taper quickly to replacement doses, but in the next week she developed immune-mediated colitis. She received a single dose of infliximab and underwent a long prednisone taper. Her colitis resolved, but she was unable to be tapered off prednisone. Since then, due to patient preference for once-daily dosing she has continued 2.5 to 5 mg of prednisone daily at a stable body weight of 57 kg.
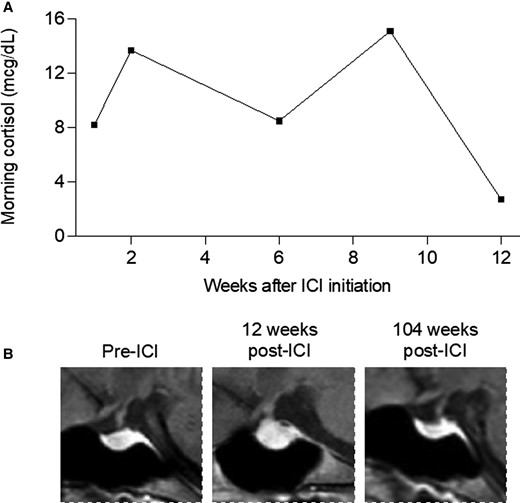
Serial monitoring of morning cortisol levels and MRI images in ICI-hypophysitis. A, Morning serum cortisol was low 12 weeks after ICI initiation, associated with symptoms of fatigue and headache. B, Sagittal plane MRI images of the pituitary, marked with red asterisk, taken before ICI, 12 weeks after ICI while symptomatic, and 104 weeks after ICI with symptoms and hypophysitis resolved.
Clinical Presentation
Hypophysitis, or inflammation of the pituitary gland, is exceedingly rare and has a broad differential diagnosis in in the general population. In contrast, it is relatively common among people receiving ICI. Between 1.8% and 17% of individuals who receive anti-CTLA-4 therapy develop hypophysitis, depending on the doses given (28, 51-55), while hypophysitis due to anti-PD-1/PD-L1 monotherapy is relatively uncommon (27, 56). This specificity for anti-CTLA-4 therapy is likely a result of CTLA-4 expression in some pituitary endocrine cells, resulting in a mixed type II (IgG dependent) and type IV (T cell mediated) hypersensitivity reaction targeting the CTLA-4 expressing cells (57).
Median time to hypophysitis onset is approximately 12 weeks after ICI initiation, with reported cases ranging from 3 to 76 weeks (29). Typical presenting symptoms of hypophysitis can be attributed either to pituitary hormone deficiencies or to the mass effects of pituitary enlargement. ICI-hypophysitis almost exclusively affects the anterior pituitary. As few as 11 cases of ICI-induced diabetes insipidus have been reported (58), so posterior pituitary involvement is possible but should motivate investigation into other causes, such as metastatic disease. Corticotrophs and thyrotrophs, which produce ACTH and TSH respectively, are the most commonly affected cell types and have the greatest clinical significance (54, 55, 59, 60). Gonadotrophs, lactotrophs, and somatotrophs may also be affected (19, 29, 54, 55, 59, 60), but deficiencies of sex hormones, prolactin, and growth hormone are less clinically relevant, and will not be a major focus in this review. Hypophysitis due to PD-1/PD-L1 monotherapy tends to occur later than in CTLA-4 therapy, is more likely to involve only a single hormonal axis, and is less likely to cause headache or gland enlargement on MRI (54, 56).
The endocrine-related symptoms of hypophysitis are nonspecific, the most common being fatigue, occurring in 59% to 73% (51, 54, 59, 61). Nausea, anorexia, weight loss, lethargy, confusion, or coma may result from adrenal insufficiency, while amenorrhea and sexual dysfunction may result from disruption of the gonadal axis. Symptoms of thyroid dysfunction are described above. Headache is the most common mass effect symptom, occurring in 32% to 87% of cases (51, 55, 61). Visual field defects such as tunnel vision or bitemporal hemianopsia, common presentations in non-ICI-hypophysitis, are rare in ICI-hypophysitis due to the relatively mild degree of pituitary enlargement (54). Physical exam may be unrevealing in mild cases of hypophysitis, whereas severe cases may present with frank hypotension, fever, and shock. In general, hypotension in secondary adrenal insufficiency is less severe than that seen in primary adrenal insufficiency since mineralocorticoid function is intact. Hyperpigmentation, as may be seen in primary adrenal insufficiency, is absent.
