





Abstract
Pheochromocytomas and paragangliomas (PPGL) are rare neuroendocrine tumors. Metastases develop in 15% to 20%. The American Joint Committee on Cancer (AJCC) established inaugural guidelines for PPGL tumor-node-metastasis (TNM) staging.
The objective of this analysis is to investigate the associations between TNM staging and overall survival (OS).
We retrospectively applied the TNM staging at the time of diagnosis of the primary tumor. The primary outcome was OS. Unadjusted survival rates were estimated by the Kaplan–Meier method. Cox proportional hazards regression models were used to evaluate the associations between OS and covariates of interest.
The study included 458 patients. Median OS was 18.0 (95% CI, 15.6-not reached) years. At diagnosis, 126 (27.5%) tumors were stage I, 213 (46.5%) were stage II, 47 (10.3%) were stage III, and 72 (15.7%) were stage IV. The 10-year OS probabilities were 0.844 (95% CI, 0.768-0.928) for patients with stage I tumors, 0.792 (95% CI, 0.726-0.865) for stage II, 0.595 (95% CI, 0.435-0.813) for stage III, and 0.221 (95% CI, 0.127-0.384) for stage IV. Compared with stage I, the hazard ratios (HR) for death were 1.50 (0.87-2.57) for stage II, 2.85 (1.45-5.63) for stage III, and 8.88 (5.16-15.29) for stage IV (P < 0.001). Compared with patients with no germline mutations, those with RET 634/918 had better OS (HR: 0.28; 95% CI, 0.12-0.69). Other germline mutations, including SDHB, did not exhibit worse OS than patients with metastasis and sporadic disease.
OS rates correlated with the recently developed AJCC TNM staging and were not worse in hereditary disease. Stage IV disease exhibited a significantly shorter OS compared with stages I-III. Future staging systems could be adjusted to better separate stages I and II.
Pheochromocytomas (PHEO) and paragangliomas (PGL), collectively referred to as PPGL, are rare neuroendocrine tumors that originate in the adrenal medulla or the extra-adrenal paraganglia, respectively. PPGL are classified as either parasympathetic or sympathetic depending on the autonomic nervous system paraganglion of origin (1). Parasympathetic PGL are usually located in the head and neck, do not produce catecholamines, and rarely metastasize. Conversely, sympathetic PGL usually originate in the chest, abdomen, or pelvis, typically produce excessive amounts of noradrenaline, predisposing patients to cardiovascular and gastrointestinal disease, and are more frequently metastatic (2, 3).
No histological features reliably predict the metastatic potential of a PPGL (1, 4). The size and location of the primary tumor and the presence of germline pathogenic variants in the succinate dehydrogenase subunit B gene (SDHB) are recognized as the best clinical predictors of metastasis and survival (2, 5). Metastases usually occur in the lymph nodes, bones, lungs, and liver and are associated with decreased overall survival (OS) (6). Metastatic PPGL exhibit variable clinical outcomes. Some patients have very aggressive disease and live no longer than 1 year from the time of diagnosis (7). In others, the disease exhibits minimal or no growth over time, and consequently, these patients may have a prolonged or even normal lifespan (7, 8). However, most patients fall in between these 2 groups; these patients exhibit disease progression and may require systemic therapy (9).
The American Joint Committee on Cancer (AJCC) and the Union for International Cancer Control recently established the first tumor-nodes-metastases (TNM) staging for patients with PPGL (Table 1) (10, 11). The World Health Organization indicates that every PPGL has the potential to spread (1, 12). Like any cancer, the degree of local and distant spread of a PPGL at the time of diagnosis is expected to predict survival and help to determine management. The TNM staging codifies the extent of a PPGL to provide clinicians and patients the means to quantify prognosis, determine treatment and follow-up, and allow comparisons of participants in clinical trials (10, 13, 14). The current TNM staging excludes patients with head and neck PGL, given their very low metastatic potential and the lack of studies on the clinical predictors of metastasis and survival in that population (13).
AJCC TNM and staging definitions for PPGLa
| Primary tumor (T) |
| ȃ• TX: primary tumor cannot be assessed |
| ȃ• T1: PHEO < 5 cm in greatest dimension, no extra-adrenal invasion |
| ȃ• T2: PHEO ≥ 5 cm OR sympathetic PGL of any size, no extra-adrenal invasion |
| ȃ• T3: Tumor of any size with invasion into surrounding tissuesb |
| Regional lymph nodes (N) |
| ȃ• NX: regional lymph nodes cannot be assessed |
| ȃ• N0: no regional lymph node metastasis |
| ȃ• N1: regional lymph node metastasis |
| Distant metastasis (M) |
| ȃ• M0: no distant metastasis |
| ȃ• M1: distant metastasis |
| ȃȃ• M1a: metastasis to bone only |
| ȃȃ• M1b: metastasis to non-regional lymph node(s), liver, and/or lung; no skeletal metastases |
| ȃȃ• M1c: metastasis to bone and multiple other sites |
| AJCC prognostic stage groups |
| ȃ• Stage I: T1 N0 M0 |
| ȃ• Stage II: T2 N0 M0 |
| ȃ• Stage III: T1-2 N1 M0 or T3 N0-1 M0 |
| ȃ• Stage IV: T1-3 N0-1 M1 |
| Primary tumor (T) |
| ȃ• TX: primary tumor cannot be assessed |
| ȃ• T1: PHEO < 5 cm in greatest dimension, no extra-adrenal invasion |
| ȃ• T2: PHEO ≥ 5 cm OR sympathetic PGL of any size, no extra-adrenal invasion |
| ȃ• T3: Tumor of any size with invasion into surrounding tissuesb |
| Regional lymph nodes (N) |
| ȃ• NX: regional lymph nodes cannot be assessed |
| ȃ• N0: no regional lymph node metastasis |
| ȃ• N1: regional lymph node metastasis |
| Distant metastasis (M) |
| ȃ• M0: no distant metastasis |
| ȃ• M1: distant metastasis |
| ȃȃ• M1a: metastasis to bone only |
| ȃȃ• M1b: metastasis to non-regional lymph node(s), liver, and/or lung; no skeletal metastases |
| ȃȃ• M1c: metastasis to bone and multiple other sites |
| AJCC prognostic stage groups |
| ȃ• Stage I: T1 N0 M0 |
| ȃ• Stage II: T2 N0 M0 |
| ȃ• Stage III: T1-2 N1 M0 or T3 N0-1 M0 |
| ȃ• Stage IV: T1-3 N0-1 M1 |
Abbreviations: PGL, paraganglioma; PHEO, pheochromocytoma; PPGL, pheochromocytoma/paraganglioma.
Jimenez et al (11).
As per AJCC definitions, invasion into surrounding tissues includes any involvement outside the wall/capsule of any organ. For adrenal tumors, this may include infiltration of extra-adrenal adipose tissue and other soft tissues and/or adjacent organs such as the kidneys, pancreas, liver, and spleen.
AJCC TNM and staging definitions for PPGLa
| Primary tumor (T) |
| ȃ• TX: primary tumor cannot be assessed |
| ȃ• T1: PHEO < 5 cm in greatest dimension, no extra-adrenal invasion |
| ȃ• T2: PHEO ≥ 5 cm OR sympathetic PGL of any size, no extra-adrenal invasion |
| ȃ• T3: Tumor of any size with invasion into surrounding tissuesb |
| Regional lymph nodes (N) |
| ȃ• NX: regional lymph nodes cannot be assessed |
| ȃ• N0: no regional lymph node metastasis |
| ȃ• N1: regional lymph node metastasis |
| Distant metastasis (M) |
| ȃ• M0: no distant metastasis |
| ȃ• M1: distant metastasis |
| ȃȃ• M1a: metastasis to bone only |
| ȃȃ• M1b: metastasis to non-regional lymph node(s), liver, and/or lung; no skeletal metastases |
| ȃȃ• M1c: metastasis to bone and multiple other sites |
| AJCC prognostic stage groups |
| ȃ• Stage I: T1 N0 M0 |
| ȃ• Stage II: T2 N0 M0 |
| ȃ• Stage III: T1-2 N1 M0 or T3 N0-1 M0 |
| ȃ• Stage IV: T1-3 N0-1 M1 |
| Primary tumor (T) |
| ȃ• TX: primary tumor cannot be assessed |
| ȃ• T1: PHEO < 5 cm in greatest dimension, no extra-adrenal invasion |
| ȃ• T2: PHEO ≥ 5 cm OR sympathetic PGL of any size, no extra-adrenal invasion |
| ȃ• T3: Tumor of any size with invasion into surrounding tissuesb |
| Regional lymph nodes (N) |
| ȃ• NX: regional lymph nodes cannot be assessed |
| ȃ• N0: no regional lymph node metastasis |
| ȃ• N1: regional lymph node metastasis |
| Distant metastasis (M) |
| ȃ• M0: no distant metastasis |
| ȃ• M1: distant metastasis |
| ȃȃ• M1a: metastasis to bone only |
| ȃȃ• M1b: metastasis to non-regional lymph node(s), liver, and/or lung; no skeletal metastases |
| ȃȃ• M1c: metastasis to bone and multiple other sites |
| AJCC prognostic stage groups |
| ȃ• Stage I: T1 N0 M0 |
| ȃ• Stage II: T2 N0 M0 |
| ȃ• Stage III: T1-2 N1 M0 or T3 N0-1 M0 |
| ȃ• Stage IV: T1-3 N0-1 M1 |
Abbreviations: PGL, paraganglioma; PHEO, pheochromocytoma; PPGL, pheochromocytoma/paraganglioma.
Jimenez et al (11).
