





Abstract
The first step in the evaluation of the short child is to decide whether growth parameters in the context of the history are abnormal or a variant of normal. If growth is considered abnormal, system and hormonal tests are likely to be required, followed by more directed testing, such as skeletal survey and/or genetic screening with karyotype or microarray. In a small percentage of short children in whom a diagnosis has not been reached, this will need to be followed by detailed genetic analysis; currently, exome sequencing using targeted panels relevant to the phenotype is the commonly used test. Clinical scenarios are presented that illustrate how such genetic testing can be used to establish a molecular diagnosis, and how that diagnosis contributes to the management of the short child. New genetic causes for short stature are being recognized on a frequent basis, while the clinical spectrum for known genes is being extended. We recommend that an international repository for short stature conditions is established for new findings to aid dissemination of knowledge, but also to help in the definition of the clinical spectrum both for new and established conditions.
Background
Short stature is one of the commonest presentations to the pediatric endocrinologist. The approach to establishing a clinical diagnosis in the short and/or slowly growing child starts with a detailed history, with particular focus on growth patterns and pubertal development in the direct and wider family. Physical examination should focus on looking for evidence of endocrine or system disease and signs of recognized growth-related syndromes. Auxological measurements in the proband, parent, and sometimes siblings on standardized equipment are essential. Pubertal assessment graded against standard criteria is required. The current growth data (standing height, sitting height, arm span, weight, body mass index) and any preceding measurements should be plotted on country-specific charts if available, or World Health Organization (WHO) charts for children younger than 2 years and Centre for Disease Control charts for those aged 2-18 years. A bone age, based on skeletal maturity in the nondominant hand and wrist, completes the physical assessment. These data will provide a diagnosis based on growth pattern (constitutional delay in growth, familial short stature, short following being born small for gestational age [SGA]) in 50% to 80% of children (1, 2) with the higher percentage from the 1994 study.
The next phase of evaluation requires a combination of the following tests, guided by history and examination: general tests for system disease; a screen for endocrine disorders or, if suspicion is high, detailed hormone axis stimulation testing; a skeletal survey for those with obvious or measurement identified skeletal disproportion; a karyotype or a microarray analysis looking for chromosomal abnormalities, with microarray being an important first-line test for short children with developmental delay; and directed genetic testing for those with clear signs of a monogenic disorder. This will lead to a specific diagnosis in 35% of children (2).
This will leave 15% of children with short stature of undefined etiology (2) in whom further genetic investigations could reveal a diagnosis, recognizing that the chance of identifying a molecular diagnosis increases as the severity of the short stature (<−3 SDS) increases (3). This includes those with “idiopathic short stature,” a commonly used term used to describe those children with a height below −2 SDS without findings of disease following a complete evaluation by a pediatric endocrinologist (4). Other groups of short children in whom a molecular diagnosis can be found include (1) those short following being born small for gestational age (S-SGA), (2) those in whom there is a suspicion of a genetic disorder, but signs of a specific disorder are not obvious, and (3) those with an endocrine disorder in which a molecular diagnosis can be reached (eg, familial growth hormone [GH] deficiency or congenital hypopituitarism). It is within these groups that further genetic (DNA) analysis can be carried out using approaches such as next-generation sequencing of exomes (ES) or genomes. Analysis of ES can focus on “targeted panels,” based on genes known to cause similar phenotypes (eg, pituitary hormone deficiency, growth failure in early childhood, skeletal dysplasia; these panels can be extensive and as an example for skeletal dysplasia the UK panel, available through the NHS National Genomic Test Directory, includes 386 genes), or use all the exome sequence data to find novel targets. The availability of DNA samples from parents and unaffected siblings is helpful in determining the clinical significance of DNA variants.