Asymptomatic Screening
The value of screening asymptomatic individuals for pituitary dysfunction is controversial. Due to life-threatening consequences of untreated adrenal insufficiency, we screen all patients receiving anti-CTLA-4 therapy with a random cortisol +/− ACTH at every cycle. While 8 Am cortisol levels are more reliable, the ability to schedule all patients for labs at 8 Am in a busy cancer clinic precludes the ability to do this routinely for patients with low clinical suspicion. Low or declining random cortisol levels prompt follow up evaluation with morning levels. People with definitively low morning cortisol levels (< 3 µg/dL on older polyclonal antibody–based immunoassays) are started on therapy. Levels > 15 µg/dL are very reassuring that adrenal function is not low. Morning serum cortisol cutoff levels for newer monoclonal antibody–based assays have not been established, but they may be lower, as they are for ACTH-stimulated levels (62, 63). Asymptomatic patients with intermediate levels are sent for ACTH stimulation testing if there is any clinical concern. Patients on steroids for other irAEs will also have altered cortisol and ACTH values, rendering these screening tests unreliable. Thyroid screening is indicated and is described above. Routine screening of other axes, including sex hormones, prolactin, and growth hormone is not necessary, but can help to diagnose pituitary disease when cortisol levels or ACTH stimulation results are equivocal.
Symptomatic Testing and Diagnosis
Patients with symptoms concerning for adrenal insufficiency should be evaluated promptly. Those with severe symptoms or with hemodynamic instability should be admitted to the hospital. Initial laboratory tests upon admission should include a basic metabolic panel, cortisol, ACTH, TSH, and FT4, in addition to other laboratory tests investigating other nonendocrine causes and precipitating illnesses. Since untreated adrenal insufficiency can be life-threatening, treatment with intravenous (IV) glucocorticoids and IV fluids should begin empirically, without waiting for results of initial laboratory evaluation. Clinical response to therapy and laboratory results will then help guide treatment going forward.
Initial evaluation of patients with mild or moderate symptoms should include morning cortisol and ACTH levels. As with asymptomatic screening, definitively low morning cortisol (< 3 µg/dL) should prompt immediate initiation of glucocorticoid replacement. Morning cortisol greater than 15 µg/dL is very reassuring that adrenal function is not impaired. Intermediate morning cortisol levels (3-15 µg/dL) are more difficult to interpret. An abnormal ACTH stimulation test (< 18 µg/dL on older assays, < 14.6 µg/dL on newer assays) (62, 63) will identify adrenal insufficiency in these cases, but a normal result can be falsely reassuring, since the adrenal cortex takes weeks to atrophy in the absence of endogenous ACTH. Insulin-induced hypoglycemia (64) and metyrapone (65) tests can more reliably diagnose secondary adrenal insufficiency, but issues with safety and availability limit their real-world utility. Thus, in cases with indeterminate results and a reasonable clinical suspicion of adrenal insufficiency, we initiate hydrocortisone replacement and re-evaluate ongoing need for replacement in 1 to 2 months. ICI almost exclusively causes secondary adrenal insufficiency, characterized by low or inappropriately normal ACTH in conjunction with low cortisol production. A finding of elevated ACTH suggests primary adrenal insufficiency and should prompt evaluation for mineralocorticoid deficiency and consideration of other causes of primary adrenal insufficiency, such as metastatic disease. See Fig. 4 for summary of approach to ICI-induced adrenal insufficiency.
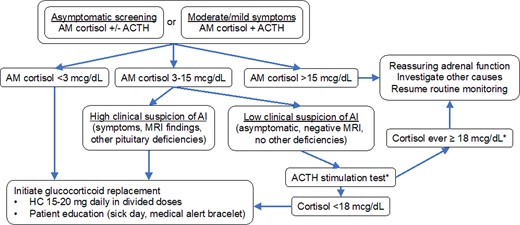
Algorithm for management of suspected adrenal insufficiency in ICI patients. Our approach favors empiric glucocorticoid replacement when cortisol levels are indeterminate and suspicion is high. *ACTH stimulation is only helpful to rule in, not to rule out, acute-onset secondary adrenal insufficiency.