As per AJCC definitions, invasion into surrounding tissues includes any involvement outside the wall/capsule of any organ. For adrenal tumors, this may include infiltration of extra-adrenal adipose tissue and other soft tissues and/or adjacent organs such as the kidneys, pancreas, liver, and spleen.
Like with many other solid tumors, the PPGL TNM staging establishes that the size of the primary tumor (T) is a clinical predictor of metastasis (14). A cutoff size of 5 cm was elected to raise the stage of a PHEO from a T1 to T2 category based upon comprehensive studies on risk factors for metastasis and survivorship (5, 15, 16). The TNM staging also recognizes the extra-adrenal sympathetic location as a powerful clinical predictor of metastases (13). Because of this, all sympathetic paragangliomas, including tumors smaller than 5 cm, are at least T2, stage II tumors. A very interesting aspect is that PHEO invasion into surrounding tissues, including the periadrenal fat, is also considered a predictor of a decreased overall survival; as per the AJCC, and based on expert panel experience, T3 is a more important predictor of survivorship than the size or location of the primary tumor (13). Nevertheless, no systematic studies have confirmed the predictive value of this finding when compared with the other clinical predictors of metastasis and survivorship. The staging of the regional lymph node involvement is straightforward and any pathologically or clinically proven regional lymph node metastasis classifies the tumor as N1. The presence of regional lymph node metastasis (N1) raises the staging to stage III, making it equal to primary tumor invasion into surrounding tissues (T3). Staging for distant metastasis (M) is also straightforward and a tumor with distant metastasis is always stage IV. Nevertheless, M staging is subdivided in 3 groups depending on the distribution of metastases (Table 1). The M staging stratifies the prognosis of these patients as the results of a previous study showed that up to 20% of metastatic PPGL present with bone metastasis only and that these patients exhibited longer OS when compared with patients with metastasis to other sites (6).
The primary objective of our study was to classify patients with PPGL according to the inaugural AJCC TNM staging system and to correlate staging at diagnosis to overall survival. A secondary objective was to determine how clinical factors such as age, sex, biochemical phenotype, tumor location, and genotype impact TNM staging and survival.
Materials and Methods
Study Population
With approval from The University of Texas MD Anderson Cancer Center institutional review board, we conducted a retrospective study of the MD Anderson Cancer Center PPGL database to identify and retrospectively stage all patients diagnosed with PPGL from January 1987 through December 2018. With 514 patients, the database contains 156 variables, including demographic, clinical, biochemical, genetic, imaging, pathological, surgical, and survival information. All patients with head and neck PGL and those with inadequate clinical information to calculate the prognostic stage group were excluded.
All patients were retrospectively staged by 2 independent observers applying the current TNM staging at initial diagnosis. Operative reports and pathology specimens were reviewed to determine resection status. Tumor diameter was reported based on pathology assessment in patients in whom the primary tumor was removed; in patients who did not undergo resection of the primary tumor, the primary tumor size was determined by conventional radiographic studies such as computed tomography (CT) and magnetic resonance imaging (MRI). As is the norm, malignancy was defined by the presence of metastasis outside the adrenal medulla/paraganglia (17). Synchronous metastases were defined as distant disease present at the time of diagnosis or within 6 months after the diagnosis of PPGL; metachronous metastases were defined as metastatic disease identified longer than 6 months after initial diagnosis. Primary tumor location and determination of metastases were verified by pathology, surgical, and/or radiographic reports (CT, MRI, 18F-fluorodeoxyglucose positron emission tomography/computerized tomography (FDG-PET-CT), 123I-metaiodobenzylguanidine (MIBG) scintigraphy, and/or somatostatin receptor–directed PET-CT). In patients with multiple synchronous primary tumors and no evidence of metastases, the staging of the largest PHEO or PGL was used for analysis. In patients with multiple tumors and metastases, the primary tumor was considered to be either the tumor with the largest diameter and/or the tumor that was associated with adjacent lymph node metastases.
Biochemical phenotypes were determined by measurements of plasma and/or 24-hour urinary fractionated metanephrines. Testing for methoxytyramine is not available in the USA. As the secretion of dopamine is not associated with an obvious endocrine syndrome, we categorized all PPGL that did not have biochemical evidence of excessive secretion of adrenaline and/or noradrenaline as nonfunctional tumors.
Statistical Analysis
Descriptive statistics were used to summarize the data. Categorical covariates were summarized by frequencies and percentages. Continuous covariates were summarized by means, standard deviation, medians, and ranges. Overall survival (OS) was calculated using the difference between the date of diagnosis of the primary tumor and the last date of follow-up or date of death. Unadjusted survival distributions were estimated by the Kaplan–Meier method and comparisons were made with the log rank test. Cox proportional hazards regression models were used to evaluate the associations between survival outcome and covariates of interest including tumor prognostic stage and OS adjusting for age, tumor size, metastatic disease, tumor location, and adrenal or extra-adrenal location. All statistical tests used a significance level of 5%. R version 3.6.1 was used to conduct all statistical analyses.
Results
Demographics
We screened 514 patients with PPGL. The final study group included 458 patients. The demographics of the study population are summarized in Table 2. Female individuals represented 52.4% (n = 240) of the cohort and 67.7% (n = 310) were of Caucasian racial/ethnic background. The median age at diagnosis was 43.3 years (range, 5.0-84.0) and the median primary tumor size was 5.5 cm (range, 0.6-24). The most common tumor was a unilateral PHEO (56.1%) followed by a single abdominal or pelvic paraganglioma (27.7%); 40.9% of the sub-diaphragmatic paragangliomas originated in the organ of Zuckerkandl. Approximately 5% of patients had a single thoracic PGL and 1.5% had multifocal primary tumors. Most patients (80.1%) did not have synchronous metastases at initial diagnosis. 15.6% of these patients ultimately developed metachronous metastases at a median of 59 months (range, 7-473) after diagnosis. Of the 428 patients with hormonal studies, 345 patients (80.6%) had catecholamine overproduction; 83 patients (19.4%) had nonfunctional tumors.
Demographic characteristics of the study population of 458 patients with PPGL at the time of diagnosis
| N (%) | |
|---|---|
| Sex | |
| ȃFemale | 240 (52.4) |
| ȃMale | 218 (47.6) |
| Race | |
| ȃCaucasian | 310 (67.7) |
| ȃAfrican American | 61 (13.3) |
| ȃHispanic | 57 (12.4) |
| ȃAsian | 15 (3.3) |
| ȃOther | 15 (3.3) |
| Location | |
| ȃUnilateral adrenal | 257 (56.1) |
| ȃBilateral adrenal | 44 (9.6) |
| ȃAbdomen/pelvis PGL | 127 (27.7) |
| ȃThorax PGL | 22 (4.8) |
| ȃMultiple locationsa | 8 (1.5) |
| Metastasis at diagnosis | |
| ȃNo metastasis | 367 (80.1) |
| ȃMetastasis (regional N1 or distant M1) | 91 (19.9) |
| ȃȃ• Regional lymph nodes only | 19 (20.8) |
| ȃȃ• Bone only (M1a) | 20 (22.0) |
| ȃȃ• Distant lymph nodes, liver, and/or lungs, and others, but not bone (M1b) | 16 (17.6) |
| ȃȃ• Multiple sites, including bone metastases (M1c) | 36 (39.6) |
| N (%) | |
|---|---|
| Sex | |
| ȃFemale | 240 (52.4) |
| ȃMale | 218 (47.6) |
| Race | |
| ȃCaucasian | 310 (67.7) |
| ȃAfrican American | 61 (13.3) |
| ȃHispanic | 57 (12.4) |
| ȃAsian | 15 (3.3) |
| ȃOther | 15 (3.3) |
| Location | |
| ȃUnilateral adrenal | 257 (56.1) |
| ȃBilateral adrenal | 44 (9.6) |
| ȃAbdomen/pelvis PGL | 127 (27.7) |
| ȃThorax PGL | 22 (4.8) |
| ȃMultiple locationsa | 8 (1.5) |
| Metastasis at diagnosis | |
| ȃNo metastasis | 367 (80.1) |
| ȃMetastasis (regional N1 or distant M1) | 91 (19.9) |
| ȃȃ• Regional lymph nodes only | 19 (20.8) |
| ȃȃ• Bone only (M1a) | 20 (22.0) |
| ȃȃ• Distant lymph nodes, liver, and/or lungs, and others, but not bone (M1b) | 16 (17.6) |
| ȃȃ• Multiple sites, including bone metastases (M1c) | 36 (39.6) |
Abbreviation: PPGL, pheochromocytoma/paraganglioma.
These patients had 2 or more paragangliomas or at least 1 pheochromocytoma and a paraganglioma.