The value of such intensive investigation requires consideration. Achieving a specific diagnosis is a part of the overall management of the child. It provides certainty and often significant reassurance to families that there is a “real, recognized” cause, and for some children it ends a “diagnostic odyssey” in which the child has been subjected to multiple opinions and investigations. The diagnosis may provide prognostic information on likely height that will be achieved, and importantly may define risk for other features that may develop over a lifetime (eg, osteoarthritis, vascular malformations, obesity, cardiometabolic disease), for which screening, and intervention may be required. In certain conditions, there may be evidence to support the choice of growth promoting therapies (recombinant human GH [r-hGH], recombinant human insulin-like growth factor I [r-hIGF-I] or therapies that directly modulate bone growth) and knowledge about the long-term benefit of such therapies to height, metabolic health, and wellbeing. The finding of diagnoses, such as Bloom syndrome, or a chromosome breakage syndrome, can support the decision not to use such therapies because of background risk for malignancy development. There are therefore compelling reasons for undertaking such detailed testing in these groups of short children.
Targeted panels can include genes known to cause short stature and genes known to impact on hormonal and basic cellular growth pathways, including those genes within growth plates influencing cartilage/bone development and metabolism (5, 6). A whole exome approach increases the opportunity to find novel targets, but the level of analysis to achieve this and to confirm the significance of variants is considerable.
We present a range of cases that illustrate the clinical scenarios in which detailed genetic testing is indicated and how it contributes to the child's management. These include (1) well-defined endocrine disorders requiring a molecular diagnosis, (2) those with a high likelihood of a genetic disorder, (3) short stature with endocrine abnormalities in the context of a complex multisystem disorder, (4) SGA with persistent short stature and dysmorphism, and (5) idiopathic short stature. A pedigree for each scenario is shown in Fig. 1.
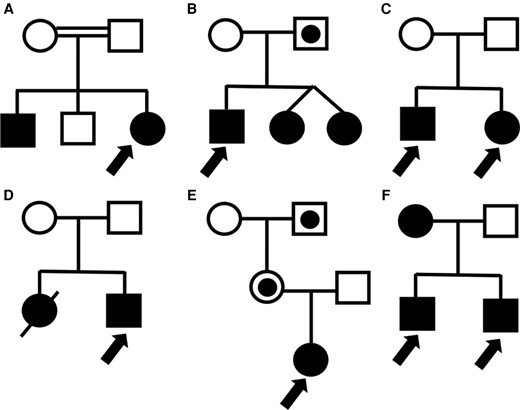
Pedigree for each of the families presented in this paper. (A) Consanguineous family with likely IGHD1A (Case 1). (B) Autosomal dominant disorder causing hypopituitarism with variable penetrance (Case 2). (C) Family with autosomal recessive mitochondrial disorder resulting in SGA children with postnatal growth impairment (Cases 3 and 4). (D) SGA with complex multisystem disorder (Case 5). (E) Silver–Russell syndrome caused by a CDKN1C mutation (Case 6). Only individuals who inherited the mutation on the maternal allele are affected. (F) Familial short stature due to SHOX duplications with autosomal dominant inheritance (Cases 7 and 8).
Clinical Scenarios
Well-Defined Endocrine Disorders Requiring a Molecular Diagnosis
Case 1
A 14-year-old was referred from her primary care physician with short stature. She had recently moved to the UK. She was born at term following an uneventful pregnancy with a birth weight of 2.25 kg. There was a history of admission to the neonatal unit for respiratory distress but no history of hypoglycemia or jaundice. Concerns about growth were raised from early life and r-hGH treatment was started at 6 months of age. She was treated intermittently, and full details were not available. Her brother, now an adult, had been treated with r-hGH for GH deficiency, but only achieved a height of 157 cm, −3.0 SDS. His adult phenotype was consistent with GH deficiency. An unaffected brother, now adult, had a final height of 172 cm, −0.8 SDS.
At presentation in the UK at 14.3 years her height was 96 cm (−10.6 SDS) and weight 13.1 kg (−10.8 SDS). She was prepubertal. There was mid-facial hypoplasia. Maternal height was 152 cm and paternal height 168 cm. Parents were first cousins. Initial baseline testing was remarkable for extremely low levels of growth factors: IGFBP-3 was 0.59 mg/L (normal range [NR] 2.58-6.40) and IGF-I was 11 μg/L (NR 123-552). There was no evidence of other pituitary hormone deficits, with thyroid stimulating hormone, free thyroxine and cortisol concentrations all normal and gonadotrophins low, consistent with her prepubertal status. Karyotype was 46XX.