There are some important caveats to the diagnosis of ICI-induced adrenal insufficiency. First, these patients often receive glucocorticoids as part of prior cancer therapies or treatment of other complications. Thus, a thorough medical history is critical, and distinguishing iatrogenic from ICI-induced adrenal insufficiency can be difficult. Since exogenous steroids would not be expected to cause growth hormone deficiency, prolactin deficiency, or gland enlargement on MRI, measurement of insulin-like growth factor 1, prolactin, and pituitary imaging can be helpful. Symptomatic patients with low cortisol should be treated regardless of underlying cause. Subsequent ACTH stimulation testing can help to detect recovery of the adrenal axis, which would be expected in iatrogenic adrenal insufficiency but not in ICI-induced adrenal insufficiency. Second, ICI-induced adrenal insufficiency can be relatively acute in onset. ACTH stimulation testing may not sensitively detect acute cases of secondary adrenal insufficiency since the adrenal cortex may remain sensitive to ACTH for days to weeks after a pituitary insult. Thus, clinical context and severity of symptoms should be considered when deciding whether to treat individuals with suspected ICI-induced adrenal insufficiency and borderline morning cortisol or ACTH stimulation results.
Individuals with mass effects, especially headache, should undergo MRI imaging of their pituitary and brain to evaluate for hypophysitis or other causes such as metastatic disease. Normal pituitary imaging does not rule out pituitary dysfunction, especially in the case of PD-1/PD-L1 monotherapy (54, 56, 59), but it can help distinguish the need for pulse-dose or replacement-dose steroids. Pituitary imaging is also helpful in establishing the diagnosis of hypophysitis when laboratory findings are borderline or indeterminate (54, 60). Pituitary enlargement on MRI typically resolves within 3 months (56, 60, 66); longer duration of symptoms or gland enlargement should prompt consideration of other potential etiologies.
Assessment of other pituitary axes can help to identify pituitary pathology when adrenal and thyroid testing is equivocal but is less urgent and less beneficial in patients receiving ICI. While ICI-induced hypogonadism has been reported (29), most cases of ICI-induced hypogonadism recover gonadal function (60), suggesting that this may represent physiologic suppression of function during acute illness rather than immune-mediated destruction of gonadotrophs. If symptoms of hypogonadism persist past the acute phase of illness, standard diagnostic tests should be obtained if estrogen or testosterone replacement is not otherwise contraindicated. Growth hormone replacement is generally contraindicated in active malignancy, and treatment in the adult population can be of unclear benefit, so growth hormone is only necessary if symptoms are convincing and if treatment is not contraindicated. Central diabetes insipidus is very rarely caused by ICI (58), so diagnosis of diabetes insipidus should prompt evaluation of other causes, such as metastatic disease.
Management
Treatment of ICI-hypophysitis depends on the severity of symptoms and the presence of mass effects. Individuals with headache and/or vision changes and evidence of hypophysitis on imaging should receive high-dose glucocorticoids (often prednisone 1 mg/kg/day) to reduce pituitary inflammation and possible mass effect, tapered over 1 to 2 weeks to physiologic replacement dosing. If adrenal function was intact prior to initiating high-dose steroids, adrenal function can be reassessed by morning cortisol and/or ACTH stimulation after 3 months of physiologic cortisol replacement, the time at which gland enlargement is expected to have resolved (56, 60, 66). Adrenal function does not generally recover after ICI-hypophysitis, but recovery of thyroid function has been reported (54, 60, 66). Patients admitted to the hospital with signs or symptoms of adrenal crisis should receive aggressive IV fluids and stress-dose IV steroids, generally hydrocortisone 50 to 100 mg every 8 hours, tapered with clinical improvement to physiologic doses. Maintenance doses of hydrocortisone 15 to 20 mg daily in divided doses (often 10-15 mg in the morning and 5 mg in the afternoon) approximate diurnal levels; prednisone 3 to 7.5 mg daily is appropriate for patients who prefer once-daily dosing. Fludrocortisone is only necessary in the rare cases of primary adrenal insufficiency. Glucocorticoid dosing is adjusted based on symptoms and physical exam, rather than on laboratory measures and should be titrated to the minimum dose that relieves symptoms. Patients should also be educated on stress dosing for sick days, typically doubling their dose when acutely ill for 3 days. If they are unable to tolerate oral intake or are too nauseous to take their pills, urgent intramuscular injection or treatment in the emergency department is needed. They should also be advised to obtain a medical alert bracelet. ICI should be temporarily held until patients are stably treated on replacement hormones.