Demographic characteristics of the study population of 458 patients with PPGL at the time of diagnosis
| N (%) | |
|---|---|
| Sex | |
| ȃFemale | 240 (52.4) |
| ȃMale | 218 (47.6) |
| Race | |
| ȃCaucasian | 310 (67.7) |
| ȃAfrican American | 61 (13.3) |
| ȃHispanic | 57 (12.4) |
| ȃAsian | 15 (3.3) |
| ȃOther | 15 (3.3) |
| Location | |
| ȃUnilateral adrenal | 257 (56.1) |
| ȃBilateral adrenal | 44 (9.6) |
| ȃAbdomen/pelvis PGL | 127 (27.7) |
| ȃThorax PGL | 22 (4.8) |
| ȃMultiple locationsa | 8 (1.5) |
| Metastasis at diagnosis | |
| ȃNo metastasis | 367 (80.1) |
| ȃMetastasis (regional N1 or distant M1) | 91 (19.9) |
| ȃȃ• Regional lymph nodes only | 19 (20.8) |
| ȃȃ• Bone only (M1a) | 20 (22.0) |
| ȃȃ• Distant lymph nodes, liver, and/or lungs, and others, but not bone (M1b) | 16 (17.6) |
| ȃȃ• Multiple sites, including bone metastases (M1c) | 36 (39.6) |
| N (%) | |
|---|---|
| Sex | |
| ȃFemale | 240 (52.4) |
| ȃMale | 218 (47.6) |
| Race | |
| ȃCaucasian | 310 (67.7) |
| ȃAfrican American | 61 (13.3) |
| ȃHispanic | 57 (12.4) |
| ȃAsian | 15 (3.3) |
| ȃOther | 15 (3.3) |
| Location | |
| ȃUnilateral adrenal | 257 (56.1) |
| ȃBilateral adrenal | 44 (9.6) |
| ȃAbdomen/pelvis PGL | 127 (27.7) |
| ȃThorax PGL | 22 (4.8) |
| ȃMultiple locationsa | 8 (1.5) |
| Metastasis at diagnosis | |
| ȃNo metastasis | 367 (80.1) |
| ȃMetastasis (regional N1 or distant M1) | 91 (19.9) |
| ȃȃ• Regional lymph nodes only | 19 (20.8) |
| ȃȃ• Bone only (M1a) | 20 (22.0) |
| ȃȃ• Distant lymph nodes, liver, and/or lungs, and others, but not bone (M1b) | 16 (17.6) |
| ȃȃ• Multiple sites, including bone metastases (M1c) | 36 (39.6) |
Abbreviation: PPGL, pheochromocytoma/paraganglioma.
These patients had 2 or more paragangliomas or at least 1 pheochromocytoma and a paraganglioma.
Germline Testing for PPGL Susceptibility Gene Variants
Of the 458 patients, 276 had genetic testing; 162 of these patients (59%) tested positive for a germline pathogenic DNA variant in one of the known PPGL susceptibility genes. The most common mutations were found in the rearranged during transfection (RET) proto-oncogene (38%), followed by the SDHB (28%), Von Hippel-Lindau (VHL) (17%), and Neurofibromatosis Type 1 (NF1) (7%) genes. Succinate dehydrogenase subunit D (SDHD) mutations were found in 4.3% of patients. Three patients had mutations of the succinate dehydrogenase subunit C (SDHC) gene (1.9%). There were single carriers of a pathogenic variant in the succinate dehydrogenase complex assembly factor 2 (SDHAF2), succinate dehydrogenase subunit A (SDHA), fumarate hydratase (FH), MYC-associated factor X (MAX), transmembrane protein 127 (TMEM127), and multiple endocrine neoplasia type 1 (MEN1) genes.
TNM Staging
Of the 458 study subjects, 126 (27.5%) were grouped as stage I, 213 (46.5%) were stage II, 47 (10.3%) were stage III, and 72 (15.7%) were stage IV. Table 3 describes the TNM categories and staging of the patients.
TNM categories and staging groups in 458 patients diagnosed with pheochromocytoma/paraganglioma
| Category | Total (n) | Stage I (n) | Stage II (n) | Stage III (n) | Stage IV (n) |
|---|---|---|---|---|---|
| T1 (%) | 127 (27.7%) | 126 (99.2%) | N/A | 0 (0%) | 1 (0.8%) |
| T2 (%) | 271 (59.2%) | N/A | 213 (78.6%) | 13 (4.8%) | 45 (16.6%) |
| T3 (%) | 60 (13.1%) | N/A | N/A | 34 (56.7%) | 26 (43.3%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| NX | 13 (2.8%) | N/A | N/A | 4 (30%) | 9 (70%) |
| N0 (%) | 406 (88.7%) | 126 (31.0%) | 213 (52.5%) | 24 (5.9%) | 43 (10.6%) |
| N1 (%) | 39 (8.5%) | N/A | N/A | 19 (48.7%) | 20 (51.3%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| M0 | 386 (84.3%) | 126 (32.6%) | 213 (55.2%) | 47 (12.2%) | 0 (0%) |
| M1 | 72 (15.7%) | N/A | N/A | N/A | 72 (15.7%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| Category | Total (n) | Stage I (n) | Stage II (n) | Stage III (n) | Stage IV (n) |
|---|---|---|---|---|---|
| T1 (%) | 127 (27.7%) | 126 (99.2%) | N/A | 0 (0%) | 1 (0.8%) |
| T2 (%) | 271 (59.2%) | N/A | 213 (78.6%) | 13 (4.8%) | 45 (16.6%) |
| T3 (%) | 60 (13.1%) | N/A | N/A | 34 (56.7%) | 26 (43.3%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| NX | 13 (2.8%) | N/A | N/A | 4 (30%) | 9 (70%) |
| N0 (%) | 406 (88.7%) | 126 (31.0%) | 213 (52.5%) | 24 (5.9%) | 43 (10.6%) |
| N1 (%) | 39 (8.5%) | N/A | N/A | 19 (48.7%) | 20 (51.3%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| M0 | 386 (84.3%) | 126 (32.6%) | 213 (55.2%) | 47 (12.2%) | 0 (0%) |
| M1 | 72 (15.7%) | N/A | N/A | N/A | 72 (15.7%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
TNM categories and staging groups in 458 patients diagnosed with pheochromocytoma/paraganglioma
| Category | Total (n) | Stage I (n) | Stage II (n) | Stage III (n) | Stage IV (n) |
|---|---|---|---|---|---|
| T1 (%) | 127 (27.7%) | 126 (99.2%) | N/A | 0 (0%) | 1 (0.8%) |
| T2 (%) | 271 (59.2%) | N/A | 213 (78.6%) | 13 (4.8%) | 45 (16.6%) |
| T3 (%) | 60 (13.1%) | N/A | N/A | 34 (56.7%) | 26 (43.3%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| NX | 13 (2.8%) | N/A | N/A | 4 (30%) | 9 (70%) |
| N0 (%) | 406 (88.7%) | 126 (31.0%) | 213 (52.5%) | 24 (5.9%) | 43 (10.6%) |
| N1 (%) | 39 (8.5%) | N/A | N/A | 19 (48.7%) | 20 (51.3%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| M0 | 386 (84.3%) | 126 (32.6%) | 213 (55.2%) | 47 (12.2%) | 0 (0%) |
| M1 | 72 (15.7%) | N/A | N/A | N/A | 72 (15.7%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| Category | Total (n) | Stage I (n) | Stage II (n) | Stage III (n) | Stage IV (n) |
|---|---|---|---|---|---|
| T1 (%) | 127 (27.7%) | 126 (99.2%) | N/A | 0 (0%) | 1 (0.8%) |
| T2 (%) | 271 (59.2%) | N/A | 213 (78.6%) | 13 (4.8%) | 45 (16.6%) |
| T3 (%) | 60 (13.1%) | N/A | N/A | 34 (56.7%) | 26 (43.3%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| NX | 13 (2.8%) | N/A | N/A | 4 (30%) | 9 (70%) |
| N0 (%) | 406 (88.7%) | 126 (31.0%) | 213 (52.5%) | 24 (5.9%) | 43 (10.6%) |
| N1 (%) | 39 (8.5%) | N/A | N/A | 19 (48.7%) | 20 (51.3%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
| M0 | 386 (84.3%) | 126 (32.6%) | 213 (55.2%) | 47 (12.2%) | 0 (0%) |
| M1 | 72 (15.7%) | N/A | N/A | N/A | 72 (15.7%) |
| Total | 458 (100%) | 126 (27.5%) | 213 (46.5%) | 47 (10.3%) | 72 (15.7%) |
Tumor category (T)
One hundred and twenty-seven patients (27.7%) had PHEO smaller than 5 cm and were categorized as T1; 126 (99.2%) of these pheochromocytomas were classified as stage I disease. In general, pheochromocytomas smaller than 5 cm did not have invasion into surrounding tissues or regional lymph node metastases. Only 1 patient with a T1 SDHB (+) pheochromocytoma had stage IV disease with metastasis to the liver and the skeleton. The majority of patients (59.2%) had T2 tumors and approximately 80% of T2 patients had localized disease (stage II); however, the number of patients with metastatic disease increased when compared with stage I. In fact, almost 17% of patients with T2 tumors had stage IV disease (Table 3).
Of interest, T3 PPGL (tumors associated with infiltration of surrounding tissues) were not common (13%); however, more than 40% of patients with T3 PPGL had stage IV disease. The median primary tumor size in patients with T3 disease was 10 cm (range, 4.1-19, n = 25) for pheochromocytomas and 6.5 cm (range, 1.8-21, n = 35) for paragangliomas.
Regional lymph node category (N)
Most patients (∼90%) did not have regional lymph node metastases (N0). About 10% of patients had locoregional metastatic lymphadenopathy (N1), 50% of whom also had distant metastases (stage IV). The median primary pheochromocytoma size in patients with regional lymph node metastases was 9.5 cm (range, 5.5-21, n = 16). The median primary paraganglioma size for N1 disease was 6.8 cm (2-24 cm, n = 23). Thirteen patients (2.8%) were not evaluable for lymph node metastases (NX). Of these, 9 patients with distant metastases did not have resection of the primary tumor and regional lymph nodes. These patients had conventional radiographic studies that identified enlarged but nonspecific regional lymph nodes that were not abnormal on nuclear medicine studies. Four patients had removal of primary tumors associated with peritumoral adipose tissue infiltration but did not have resection of peritumoral lymph nodes; however, conventional preoperative radiographic studies described enlarged, nonspecific peritumoral lymph nodes that were not apparent on an MIBG scan. These patients did not have obvious distant metastases.
Distant metastasis category (M)
Most patients (n = 367, 80.1%) did not have distant metastatic disease (stage IV) at diagnosis. Approximately 20% of patients (n = 91) had either lymph node and/or distant metastatic disease at diagnosis (Table 2).