A primed glucagon GH stimulation test identified severe GH deficiency with a peak GH concentration <0.1 μg/L while the pituitary magnetic resonance imaging (MRI) demonstrated anterior pituitary hypoplasia. Treatment with r-hGH at 30 μg/kg/day for 6 weeks did not result in any improvement in IGF-I levels (<10 μg/L) or IGFBP-3 level (<0.5 mg/L). The diagnosis was therefore isolated GH deficiency type 1A (IGHD1A), and treatment with r-hIGF-1 was started with a moderate improvement in height SDS to −8.8 at 17 years of age.
The differential diagnosis at presentation with severe short stature and low IGF-I levels was between GH resistance and severe isolated GH deficiency but the glucagon stimulation test was able to define the biochemical phenotype. This in turn provides a very strong pointer to target genetic investigation. Genetic forms of isolated GH deficiency are most commonly caused by mutations within GH1 or GHRHR genes. Isolated GH deficiency type 1A (IGHD1A) is an autosomal recessive disorder caused by complete loss of GH protein due to homozygous deletions or nonsense mutations in GH1 (7). The clinical presentation is that of severe GH deficiency with patients initially responding well to r-hGH therapy but this response can falter due to the development of anti-GH antibodies. GH antibodies had not been measured in this case: this would have been an important investigation, but the assay is not routinely available in many centers.
In this family as in other cases of monogenic disorders affecting the GH–IGF-I axis defining the clinical phenotype and biochemistry can lead to the likely genetic etiology. The phenotype of severe GH deficiency with a profound lack of response to treatment with r-hGH strongly pointed toward a mutation in the GH1 gene. Other disorders with a similar clinical presentation include resistance to GH which presents with high levels of GH and low levels of IGF-I and IGFBP-3 and can be caused by mutations in the GHR, STAT5B, or IGF-I genes. Both Laron syndrome (GHR mutations) (8) and STAT5B defects (9) present with a clinical picture similar to that of GH deficiency with postnatal growth failure with relatively preserved head size. GH binding protein measurement can be useful in identifying the etiology of GH resistance, but assays are not routinely available in most centers, with more accessible targeted genetic testing providing a specific diagnosis.
These genetic disorders can therefore still be approached through targeted sequencing of a single gene following definition of the biochemistry or where gene panels are easier to access, the clinician may choose to ask the laboratory to examine a single gene initially. This approach saves time and reduces the probability of identifying variants of unknown significance. In this case, we are awaiting the result of sequencing of the GH1 gene.
Case 2
A boy was brought to the endocrine clinic at 2 months of age following a complicated neonatal period. He was born by forceps delivery at term with a birth weight of 3.9 kg. He had a cleft lip and palate which had been identified antenatally on the 20-week fetal anomaly scan. At 6 hours of age, he was admitted to the neonatal intensive care unit with hypoglycemia and hyponatremia. Cortisol levels taken when the blood glucose was 27 mg/dL (2.1 mmol/L) were suboptimal at 1.1 μg/dL (30 nmol/L). Thyroid function testing demonstrated a low free thyroxine level of 0.6 μg/dL (7.2 pmol/L) (NRs 1.2-2.4 μg/dL (15-31pmol/L)) with a normal thyrotropin level of 3.4 mU/L. He had an undescended left testis and micropenis with undetectable gonadotrophins. Serum IGF-I concentration was undetectable, and the MRI scan of the pituitary identified the presence of an ectopic posterior pituitary with absent pituitary stalk and severe anterior pituitary hypoplasia.