Hypophysitis is rare in the general population but may occur in up to 10% of patients treated with combination ICI. Diagnosis is performed with morning serum cortisol and ACTH, as well as pituitary MRI.
ACTH stimulation testing can be falsely normal in acute/subacute secondary adrenal insufficiency; iatrogenic adrenal insufficiency may also alter diagnostic testing.
Clinical onset may be rapid, and clinicians should have a low threshold to provide replacement glucocorticoids while completing the evaluation.
Treatment for ICI-induced hypophysitis is replacement-dose steroids (possibly following a short-term course of high-dose steroids for symptoms of pituitary inflammation); permanent discontinuation of ICI is NOT required.
Clinical Case 3: ICI-Associated Diabetes Mellitus
A 55-year-old man with history of hypothyroidism due to presumed Hashimoto thyroiditis was diagnosed with metastatic duodenal adenocarcinoma. After progression of disease on chemotherapy and palliative radiation for bony metastases, he was treated with avelumab (anti-PD-L1), approximately 9 months after initial diagnosis. At each ICI infusion, random blood glucose was within normal limits (< 200 mg/dL), but in retrospect, a trend of increasing random glucose 2 and 4 weeks after ICI initiation was noted (Fig. 5). Ten days after his third ICI dose, he presented to the emergency department with 2 days of increasing thirst and urination, lethargy, and confusion. Laboratory evaluation upon admission to the hospital demonstrated diabetic ketoacidosis (DKA) with bicarbonate < 5 mmol/L, anion gap > 28, blood glucose 756 mg/dL, and elevated urine ketones. Hemoglobin A1c (HbA1c) was 8.8% and C-peptide was undetectable. Islet cell antibodies (< 1:4), GAD (< 1:600), and IA-2 antibodies (< 0.8 U/mL) were negative, while insulin antibodies were elevated at 3.0 units/mL (reference range ≤0.4 units/mL). Ketoacidosis resolved with aggressive IV fluids and insulin. He was given training on self-injection and glucose management with a Certified Diabetes Care and Education Specialists (CDCES), and was discharged on multiple daily insulin injections, with basal insulin glargine and mealtime insulin aspart. He received ongoing diabetes care in endocrinology clinic, but his cancer continued to progress, and he passed away within one year.
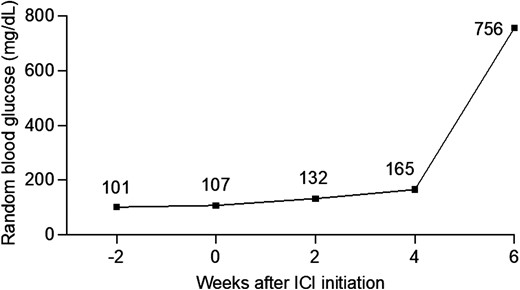
Serial random blood glucose monitoring ICI-DM. Individual values in mg/dL are shown above each point. Patient had a trend of increasing random blood glucose until 4 weeks after ICI initiation. At 6 weeks, he had sudden onset of symptoms and presented in DKA.
Clinical Presentation
ICI-induced diabetes mellitus (ICI-DM) is a relatively rare complication of ICI, occurring in approximately 1% of patients treated with ICI (28, 67). ICI-DM is caused almost exclusively by anti-PD-1/PD-L1 therapy. Very few cases, if any, are thought to have been caused by CTLA-4 inhibitor therapy (68-70). PD-L1 is expressed in human islet beta cells (71), which likely contributes to its pathogenic mechanism.