Metachronous Distant Metastases According to the Stage
Metachronous metastases occurred in 57 (12.4%) of the subjects and the median time to development was 59 months (range, 8-211) Only 2 patients with a T1 pheochromocytoma, both carriers of an SDHB mutation, developed distant metachronous metastases (1.6% of stage I patients) (Table 4). Median time to development of metachronous metastases was 55.5 months (range, 37-74). Thirty-eight patients (17.8%) with stage II tumors developed metachronous metastases. Median time to development of metachronous metastases in patients with stage II tumors was 59.5 months (range, 11-211). Five of these patients had SDHB mutations and one had a MAX mutation. The other 32 patients had apparently sporadic tumors based on comprehensive gene testing and negative family history.
Rate of distant metachronous metastases and genetic background
| Initial stage | Total (n) | Patients with distant metachronous metastases N (%) | Genetic background |
|---|---|---|---|
| Stage I | 126 | 2 (1.6%) | SDHB (+) n = 2 |
| Stage II | 213 | 38 (17.8%) | SDHB (+) n = 5 |
| MAX n = 1 | |||
| Apparently sporadic n = 32 | |||
| Stage III | 47 | 17 (36.2%) | SDHB (+) n = 7 |
| SDHC (+) n = 1 | |||
| SDHD (+) n = 1 | |||
| RET (+) n = 1 | |||
| VHL (+) n = 1 | |||
| Apparently sporadic n = 6 |
| Initial stage | Total (n) | Patients with distant metachronous metastases N (%) | Genetic background |
|---|---|---|---|
| Stage I | 126 | 2 (1.6%) | SDHB (+) n = 2 |
| Stage II | 213 | 38 (17.8%) | SDHB (+) n = 5 |
| MAX n = 1 | |||
| Apparently sporadic n = 32 | |||
| Stage III | 47 | 17 (36.2%) | SDHB (+) n = 7 |
| SDHC (+) n = 1 | |||
| SDHD (+) n = 1 | |||
| RET (+) n = 1 | |||
| VHL (+) n = 1 | |||
| Apparently sporadic n = 6 |
Abbreviations: MAX, MYC-associated factor X; RET, rearranged during transfection; SDHB, succinate dehydrogenase subunit B; SDHC, succinate dehydrogenase subunit C; SDHD, succinate dehydrogenase subunit D; VHL, von Hippel-Lindau.
Rate of distant metachronous metastases and genetic background
| Initial stage | Total (n) | Patients with distant metachronous metastases N (%) | Genetic background |
|---|---|---|---|
| Stage I | 126 | 2 (1.6%) | SDHB (+) n = 2 |
| Stage II | 213 | 38 (17.8%) | SDHB (+) n = 5 |
| MAX n = 1 | |||
| Apparently sporadic n = 32 | |||
| Stage III | 47 | 17 (36.2%) | SDHB (+) n = 7 |
| SDHC (+) n = 1 | |||
| SDHD (+) n = 1 | |||
| RET (+) n = 1 | |||
| VHL (+) n = 1 | |||
| Apparently sporadic n = 6 |
| Initial stage | Total (n) | Patients with distant metachronous metastases N (%) | Genetic background |
|---|---|---|---|
| Stage I | 126 | 2 (1.6%) | SDHB (+) n = 2 |
| Stage II | 213 | 38 (17.8%) | SDHB (+) n = 5 |
| MAX n = 1 | |||
| Apparently sporadic n = 32 | |||
| Stage III | 47 | 17 (36.2%) | SDHB (+) n = 7 |
| SDHC (+) n = 1 | |||
| SDHD (+) n = 1 | |||
| RET (+) n = 1 | |||
| VHL (+) n = 1 | |||
| Apparently sporadic n = 6 |
Abbreviations: MAX, MYC-associated factor X; RET, rearranged during transfection; SDHB, succinate dehydrogenase subunit B; SDHC, succinate dehydrogenase subunit C; SDHD, succinate dehydrogenase subunit D; VHL, von Hippel-Lindau.
Seventeen patients (36.2%) with stage III PPGL developed metachronous distant metastases. Median time to development of metachronous metastases in patients with stage III disease was 54.5 months (range, 8-189). Of these, 35% had apparently sporadic tumors and 41% carried an SDHB mutation (Table 4). Only 1 patient with a germline 634 RET mutation developed metachronous metastases.
We did not observe a statistically significant difference in the median time of diagnosis of distant metachronous metastases between stages I, II, and III.
TNM Categories and Staging Overall Survival
The T categories correlated with OS. Compared with T1 stage, the hazard ratio (HR) for death in the T2 category was 2.22 (95% CI, 1.35-3.63) and for T3 it was 3.96 (95% CI, 2.21-7.10) (P < 0.001). Subjects with T1 tumors had longer OS when compared with T2 (T1 median survival time [MST]: 25.5 years [95% CI, 20.9-not reached]) vs T2 MST: 15.6 years [95% CI, 12.5-not reached]). T2 had a longer OS when compared with T3 (T3 MST: 7.1 years [95% CI, 5.6-not reached]). Figure 1A shows the OS for each T category, along with MST. Also, patients with N1 disease had worse survival compared with N0 disease (Fig. 1B). Compared with N0 disease, the HR for death in N1 disease was 2.98 (95% CI, 1.81-4.90; P = 0.001).
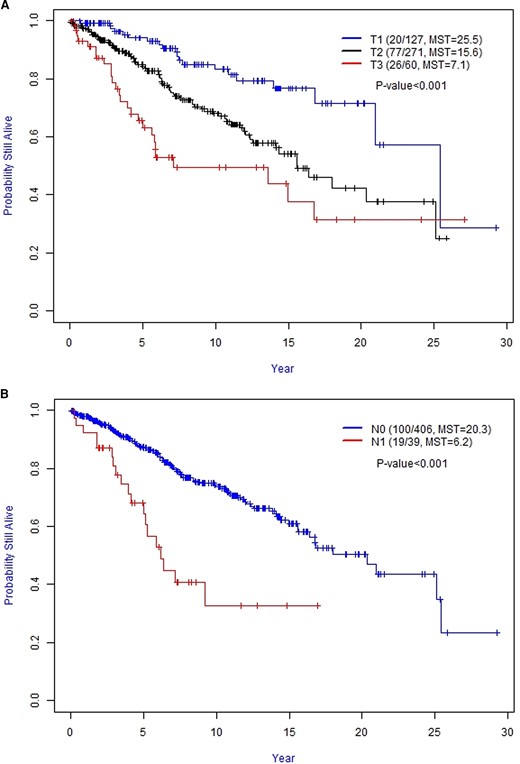
A, Kaplan–Meier curves of the overall survival for T category. B, Kaplan–Meier curves of the overall survival for N category. Abbreviation: MST, median survival time (in years).
Median OS for stage I was 25.2 years (95% CI, 20.9-not reached), for stage II was 20.3 years (95% CI, 18.0-not reached), for stage III was 13.6 years (95% CI, 9.2-not reached), and for stage IV was 5.1 years (95% CI, 3.7-6.4) (P = 0.001). Patients with distant metastasis at diagnosis had worse survival (stage IV) compared with patients without distant metastases (stages I-III) (Fig. 2). Compared with M1, the HR for death in patients with M0 was 0.16 (95% CI, 0.11-0.24) and the HR for death in patients with stage IV was 8.88 (95% CI, 5.16-15.29) compared with stage I patients.
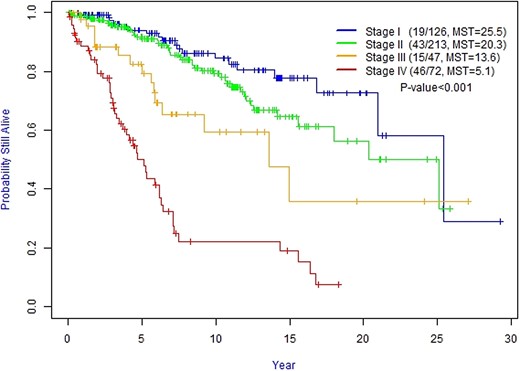
Kaplan–Meier curves of the overall survival for staging groups. Abbreviation: MST, median survival time (in years).
Of interest, we did not observe a statistically significant difference in OS between Stage II PGL and PHEO (Fig. 3A, P = 0.476). In addition, we did not observe a statistically significant difference in OS between Stage III N0 and Stage III N1 patients (Fig. 3B, P = 0.865).
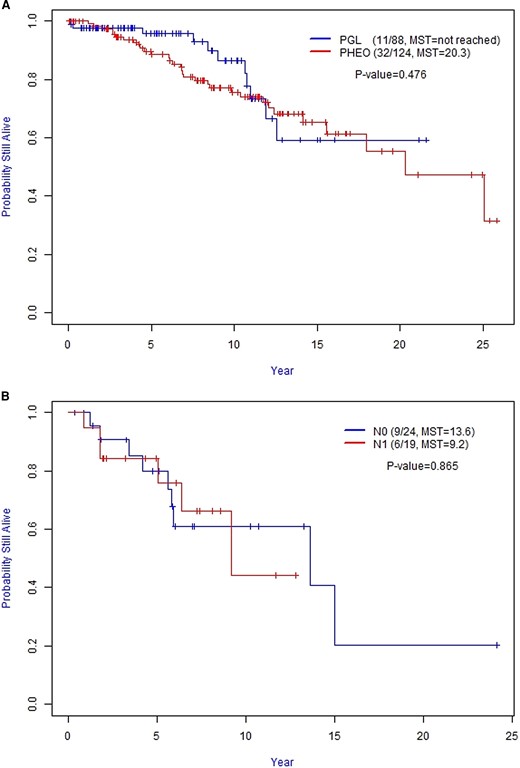
A, Overall Survival (OS) between Stage II PGL and PHEO. B, Overall Survival (OS) between Stage III N0 and N1 patients. Abbreviation: MST, median survival time (in years); PGL, paraganglioma; PHEO, pheochromocytoma.