The diagnosis of congenital hypopituitarism with multiple pituitary hormone deficiencies was clear but unlike the first case, it is more challenging to diagnose the genetic etiology through the clinical and biochemical findings. A complex cascade of spatiotemporal expressed transcription factors and signaling molecules control development of the pituitary gland (10). While congenital hypopituitarism with multiple pituitary hormone deficits can be subclassified into (1) children without midline deficits, (2) those on the septo-optic dysplasia spectrum, (3) those with midline deficits, and (4) other syndromic congenital hypopituitarism, the number of genes associated with each group is high and there is overlap in the genes associated with each of these groups. In this scenario, a wide panel-based approach should be applied. In 170 congenital hypopituitarism patients, Vishnopolska et al identified a genetic etiology in 15% using a panel of 67 genes (11); there is therefore a relatively low yield of positive molecular diagnoses in the subgroups of congenital hypopituitarism. However in our child, a heterozygous frameshift mutation in GLI2 was identified (c.922delT, p.[Ser308Glnfs]) giving a diagnosis of Culler–Jones syndrome (12). Younger twin siblings have now also been diagnosed with this mutation.
Those With a High Likelihood of a Genetic Disorder
Cases 3 and 4
Two siblings of Irish ancestry were referred for evaluation following a clinical diagnosis of 3-M syndrome. The older male sibling was born at term with a birthweight of 2.5 kg (−1.9 SDS) and a history of poor feeding in infancy. Short stature prompted a pediatric endocrinology referral at 2.5 years of age with height of −3.6 SDS and weight of −3.1 SDS. His sister was born at term, following intrauterine growth restriction (IUGR) with a birthweight of 2.27 kg (−2.3 SDS). At 3 years of age her height was −3.4 SDS and weight −3.1 SDS. Both siblings had a prominent forehead, a long thin philtrum, deep set eyes and low set ears. Their facial features associated with short stature had initially raised the suspicion of 3-M syndrome (13). However, gene sequencing to look for mutations in CUL7, OBSL1 (exons 1-9) and CCDC8 (14) did not reveal any mutation. Therefore, ES was undertaken, which revealed a homozygous pathogenic mutation in NDUFB3 [c.64T>C, p.Trp22Arg] (13), indicating that these siblings had a mitochondrial disorder. Further investigations related to this diagnosis demonstrated that the male sibling had a high signal in the globus pallidus on MRI scan and Wolff–Parkinson–White syndrome on electrocardiogram. His sister had a normal cranial MRI and electrocardiogram .
NDUFB3 encodes for a subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase (Complex I). Although other patients have presented with features of mitochondrial disease such as lactic acidosis (13), these siblings were well with no obvious metabolic dysfunction and normal lactate levels. Therefore, a diagnosis of mitochondrial disease would not have been suspected and would not have been diagnosed unless the ES had been undertaken. Making this diagnosis allowed the detection of Wolff–Parkinson–White syndrome in the male sibling. This is known to be overrepresented in patients with mitochondrial disease (15). In some patients, the first presentation of Wolff–Parkinson–White is with sudden death, but if screened for (following a genetic diagnosis) and detected early, therapeutic ablation is possible (16).
The sister had IUGR and was born SGA (13). This can be a marker for a possible genetic cause for short stature, and if fetal measurements are available, this may aid the clinician in deciding which children should have genetic testing, even in the absence of dysmorphic features. In addition, more than one sibling with the same clinical features increases the chances of finding a genetic condition. It is also important to note that in studies looking for genetic etiologies in short SGA children, being SGA may not be a recognized feature of the monogenic disorder identified (17). Thus, in some cases SGA may be an incidental finding.
These siblings demonstrate that there may be a degree of overlap in the clinical features of very different conditions, and although clinical signs may point toward one diagnosis, an absence of known mutations should lead the clinician to consider ES. As in this case, an important unexpected diagnosis was found, that has extended the phenotype of the mitochondrial disorder, previously recognized as a severe condition resulting in death in early life (18), and provided another condition to add to the differential diagnosis of those with clinical features of 3-M syndrome. This syndrome is a good example of how genetic testing has markedly increased the range of differential diagnoses (Table 1) and will continue to do so.