ICI-DM presents similarly to autoimmune type 1 diabetes and is due to insulin deficiency. Peri-islet infiltration of T lymphocytes was noted in the only published histologic examination of the pancreas from a donor with ICI-DM (72). Thus, ICI-DM is thought to be due to near-complete T-cell-mediated destruction of insulin-producing pancreatic beta cells, although differences in detectable islet auto antibodies, which are less common in ICI-DM, and in HLA predominance suggest some possible mechanistic differences between ICI-DM and type 1 diabetes mellitus (19, 73). Worsening of underlying type 2 diabetes (74, 75), mild hyperglycemia associated with alterations in incretin signaling (76), and diabetes mellitus due to acquired generalized lipodystrophy (77-79) have also been described associated with ICI therapy.
Median time to onset from ICI initiation and DM onset is 7 to 17 weeks, though cases have been reported as late as 228 weeks (80), and some cases have been reported after ICI discontinuation (68). Between 38% and 71% of cases present in DKA with relatively mild elevation of HbA1c (67-70, 75), suggesting a precipitous drop in insulin secretory capacity and rapid elevation in blood glucose. The patient in our case (Fig. 5) had HbA1c of 8.8%, suggestive of a subacute onset. The trend of increasing glucose at 2 and 4 weeks, although technically within normal limits for random blood glucose, may have been the earliest manifestation of declining insulin secretory capacity. Polyuria and polydipsia are common symptoms, and weight loss, although present (75), may be milder than in type 1 diabetes due to the acuity of onset. Mild or very early cases may be relatively asymptomatic, whereas severe cases often have typical signs and symptoms of DKA, including lethargy, confusion, obtundation, abdominal pain, decreased skin turgor, tachycardia, fruity odor to their breath, and deep respirations (called Kussmaul respirations). Physical exam is often unrevealing, though severe cases presenting with DKA may exhibit neurologic changes, including lethargy or obtundation.
Asymptomatic Screening
Due to the acuity of onset, HbA1c is of little use in screening for ICI-DM. We measure random or fasting glucose at every ICI cycle, although even this may fail to detect hyperglycemia prior to DKA onset. The fact that our patient's random glucose was rising within 2 weeks of ICI likely suggested worsening islet function, but since it fell within normal limits, it was not noticed at that time. Fasting blood glucose is preferable but may be difficult to obtain routinely in some cases. This further emphasizes the importance of monitoring blood glucose trends, even when normal, and responding to mild hyperglycemia. In addition, patient education and training about signs and symptoms of hyperglycemia and early DKA are critical to detect early cases and prevent life-threatening severe DKA. Fasting glucose greater than 125 mg/dL, random glucose greater than 199 mg/dL, or increasing glucose levels within the “normal” range should prompt confirmatory testing with a repeat fasting glucose, C-peptide, and urine ketones (Fig. 6). Islet autoantibodies (IAA) cannot exclude ICI-DM but may predict faster progression and risk of DKA (75). Some clinicians recommend home glucose monitoring, either with continuous glucose monitors or glucometers, although we have not adopted this practice broadly.
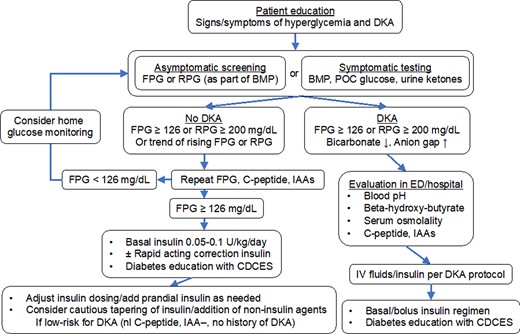
Algorithm for screening and management of ICI-DM. Abbreviations: CDCES, Certified Diabetes Care and Education Specialist; FPG, fasting plasma glucose; IAA, islet auto-antibodies; RPG, random plasma glucose. See ref (81) for detailed algorithm.
Symptomatic Testing and Diagnosis
Individuals presenting with symptoms suggestive of hyperglycemia or DKA should have urgent laboratory evaluation, including in-office capillary glucose, basic metabolic panel, C-peptide, HbA1c, and urine ketones. Patients with laboratory results suggestive of DKA (low bicarbonate, elevated glucose, high anion gap, positive ketones) should be admitted to the hospital for initial inpatient management and evaluation. Those with new-onset hyperglycemia, as defined above, without DKA should be started on insulin replacement therapy (typically starting dose of 0.05 to 0.1 units/kg/day long-acting insulin, with or without rapid-acting correction insulin) in consultation with an endocrinologist to prevent DKA. In this patient population, other causes of hyperglycemia should also be considered (steroid-induced, stress-induced, progression of underlying type 2 diabetes, etc.), but given the high rate of DKA in patients on ICI, with its associated high rate of morbidity and mortality, we recommend having a low threshold to initiate insulin. Subsequent evaluation can help determine appropriate therapy going forward, based on DKA risk assessment that includes C-peptide levels and islet autoantibody measurement (81). See reference (81) for an excellent, in-depth review of screening and management of ICI-DM.