In addition, older age (HR 1.03; 95% CI, 1.02-1.04; P < 0.001), larger tumor size (HR 1.11; 95% CI, 1.07-1.15; P < 0.001), and synchronous metastatic disease (HR 2.51; 95% CI, 1.56-4.05; P < 0.001) were associated with worse overall survival. OS was not associated with PGL location, gender, race, or hormonal secretion. Most germline mutations were not associated with higher rates of death compared with apparently sporadic cases; however, RET mutations were associated with a better prognosis (HR 0.28; 95% CI, 0.12-0.69; P = 0.0026). The HR for death for NF1 was 1.79 (95% CI, 0.42-7.63), for SDHB was 1.14 (95% CI, 0.58-2.21), for VHL was 0.64 (95% CI, 0.26-1.56) (Table 5). Of interest, the mortality in NF1 PPGL patients was related to the presence of distant metastases (stage IV) rather than complications related to other NF1-related tumors.
Univariate Cox analysis of prognostic factors (whole cohort)
| Prognostic factor | N | Events | HR | 95% CI for HR | P |
|---|---|---|---|---|---|
| Age at diagnosis | 458 | 123 | 1.03 | (1.02, 1.04) | <0.001 |
| Tumor size | 458 | 123 | 1.11 | (1.07, 1.15) | <0.001 |
| T stage | 458 | 123 | <0.001 | ||
| ȃT1 (ref)a | 127 | 20 | — | — | |
| ȃT2 | 271 | 77 | 2.22 | (1.35, 3.63) | |
| ȃT3 | 60 | 26 | 3.96 | (2.21, 7.10) | |
| N stage | 458 | 123 | 0.001 | ||
| ȃN0 (ref)a | 406 | 100 | — | — | |
| ȃN1 | 39 | 19 | 2.98 | (1.81, 4.90) | |
| ȃNX | 13 | 4 | 1.35 | (0.49, 3.75) | |
| M stage | 458 | 123 | <0.001 | ||
| ȃM0 (ref)a | 386 | 77 | — | — | |
| ȃM1 | |||||
| ȃȃM1a | 20 | 13 | 5.93 | (3.26, 10.80) | |
| ȃȃM1b | 16 | 9 | 3.98 | (1.99, 7.98) | |
| ȃȃM1c | 36 | 24 | 7.88 | (4.91, 12.66) | |
| Stage | 458 | 123 | <0.001 | ||
| ȃStage I (ref)a | 126 | 19 | — | — | |
| ȃStage II | 213 | 43 | 1.50 | (0.87, 2.57) | |
| ȃStage III | 47 | 15 | 2.85 | (1.45, 5.63) | |
| ȃStage IV | 72 | 46 | 8.88 | (5.16, 15.29) | |
| Timing of metastasis | 148 | 79 | <0.001 | ||
| ȃMetachronous (ref)a | 57 | 26 | — | — | |
| ȃSynchronous | 91 | 53 | 2.51 | (1.56, 4.05) | |
| Genetic | 276 | 59 | 0.026 | ||
| ȃNegative (ref)a | 114 | 27 | — | — | |
| ȃNF1 | 11 | 2 | 1.79 | (0.42, 7.63) | |
| ȃOther mutation | 18 | 4 | 0.57 | (0.20, 1.63) | |
| ȃRET | 48 | 6 | 0.28 | (0.12, 0.69) | |
| ȃSDHX | 57 | 14 | 1.05 | (0.55, 2.01) | |
| ȃVHL | 28 | 6 | 0.64 | (0.26, 1.56) | |
| Tumor location | 458 | 123 | 0.196 | ||
| ȃAdrenal (ref)a | 257 | 72 | — | — | |
| ȃAbdomen/pelvis PGL | 127 | 34 | 1.25 | (0.83, 1.88) | |
| ȃBilateral adrenal | 44 | 9 | 0.53 | (0.27, 1.07) | |
| ȃMultiple locations | 8 | 2 | 1.01 | (0.25, 4.13) | |
| ȃThorax PGL | 22 | 6 | 1.17 | (0.51, 2.69) | |
| Adrenal vs not adrenal | 458 | 123 | 0.198 | ||
| ȃAdrenal (ref)a | 300 | 82 | — | — | |
| ȃNot adrenal | 157 | 41 | 1.29 | (0.88, 1.88) | |
| Gender | 458 | 123 | 0.365 | ||
| ȃF (ref)a | 240 | 60 | — | — | |
| ȃM | 218 | 63 | 1.18 | (0.83, 1.68) | |
| Race | 458 | 122 | 0.553 | ||
| ȃAsian (ref)a | 15 | 1 | — | — | |
| ȃBlack | 61 | 19 | 2.50 | (0.33, 18.67) | |
| ȃHispanic | 57 | 17 | 2.52 | (0.34, 18.98) | |
| ȃOther | 15 | 2 | 1.41 | (0.13, 15.58) | |
| ȃWhite | 310 | 83 | 1.86 | (0.26, 13.39) | |
| Zuckerkandl | 458 | 122 | 0.287 | ||
| ȃNo (ref)a | 396 | 107 | — | — | |
| ȃYes | 52 | 15 | 1.36 | (0.79, 2.33) | |
| SDH | 276 | 59 | 0.199 | ||
| ȃNo (ref)a | 219 | 45 | — | — | |
| ȃYes | 57 | 14 | 1.51 | (0.82, 2.76) | |
| Hormones | 428 | 115 | 0.034 | ||
| ȃNo secretion (ref)a | 83 | 28 | — | — | |
| ȃCatecholamine secretion | 345 | 87 | 0.62 | (0.40, 0.95) | |
| M stage | 458 | 123 | <0.001 | ||
| ȃM1 (ref)a | 72 | 46 | — | — | |
| ȃM0 | 386 | 77 | 0.16 | (0.11, 0.24) |
| Prognostic factor | N | Events | HR | 95% CI for HR | P |
|---|---|---|---|---|---|
| Age at diagnosis | 458 | 123 | 1.03 | (1.02, 1.04) | <0.001 |
| Tumor size | 458 | 123 | 1.11 | (1.07, 1.15) | <0.001 |
| T stage | 458 | 123 | <0.001 | ||
| ȃT1 (ref)a | 127 | 20 | — | — | |
| ȃT2 | 271 | 77 | 2.22 | (1.35, 3.63) | |
| ȃT3 | 60 | 26 | 3.96 | (2.21, 7.10) | |
| N stage | 458 | 123 | 0.001 | ||
| ȃN0 (ref)a | 406 | 100 | — | — | |
| ȃN1 | 39 | 19 | 2.98 | (1.81, 4.90) | |
| ȃNX | 13 | 4 | 1.35 | (0.49, 3.75) | |
| M stage | 458 | 123 | <0.001 | ||
| ȃM0 (ref)a | 386 | 77 | — | — | |
| ȃM1 | |||||
| ȃȃM1a | 20 | 13 | 5.93 | (3.26, 10.80) | |
| ȃȃM1b | 16 | 9 | 3.98 | (1.99, 7.98) | |
| ȃȃM1c | 36 | 24 | 7.88 | (4.91, 12.66) | |
| Stage | 458 | 123 | <0.001 | ||
| ȃStage I (ref)a | 126 | 19 | — | — | |
| ȃStage II | 213 | 43 | 1.50 | (0.87, 2.57) | |
| ȃStage III | 47 | 15 | 2.85 | (1.45, 5.63) | |
| ȃStage IV | 72 | 46 | 8.88 | (5.16, 15.29) | |
| Timing of metastasis | 148 | 79 | <0.001 | ||
| ȃMetachronous (ref)a | 57 | 26 | — | — | |
| ȃSynchronous | 91 | 53 | 2.51 | (1.56, 4.05) | |
| Genetic | 276 | 59 | 0.026 | ||
| ȃNegative (ref)a | 114 | 27 | — | — | |
| ȃNF1 | 11 | 2 | 1.79 | (0.42, 7.63) | |
| ȃOther mutation | 18 | 4 | 0.57 | (0.20, 1.63) | |
| ȃRET | 48 | 6 | 0.28 | (0.12, 0.69) | |
| ȃSDHX | 57 | 14 | 1.05 | (0.55, 2.01) | |
| ȃVHL | 28 | 6 | 0.64 | (0.26, 1.56) | |
| Tumor location | 458 | 123 | 0.196 | ||
| ȃAdrenal (ref)a | 257 | 72 | — | — | |
| ȃAbdomen/pelvis PGL | 127 | 34 | 1.25 | (0.83, 1.88) | |
| ȃBilateral adrenal | 44 | 9 | 0.53 | (0.27, 1.07) | |
| ȃMultiple locations | 8 | 2 | 1.01 | (0.25, 4.13) | |
| ȃThorax PGL | 22 | 6 | 1.17 | (0.51, 2.69) | |
| Adrenal vs not adrenal | 458 | 123 | 0.198 | ||
| ȃAdrenal (ref)a | 300 | 82 | — | — | |
| ȃNot adrenal | 157 | 41 | 1.29 | (0.88, 1.88) | |
| Gender | 458 | 123 | 0.365 | ||
| ȃF (ref)a | 240 | 60 | — | — | |
| ȃM | 218 | 63 | 1.18 | (0.83, 1.68) | |
| Race | 458 | 122 | 0.553 | ||
| ȃAsian (ref)a | 15 | 1 | — | — | |
| ȃBlack | 61 | 19 | 2.50 | (0.33, 18.67) | |
| ȃHispanic | 57 | 17 | 2.52 | (0.34, 18.98) | |
| ȃOther | 15 | 2 | 1.41 | (0.13, 15.58) | |
| ȃWhite | 310 | 83 | 1.86 | (0.26, 13.39) | |
| Zuckerkandl | 458 | 122 | 0.287 | ||
| ȃNo (ref)a | 396 | 107 | — | — | |
| ȃYes | 52 | 15 | 1.36 | (0.79, 2.33) | |
| SDH | 276 | 59 | 0.199 | ||
| ȃNo (ref)a | 219 | 45 | — | — | |
| ȃYes | 57 | 14 | 1.51 | (0.82, 2.76) | |
| Hormones | 428 | 115 | 0.034 | ||
| ȃNo secretion (ref)a | 83 | 28 | — | — | |
| ȃCatecholamine secretion | 345 | 87 | 0.62 | (0.40, 0.95) | |
| M stage | 458 | 123 | <0.001 | ||
| ȃM1 (ref)a | 72 | 46 | — | — | |
| ȃM0 | 386 | 77 | 0.16 | (0.11, 0.24) |
P values are based on likelihood ratio test. Abbreviations: HR, hazard ratio; TNM, tumor, nodes, and metastasis (classification).