Differential diagnoses for 3M syndrome
| 3-M syndrome | Floating harbor syndrome | IMAGe syndrome | Isolated complex I deficiency | Mulibrey nanism | SHORT syndrome | SOFT syndrome | Silver–Russell syndrome | |
|---|---|---|---|---|---|---|---|---|
| Birthweight SDS | Mean −3.1 | Mean −2.5 | −2.0 to −4.0 | −1.3 to −2.7 | Mean −2.8 (range −4.0 to 0.5) | Mean −3.3 | −2.7 to −4.3 | Mean −2.7 |
| Adult height range (cm) | 115-150 | M: 106 to 164 F: 98 to 156 | M: 160 (n = 1) F: 142 (n = 1) | Not reported | 136-150 | Mean 154 | 112 to 127 cm | Mean 151 |
| Cognitive function | Normal | Variable Delayed speech | Normal | Normal | Mild motor and speech delay | Normal | Normal social and motor development | Variable |
| Main features | Dolichocephaly, triangular face, prominent heels, pectus deformity, short thorax, winged scapulae, hyperlordosis, hip dysplasia, slender long bones and vertebral bodies | Delayed bone age, expressive language delay, triangular face, prominent nose, deep set eyes | Metaphyseal dysplasia, congenital adrenal hypoplasia, male genital anomalies | Prominent forehead, long thin philtrum, may have raised lactate, hypertrophic cardiomyopathy, Wolf–Parkinson–White syndrome | Triangular face, short broad neck, misshapen sternum, hepatomegaly, increased tumor risk (Wilms, ovarian stromal) | S—short stature H—hyperextensibility of joints/inguinal hernia O—ocular depression R—Rieger anomaly T—teething delay | Triangular face, hypotrichosis, onychodysplasia, short long bones, hypogonadism, high pitched voice | Macrocephaly, triangular face, downturned mouth, limb asymmetry, feeding difficulties, fifth finger clinodactyly, brachydactyly, syndactyly, hypospadias, cryptorchidism, café au lait spots |
| Inheritance | AR | AD | Paternally imprinted | AR | AR | AD | AR | Most de novo |
| Genetics | CUL7, OBSL1 (exons 1-9), CCDC8 mutations | SRCAP mutations | CDKN1C mutations | c.64T>C, p.Trp22Arg NDUFB3 mutation | TRIM37 mutations High prevalence in Finnish population | PIK3R1 mutations | POC1A mutations | 11p15 loss of methylation, maternal UPD chromosome 7 |
| 3-M syndrome | Floating harbor syndrome | IMAGe syndrome | Isolated complex I deficiency | Mulibrey nanism | SHORT syndrome | SOFT syndrome | Silver–Russell syndrome | |
|---|---|---|---|---|---|---|---|---|
| Birthweight SDS | Mean −3.1 | Mean −2.5 | −2.0 to −4.0 | −1.3 to −2.7 | Mean −2.8 (range −4.0 to 0.5) | Mean −3.3 | −2.7 to −4.3 | Mean −2.7 |
| Adult height range (cm) | 115-150 | M: 106 to 164 F: 98 to 156 | M: 160 (n = 1) F: 142 (n = 1) | Not reported | 136-150 | Mean 154 | 112 to 127 cm | Mean 151 |
| Cognitive function | Normal | Variable Delayed speech | Normal | Normal | Mild motor and speech delay | Normal | Normal social and motor development | Variable |
| Main features | Dolichocephaly, triangular face, prominent heels, pectus deformity, short thorax, winged scapulae, hyperlordosis, hip dysplasia, slender long bones and vertebral bodies | Delayed bone age, expressive language delay, triangular face, prominent nose, deep set eyes | Metaphyseal dysplasia, congenital adrenal hypoplasia, male genital anomalies | Prominent forehead, long thin philtrum, may have raised lactate, hypertrophic cardiomyopathy, Wolf–Parkinson–White syndrome | Triangular face, short broad neck, misshapen sternum, hepatomegaly, increased tumor risk (Wilms, ovarian stromal) | S—short stature H—hyperextensibility of joints/inguinal hernia O—ocular depression R—Rieger anomaly T—teething delay | Triangular face, hypotrichosis, onychodysplasia, short long bones, hypogonadism, high pitched voice | Macrocephaly, triangular face, downturned mouth, limb asymmetry, feeding difficulties, fifth finger clinodactyly, brachydactyly, syndactyly, hypospadias, cryptorchidism, café au lait spots |
| Inheritance | AR | AD | Paternally imprinted | AR | AR | AD | AR | Most de novo |
| Genetics | CUL7, OBSL1 (exons 1-9), CCDC8 mutations | SRCAP mutations | CDKN1C mutations | c.64T>C, p.Trp22Arg NDUFB3 mutation | TRIM37 mutations High prevalence in Finnish population | PIK3R1 mutations | POC1A mutations | 11p15 loss of methylation, maternal UPD chromosome 7 |
3-M syndrome has a wide differential diagnosis. Syndromes with macrocephaly and normocephaly have been included. Differential diagnoses include isolated complex I deficiency, caused by a specific NDUFB3 mutation, as demonstrated by Cases 3 and 4.