Management
ICI-DM management consists of initial treatment of DKA, if present, with standardized IV fluid and insulin protocols. As with other forms of insulin-deficient diabetes, prompt insulin therapy is key to successful outcomes. Treatment of ICI-DM with steroids is not helpful and may further exacerbate hyperglycemia (82, 83). Like type 1 diabetes, individuals with ICI-DM require lifelong intensive insulin therapy, either with multiple daily injections or continuous subcutaneous infusion with an insulin pump. Initially, the treatment goal in ICI-DM is to prevent DKA and avoid severe hypoglycemia, which both can be accomplished with relatively low doses of basal insulin (0.05–0.1 units/kg/day). With time, as the patient becomes comfortable with insulin injections and glucose monitoring, the treatment goal will evolve to standard glycemic targets in diabetes mellitus, typically targeting a HbA1c less than 7%, though this may be adjusted depending on the patient's life expectancy. There is a broad range of insulin analogs, insulin delivery devices, and glucose monitoring technologies available to individuals with ICI-DM and type 1 diabetes. The details of these options are beyond the scope of this review. ICI-DM should be managed in consultation with an endocrinologist and a multidisciplinary care team that includes nurses, CDCES, and dieticians. In the short term, ICI may be temporarily held until clinical stability is achieved, then may be resumed.
ICI-DM is rare (< 1% of patients treated with ICI) but onset is rapid and often presents with diabetic ketoacidosis due to severe insulin deficiency; early initiation of insulin replacement is essential.
All ICI patients should be educated regarding signs/symptoms of hyperglycemia and diabetic ketoacidosis.
Patients should be screened with random or fasting blood glucose at every ICI cycle.
Treatment of ICI-DM is similar to type 1 diabetes, requiring lifelong intensive insulin therapy; high-dose steroids and permanent discontinuation of ICI are NOT necessary.
Conclusion
As use of ICIs continues to increase, the prevalence of ICI-induced toxicities, including endocrinopathies, will also continue to increase. Appropriate screening, diagnosis, and treatment of these conditions will improve the ability of oncologists, primary care providers, and endocrinologists to prevent acute morbidity and mortality caused by undiagnosed endocrinopathies and to improve patients’ long-term health and quality of life.
Acknowledgments
This study was supported by NIH R01CA227481 (D.B.J.), the James C. Bradford Jr. Melanoma Fund (D.B.J.), the Susan and Luke Simons Melanoma Directorship (D.B.J.), and the Doris Duke Charitable Foundation (J.J.W.). We thank Howard Baum, MD, for critical review of the manuscript.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Abbreviations
- ACTH
adrenocorticotropin hormone
- CTLA-4
cytotoxic T-lymphocyte antigen 4
- DKA
diabetic ketoacidosis
- FT4
free thyroxine
- HbA1c
glycated hemoglobin
- ICI
immune checkpoint inhibitor
- ICI-DM
ICI-induced diabetes mellitus
- irAE
immune-related adverse events
- IV
intravenous
- Lag-3
lymphocyte activating-3
- MRI
magnetic resonance imaging
- PD-1
programmed cell death protein 1
- PD-L1
programmed cell death ligand 1
- SCH
subclinical hypothyroidism
- T3
triiodothyronine
- T4
thyroxine
- TFT
thyroid function test
- TSH
thyrotropin (thyroid-stimulating hormone)
- ULN
upper limit of normal
Author notes
Conflicts of Interest D.B.J. has served on advisory boards or as a consultant for BMS, Catalyst Biopharma, Iovance, Jansen, Mallinckrodt, Merck, Mosaic ImmunoEngineering, Novartis, Oncosec, Pfizer, Targovax, and Teiko, and has received research funding from BMS and Incyte.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}