(ref): reference category to which each other category is compared.
Univariate Cox analysis of prognostic factors (whole cohort)
| Prognostic factor | N | Events | HR | 95% CI for HR | P |
|---|---|---|---|---|---|
| Age at diagnosis | 458 | 123 | 1.03 | (1.02, 1.04) | <0.001 |
| Tumor size | 458 | 123 | 1.11 | (1.07, 1.15) | <0.001 |
| T stage | 458 | 123 | <0.001 | ||
| ȃT1 (ref)a | 127 | 20 | — | — | |
| ȃT2 | 271 | 77 | 2.22 | (1.35, 3.63) | |
| ȃT3 | 60 | 26 | 3.96 | (2.21, 7.10) | |
| N stage | 458 | 123 | 0.001 | ||
| ȃN0 (ref)a | 406 | 100 | — | — | |
| ȃN1 | 39 | 19 | 2.98 | (1.81, 4.90) | |
| ȃNX | 13 | 4 | 1.35 | (0.49, 3.75) | |
| M stage | 458 | 123 | <0.001 | ||
| ȃM0 (ref)a | 386 | 77 | — | — | |
| ȃM1 | |||||
| ȃȃM1a | 20 | 13 | 5.93 | (3.26, 10.80) | |
| ȃȃM1b | 16 | 9 | 3.98 | (1.99, 7.98) | |
| ȃȃM1c | 36 | 24 | 7.88 | (4.91, 12.66) | |
| Stage | 458 | 123 | <0.001 | ||
| ȃStage I (ref)a | 126 | 19 | — | — | |
| ȃStage II | 213 | 43 | 1.50 | (0.87, 2.57) | |
| ȃStage III | 47 | 15 | 2.85 | (1.45, 5.63) | |
| ȃStage IV | 72 | 46 | 8.88 | (5.16, 15.29) | |
| Timing of metastasis | 148 | 79 | <0.001 | ||
| ȃMetachronous (ref)a | 57 | 26 | — | — | |
| ȃSynchronous | 91 | 53 | 2.51 | (1.56, 4.05) | |
| Genetic | 276 | 59 | 0.026 | ||
| ȃNegative (ref)a | 114 | 27 | — | — | |
| ȃNF1 | 11 | 2 | 1.79 | (0.42, 7.63) | |
| ȃOther mutation | 18 | 4 | 0.57 | (0.20, 1.63) | |
| ȃRET | 48 | 6 | 0.28 | (0.12, 0.69) | |
| ȃSDHX | 57 | 14 | 1.05 | (0.55, 2.01) | |
| ȃVHL | 28 | 6 | 0.64 | (0.26, 1.56) | |
| Tumor location | 458 | 123 | 0.196 | ||
| ȃAdrenal (ref)a | 257 | 72 | — | — | |
| ȃAbdomen/pelvis PGL | 127 | 34 | 1.25 | (0.83, 1.88) | |
| ȃBilateral adrenal | 44 | 9 | 0.53 | (0.27, 1.07) | |
| ȃMultiple locations | 8 | 2 | 1.01 | (0.25, 4.13) | |
| ȃThorax PGL | 22 | 6 | 1.17 | (0.51, 2.69) | |
| Adrenal vs not adrenal | 458 | 123 | 0.198 | ||
| ȃAdrenal (ref)a | 300 | 82 | — | — | |
| ȃNot adrenal | 157 | 41 | 1.29 | (0.88, 1.88) | |
| Gender | 458 | 123 | 0.365 | ||
| ȃF (ref)a | 240 | 60 | — | — | |
| ȃM | 218 | 63 | 1.18 | (0.83, 1.68) | |
| Race | 458 | 122 | 0.553 | ||
| ȃAsian (ref)a | 15 | 1 | — | — | |
| ȃBlack | 61 | 19 | 2.50 | (0.33, 18.67) | |
| ȃHispanic | 57 | 17 | 2.52 | (0.34, 18.98) | |
| ȃOther | 15 | 2 | 1.41 | (0.13, 15.58) | |
| ȃWhite | 310 | 83 | 1.86 | (0.26, 13.39) | |
| Zuckerkandl | 458 | 122 | 0.287 | ||
| ȃNo (ref)a | 396 | 107 | — | — | |
| ȃYes | 52 | 15 | 1.36 | (0.79, 2.33) | |
| SDH | 276 | 59 | 0.199 | ||
| ȃNo (ref)a | 219 | 45 | — | — | |
| ȃYes | 57 | 14 | 1.51 | (0.82, 2.76) | |
| Hormones | 428 | 115 | 0.034 | ||
| ȃNo secretion (ref)a | 83 | 28 | — | — | |
| ȃCatecholamine secretion | 345 | 87 | 0.62 | (0.40, 0.95) | |
| M stage | 458 | 123 | <0.001 | ||
| ȃM1 (ref)a | 72 | 46 | — | — | |
| ȃM0 | 386 | 77 | 0.16 | (0.11, 0.24) |
| Prognostic factor | N | Events | HR | 95% CI for HR | P |
|---|---|---|---|---|---|
| Age at diagnosis | 458 | 123 | 1.03 | (1.02, 1.04) | <0.001 |
| Tumor size | 458 | 123 | 1.11 | (1.07, 1.15) | <0.001 |
| T stage | 458 | 123 | <0.001 | ||
| ȃT1 (ref)a | 127 | 20 | — | — | |
| ȃT2 | 271 | 77 | 2.22 | (1.35, 3.63) | |
| ȃT3 | 60 | 26 | 3.96 | (2.21, 7.10) | |
| N stage | 458 | 123 | 0.001 | ||
| ȃN0 (ref)a | 406 | 100 | — | — | |
| ȃN1 | 39 | 19 | 2.98 | (1.81, 4.90) | |
| ȃNX | 13 | 4 | 1.35 | (0.49, 3.75) | |
| M stage | 458 | 123 | <0.001 | ||
| ȃM0 (ref)a | 386 | 77 | — | — | |
| ȃM1 | |||||
| ȃȃM1a | 20 | 13 | 5.93 | (3.26, 10.80) | |
| ȃȃM1b | 16 | 9 | 3.98 | (1.99, 7.98) | |
| ȃȃM1c | 36 | 24 | 7.88 | (4.91, 12.66) | |
| Stage | 458 | 123 | <0.001 | ||
| ȃStage I (ref)a | 126 | 19 | — | — | |
| ȃStage II | 213 | 43 | 1.50 | (0.87, 2.57) | |
| ȃStage III | 47 | 15 | 2.85 | (1.45, 5.63) | |
| ȃStage IV | 72 | 46 | 8.88 | (5.16, 15.29) | |
| Timing of metastasis | 148 | 79 | <0.001 | ||
| ȃMetachronous (ref)a | 57 | 26 | — | — | |
| ȃSynchronous | 91 | 53 | 2.51 | (1.56, 4.05) | |
| Genetic | 276 | 59 | 0.026 | ||
| ȃNegative (ref)a | 114 | 27 | — | — | |
| ȃNF1 | 11 | 2 | 1.79 | (0.42, 7.63) | |
| ȃOther mutation | 18 | 4 | 0.57 | (0.20, 1.63) | |
| ȃRET | 48 | 6 | 0.28 | (0.12, 0.69) | |
| ȃSDHX | 57 | 14 | 1.05 | (0.55, 2.01) | |
| ȃVHL | 28 | 6 | 0.64 | (0.26, 1.56) | |
| Tumor location | 458 | 123 | 0.196 | ||
| ȃAdrenal (ref)a | 257 | 72 | — | — | |
| ȃAbdomen/pelvis PGL | 127 | 34 | 1.25 | (0.83, 1.88) | |
| ȃBilateral adrenal | 44 | 9 | 0.53 | (0.27, 1.07) | |
| ȃMultiple locations | 8 | 2 | 1.01 | (0.25, 4.13) | |
| ȃThorax PGL | 22 | 6 | 1.17 | (0.51, 2.69) | |
| Adrenal vs not adrenal | 458 | 123 | 0.198 | ||
| ȃAdrenal (ref)a | 300 | 82 | — | — | |
| ȃNot adrenal | 157 | 41 | 1.29 | (0.88, 1.88) | |
| Gender | 458 | 123 | 0.365 | ||
| ȃF (ref)a | 240 | 60 | — | — | |
| ȃM | 218 | 63 | 1.18 | (0.83, 1.68) | |
| Race | 458 | 122 | 0.553 | ||
| ȃAsian (ref)a | 15 | 1 | — | — | |
| ȃBlack | 61 | 19 | 2.50 | (0.33, 18.67) | |
| ȃHispanic | 57 | 17 | 2.52 | (0.34, 18.98) | |
| ȃOther | 15 | 2 | 1.41 | (0.13, 15.58) | |
| ȃWhite | 310 | 83 | 1.86 | (0.26, 13.39) | |
| Zuckerkandl | 458 | 122 | 0.287 | ||
| ȃNo (ref)a | 396 | 107 | — | — | |
| ȃYes | 52 | 15 | 1.36 | (0.79, 2.33) | |
| SDH | 276 | 59 | 0.199 | ||
| ȃNo (ref)a | 219 | 45 | — | — | |
| ȃYes | 57 | 14 | 1.51 | (0.82, 2.76) | |
| Hormones | 428 | 115 | 0.034 | ||
| ȃNo secretion (ref)a | 83 | 28 | — | — | |
| ȃCatecholamine secretion | 345 | 87 | 0.62 | (0.40, 0.95) | |
| M stage | 458 | 123 | <0.001 | ||
| ȃM1 (ref)a | 72 | 46 | — | — | |
| ȃM0 | 386 | 77 | 0.16 | (0.11, 0.24) |
P values are based on likelihood ratio test. Abbreviations: HR, hazard ratio; TNM, tumor, nodes, and metastasis (classification).
(ref): reference category to which each other category is compared.
In the multivariate analysis, adjusting age, tumor size and location, only age and stage IV disease were associated with OS (Table 6).
Multivariate cox analysis for overall survival, n = 458 and n events = 123
| Variable | N | Events | HR | 95% CI for HR | P |
|---|---|---|---|---|---|
| Age at diagnosis | 458 | 123 | 1.03 | (1.02, 1.05) | <0.001 |
| Tumor size | 1.03 | (0.98, 1.08) | 0.285 | ||
| Stage | |||||
| ȃStage I (ref)a | — | — | — | ||
| ȃStage II | 0.97 | (0.50, 1.88) | 0.935 | ||
| ȃStage III | 1.32 | (0.52, 3.34) | 0.557 | ||
| ȃStage IV | 4.07 | (1.75, 9.48) | 0.001 | ||
| Tumor location | |||||
| ȃAdrenal (ref)a | — | — | — | ||
| ȃAbdomen/Pelvis PGL | 1.94 | (0.35, 10.62) | 0.444 | ||
| ȃBilateral adrenal | 0.92 | (0.45, 1.91) | 0.828 | ||
| ȃMultiple location | 2.45 | (0.48, 12.55) | 0.282 | ||
| ȃThorax PG | 1.58 | (0.26, 9.55) | 0.618 |
| Variable | N | Events | HR | 95% CI for HR | P |
|---|---|---|---|---|---|
| Age at diagnosis | 458 | 123 | 1.03 | (1.02, 1.05) | <0.001 |
| Tumor size | 1.03 | (0.98, 1.08) | 0.285 | ||
| Stage | |||||
| ȃStage I (ref)a | — | — | — | ||
| ȃStage II | 0.97 | (0.50, 1.88) | 0.935 | ||
| ȃStage III | 1.32 | (0.52, 3.34) | 0.557 | ||
| ȃStage IV | 4.07 | (1.75, 9.48) | 0.001 | ||
| Tumor location | |||||
| ȃAdrenal (ref)a | — | — | — | ||
| ȃAbdomen/Pelvis PGL | 1.94 | (0.35, 10.62) | 0.444 | ||
| ȃBilateral adrenal | 0.92 | (0.45, 1.91) | 0.828 | ||
| ȃMultiple location | 2.45 | (0.48, 12.55) | 0.282 | ||
| ȃThorax PG | 1.58 | (0.26, 9.55) | 0.618 |
Abbreviation: HR, hazard ratio.
(ref): reference category to which each other category is compared.
Multivariate cox analysis for overall survival, n = 458 and n events = 123
| Variable | N | Events | HR | 95% CI for HR | P |
|---|---|---|---|---|---|
| Age at diagnosis | 458 | 123 | 1.03 | (1.02, 1.05) | <0.001 |
| Tumor size | 1.03 | (0.98, 1.08) | 0.285 | ||
| Stage | |||||
| ȃStage I (ref)a | — | — | — | ||
| ȃStage II | 0.97 | (0.50, 1.88) | 0.935 | ||
| ȃStage III | 1.32 | (0.52, 3.34) | 0.557 | ||
| ȃStage IV | 4.07 | (1.75, 9.48) | 0.001 | ||
| Tumor location | |||||
| ȃAdrenal (ref)a | — | — | — | ||
| ȃAbdomen/Pelvis PGL | 1.94 | (0.35, 10.62) | 0.444 | ||
| ȃBilateral adrenal | 0.92 | (0.45, 1.91) | 0.828 | ||
| ȃMultiple location | 2.45 | (0.48, 12.55) | 0.282 | ||
| ȃThorax PG | 1.58 | (0.26, 9.55) | 0.618 |
| Variable | N | Events | HR | 95% CI for HR | P |
|---|---|---|---|---|---|
| Age at diagnosis | 458 | 123 | 1.03 | (1.02, 1.05) | <0.001 |
| Tumor size | 1.03 | (0.98, 1.08) | 0.285 | ||
| Stage | |||||
| ȃStage I (ref)a | — | — | — | ||
| ȃStage II | 0.97 | (0.50, 1.88) | 0.935 | ||
| ȃStage III | 1.32 | (0.52, 3.34) | 0.557 | ||
| ȃStage IV | 4.07 | (1.75, 9.48) | 0.001 | ||
| Tumor location | |||||
| ȃAdrenal (ref)a | — | — | — | ||
| ȃAbdomen/Pelvis PGL | 1.94 | (0.35, 10.62) | 0.444 | ||
| ȃBilateral adrenal | 0.92 | (0.45, 1.91) | 0.828 | ||
| ȃMultiple location | 2.45 | (0.48, 12.55) | 0.282 | ||
| ȃThorax PG | 1.58 | (0.26, 9.55) | 0.618 |
Abbreviation: HR, hazard ratio.
(ref): reference category to which each other category is compared.
Discussion
The results of our study suggest that the inaugural PPGL AJCC TNM staging system seems to correlate well with the OS rates observed in patients with PPGL. In fact, the size of the primary tumor, its location (adrenal vs extra-adrenal), the presence of regional lymph node metastases, the primary tumor's extension into adjacent tissues (fat, liver, kidney, pancreas, etc.), and the evidence of distant metastases independently correlated with OS. Accordingly, patients with distant metastases or stage IV disease had worse prognosis than patients with stage III disease (regional lymph node metastases and/or extension to adjacent tissues); furthermore, patients with stage III disease had worse prognosis when compared with patients with nonmetastatic PHEOs larger than 5 cm or nonmetastatic sympathetic PGLs (regardless of tumor size) with no extension into surrounding tissues (stage II disease). Stage I disease had the best OS rates and an almost uniform low risk for distant metachronous metastases.
Pheochromocytomas smaller than 5 cm in size had a very low risk for regional lymph node or distant metastases, and none were associated with extra-adrenal extension. These results suggest that a laparoscopic adrenalectomy for patients with stage I disease could lead to very high rates of cure with minimal risk for residual or recurrent disease. Although laparoscopic procedures offer limited visualization of the regional lymph nodes, the risk of missing regional lymphadenopathy in patients with stage I disease seems very low; in our study, no pheochromocytomas smaller than 5 cm were associated with either synchronous or metachronous regional lymph node metastases. Less than 3% of patients with pheochromocytomas smaller than 5 cm presented with distant metastases; these patients included 2 patients with stage I disease who developed metachronous metastases and 1 patient with synchronous metastases or stage IV disease. Of interest, all 3 patients were SDHB carriers. Every patient with a PPGL needs genetic counseling and testing (18); for SDHB carriers with stage I disease, highly sensitive functional imaging studies are indicated; somatostatin receptor–directed is the most sensitive imaging modality for SDHB-related disease; FDG-PET-CT is the second most sensitive imaging modality and either one may be recommended in addition to cross-sectional imaging testing to better characterize the extent of the tumor (18–22). These functional studies may identify multifocal disease, regional lymph node disease (N1) and/or distant metastases (M1), thus better determining TNM staging and, in turn, treatment planning. Furthermore, stage I SDHB carriers need long-term follow-up with periodic biochemical and radiographic studies after surgery (23). Conversely, patients with apparently sporadic stage I tumors may not need a long-term follow-up after surgery. Perhaps, SDHB (+) pheochromocytomas smaller than 5 cm should be considered at least T2 tumors; this may better separate stage I from the rest as the SDHB mutations are independent predictors of metastases. This information is now prospectively collected by the AJCC, and future updates derived from these results may help to stratify stages based on the presence of SDHB mutations.
Large pheochromocytomas (>5 cm) and sympathetic paragangliomas, regardless of tumor size, with no lymph node metastases and/or adjacent tissue infiltration (stage II disease) had higher rates of metachronous metastases and, consequently, lower OS rates when compared with stage I disease. In fact, primary tumor size and the extra-adrenal location of the primary tumor have been recognized by several studies as an important predictor of metastases and survivorship (2, 5). Our group previously showed that the risk of death associated with an extra-adrenal location (and independent of the size) doubled the risk of death when compared with primary tumors larger than 5 cm (5). Nevertheless, in the current study, the survival curves of patients with stage I and II overlapped for approximately 8 years and there was no difference in OS in between patients with stage II pheochromocytomas and stage II paragangliomas. Two recent retrospective studies of the Surveillance, Epidemiology, and End Results (SEER) database also suggest that the primary tumor site per se is not always a powerful predictor of metastases and survivorship (24, 25). Our findings suggest that most patients with nonmetastatic PPGLs have a good prognosis and that it takes time before metachronous metastases become clinically relevant. The overlap of the OS curves makes us also wonder if all paragangliomas smaller than 5 cm should be classified as stage II. Perhaps, nonmetastatic paragangliomas should be further categorized based on different tumor size cutoffs and/or genetic background (eg, non-SDHB paragangliomas < 5 cm could be categorized as T1 while SDHB paragangliomas should be categorized as T2); these patients may also benefit from functional imaging studies at diagnosis. Metachronous disease could also be disease not previously detected and with the current highly sensitive PET modalities, distant metastases could be diagnosed earlier, especially bone metastases, which are frequently missed by conventional radiographic studies (6).
Stage III disease exhibits shorter OS than stages I-II. Subsequently, an open laparotomy may be the preferred approach for patients with locoregional lymph node metastases to ensure completeness of resection (26). It is not clear if an open laparotomy would change the risk of death from distant metastases; however, it would likely decrease the risk of local recurrence. Of interest, the extension of the tumor into periadrenal adipose tissue and adjacent organs correlated with lower rates of overall survival, suggesting that it is a clinical predictor of metastases and survival. In addition, these patients exhibit similar OS rates to those of patients with stage III disease because of regional lymph node metastases. To the best of our knowledge, this is the first survivorship study that indicates that infiltration of periadrenal fat or adjacent visceral tissues impacts prognosis in comparison with other predictors of aggressiveness. Previously, Moog et al found that 22% of locally advanced PPGL defined as tumors with adipose, capsular, and/or vascular infiltration were associated with local recurrences and/or distant metastases (27). In our study, infiltration of surrounding tissues (adipose, visceral organs) was present only in patients with tumors larger than 5 cm. This also suggests that an open approach to resection should strongly be considered in patients with large tumors below the diaphragm and that functional imaging studies may complement conventional radiographic studies to determine surgical planning (26, 28). Moog et al also indicated that the presence of vascular and capsular invasion was associated with large tumors and that these parameters should be considered as part of the definition of a pathologic AJCC stage III (27). This is very important, as currently, the TNM staging is mainly clinical.
The median time of discovery of metachronous metastases was approximately 5 years irrespective of staging. This is surprising, as it would be expected that the time to develop metastases would be shorter in stage III compared with stages I and II; this finding emphasizes that PPGL are a heterogenous group of tumors and that most PPGL grow slowly when compared with other malignancies. Therefore, screening evaluation for metastatic disease in patients with stage I SDHB disease and stage II and III disease should continue for a long period of time; in fact, our experience shows that metastases may be discovered longer than 10 years after the initial diagnosis of the primary tumor. As suggested by others, optimal follow-up will require annual evaluations for at least 10 years and then periodically thereafter with clinical, biochemical, and low-irradiation radiographic assessments, depending on the patient's unique clinical situation (27, 29).
As expected, patients with distant synchronous metastases or stage IV disease had the shortest OS. In these patients, therapeutic decisions should be done on a case-by-case basis. For example, patients with stage IV disease may benefit from undergoing MIBG scintigraphy, given the recent FDA approval of high-specific-activity, carrier-free iodine-131-radiolabeled MIBG for the treatment of metastatic disease (30, 31). Clinical trials using novel therapeutics should also be considered in this population, especially for those whose tumors do not concentrate MIBG.
Patients with multiple endocrine neoplasia type 2 (MEN2) and a germline RET mutation had better OS rates compared with patients with apparently sporadic and other hereditary tumors. Depending on the genotype, up to 50% of MEN 2 patients develop pheochromocytomas (32). Paragangliomas are exceedingly rare in this population. MEN2 pheochromocytomas release both norepinephrine and epinephrine and are rarely metastatic (32). In our study, MEN2 patients had better outcomes than patients with NF1, paraganglioma syndrome type 4 (SDHB), or apparently sporadic tumors. Thus, MEN2 pheochromocytomas may need to be categorized independently of the AJCC staging. Like MEN2, NF1 predisposes to pheochromocytomas that are adrenergic and produce both norepinephrine and epinephrine. Nevertheless, it seems that NF1 pheochromocytomas may have a higher potential of metastases than MEN2 pheochromocytomas. In this study, the mortality of patients with NF1 pheochromocytomas was related to complications of distant PPGL metastases rather than other NF1-related tumors. Of interest, patients with SDHB mutations and metastatic disease did not have lower survival when compared with patients with other metastatic tumors. This finding is in agreement with other studies that have noticed that metastatic SDHB tumors do not exhibit lower survival rates when compared with apparently sporadic metastatic PPGL (33). Genetic testing is recommended to every patient with PPGL (18). However, 39.4% of our patients did not have genetic testing. Our study included patients diagnosed with PPGL during the last 13 years of the twentieth century when there was no knowledge on the existence of SDHx and other germline gene mutations; in addition, several patients diagnosed with PPGL in the first decade of the twenty-first century did not have genetic testing because of concerns of genetic discrimination and cost (34). The United States Title II of the Genetic Information Nondiscrimination Act took effect in 2009, making possible for all individuals to undergo genetic testing without fear for discrimination. Due to the large number of susceptibility genes implicated in the diagnosis of inherited PPGLs, next-generation sequencing technology was also suited for carrying out complete and affordable genetic screening. Since 2009, we have provided broad genetic testing to every patient with PPGL. We recognized that the lack of genetic testing could have influenced the results of our study as some patients with hereditary predisposition were not identified and perhaps, were diagnosed with more advanced TNM stages.
Patients with tumors associated with excessive hormonal secretion did not have shorter overall survival compared with patients who did not have hormonal secretion, a finding that was described in a previous paper (26). Although the excessive release of catecholamines leads to morbidity, it appears that the tumor burden is the more important determinant of survivorship, emphasizing the need and value of the TNM staging. Our study and others also suggest that survival correlates with age and that young patients have better clinical outcomes (33, 35). Nevertheless, deaths have been described in children, adolescents, and young adults and it is currently very difficult to determine how age may impact the TNM staging. Of note, 19.4% of our patients did not have an increased production of adrenaline and/or noradrenaline. Some of these patients may have had excessive secretion of dopamine; however, testing for methoxytyramine is not available in the United States. Whether the secretion of dopamine impacts the TNM staging is to be determined.
This study has several limitations. First, this a retrospective study hampered by the lack of systematic data collection; for instance, functional studies were not performed in all patients at diagnosis. Second, the cause of death was unknown in many cases and so we could not determine disease-specific survival, the most important long-term outcome. Finally, there is also a referral bias with a higher likelihood of more advanced tumors being referred to our institution. In fact, the median primary tumor size in our study is larger than the size usually seen in incidentally discovered pheochromocytomas (36) and 20% of the study patients had metastases. A referral bias is also suggested by the fact that up to 60% of patients who underwent genetic testing carried a germline mutation. Although genetic testing could have helped identifying patients at early stages affecting survivorship is a positive manner, the large primary tumor size and the presence of metastases may have had a negative impact on survivorship. Therefore, this study should be confirmed in other populations and by other academic centers. Similar to our study, a recent small, retrospective study from Sweden suggested that the current TNM staging correlated well with OS (37). Nevertheless, our study and others also suggest that the eighth edition of the AJCC TNM staging for PPGL could be optimized by considering genetic and histologic aspects among others (25, 27). The AJCC has started collecting staging information prospectively and this information will come from referral as well as non-referral centers. Its analysis in several years from now will provide us with the optimal guidance as to how to refine the TNM system if needed.
In conclusion, the inaugural TNM staging system for PPGL seems to correlate with overall survival. A large primary tumor size, an extra-adrenal location, the infiltration of surrounding tissues by the primary tumor, and the presence of regional lymph node metastases are associated with a higher risk for distant metastases and, subsequently, a decreased OS. Not surprisingly, patients with distant metastases (stage IV) exhibit the worse prognosis. Prognosis appears independent of gender, race, hormonal secretion status, and the presence of germline mutations in the NF1, SDHB, and VHL genes. Patients with a RET germline mutation exhibit a better prognosis than those with apparently sporadic and other hereditary tumors and may need to be categorized independently in the TNM staging. With the advent of better functional studies and preoperative staging, the AJCC PPGL staging can be further refined and TNM categories better defined to permit better prognostication for PPGL patients, especially those with stage I and II disease.
Data Availability
Restrictions apply to the availability of some or all data generated or analyzed during this study to preserve patient confidentiality or because they were used under license. The corresponding author will, on request, detail the restrictions and any conditions under which access to some data may be provided.
References
Abbreviations
- AJCC
American Joint Committee on Cancer
- CT
computed tomography
- FDG
18F-fluorodeoxyglucose
- HR
hazard ratio
- MIBG
123I-metaiodobenzylguanidine
- MRI
magnetic resonance imaging
- MST
median survival time
- OS
overall survival
- PET
positron emission tomography
- PGL
paraganglioma
- PHEO
pheochromocytoma
- PPGL
pheochromocytoma/paraganglioma
- SDHx
succinate dehydrogenase subunit x
- TNM
tumor, nodes, and metastasis (classification)
Author notes
Conflict of Interest C.J. has received research support from Lantheus Pharmaceuticals, Progenics Pharmaceuticals, Exelixis, Pfizer, and Merck Sharp and Dohme. Advisory Board for Lantheus, Pfizer, HRA Pharma. A.R.G. has received research support from Novartis and lecture fees from Pfizer, Sanofi, and Ipsen. J.M., J.V., M.Z., N.P., M.A.H., P.G., and S.G.W. have nothing to disclose.

{kind=link}
{kind=link}
{kind=link}