Abbreviations: AR, autosomal recessive; AD, autosomal dominant.
Differential diagnoses for 3M syndrome
| 3-M syndrome | Floating harbor syndrome | IMAGe syndrome | Isolated complex I deficiency | Mulibrey nanism | SHORT syndrome | SOFT syndrome | Silver–Russell syndrome | |
|---|---|---|---|---|---|---|---|---|
| Birthweight SDS | Mean −3.1 | Mean −2.5 | −2.0 to −4.0 | −1.3 to −2.7 | Mean −2.8 (range −4.0 to 0.5) | Mean −3.3 | −2.7 to −4.3 | Mean −2.7 |
| Adult height range (cm) | 115-150 | M: 106 to 164 F: 98 to 156 | M: 160 (n = 1) F: 142 (n = 1) | Not reported | 136-150 | Mean 154 | 112 to 127 cm | Mean 151 |
| Cognitive function | Normal | Variable Delayed speech | Normal | Normal | Mild motor and speech delay | Normal | Normal social and motor development | Variable |
| Main features | Dolichocephaly, triangular face, prominent heels, pectus deformity, short thorax, winged scapulae, hyperlordosis, hip dysplasia, slender long bones and vertebral bodies | Delayed bone age, expressive language delay, triangular face, prominent nose, deep set eyes | Metaphyseal dysplasia, congenital adrenal hypoplasia, male genital anomalies | Prominent forehead, long thin philtrum, may have raised lactate, hypertrophic cardiomyopathy, Wolf–Parkinson–White syndrome | Triangular face, short broad neck, misshapen sternum, hepatomegaly, increased tumor risk (Wilms, ovarian stromal) | S—short stature H—hyperextensibility of joints/inguinal hernia O—ocular depression R—Rieger anomaly T—teething delay | Triangular face, hypotrichosis, onychodysplasia, short long bones, hypogonadism, high pitched voice | Macrocephaly, triangular face, downturned mouth, limb asymmetry, feeding difficulties, fifth finger clinodactyly, brachydactyly, syndactyly, hypospadias, cryptorchidism, café au lait spots |
| Inheritance | AR | AD | Paternally imprinted | AR | AR | AD | AR | Most de novo |
| Genetics | CUL7, OBSL1 (exons 1-9), CCDC8 mutations | SRCAP mutations | CDKN1C mutations | c.64T>C, p.Trp22Arg NDUFB3 mutation | TRIM37 mutations High prevalence in Finnish population | PIK3R1 mutations | POC1A mutations | 11p15 loss of methylation, maternal UPD chromosome 7 |
| 3-M syndrome | Floating harbor syndrome | IMAGe syndrome | Isolated complex I deficiency | Mulibrey nanism | SHORT syndrome | SOFT syndrome | Silver–Russell syndrome | |
|---|---|---|---|---|---|---|---|---|
| Birthweight SDS | Mean −3.1 | Mean −2.5 | −2.0 to −4.0 | −1.3 to −2.7 | Mean −2.8 (range −4.0 to 0.5) | Mean −3.3 | −2.7 to −4.3 | Mean −2.7 |
| Adult height range (cm) | 115-150 | M: 106 to 164 F: 98 to 156 | M: 160 (n = 1) F: 142 (n = 1) | Not reported | 136-150 | Mean 154 | 112 to 127 cm | Mean 151 |
| Cognitive function | Normal | Variable Delayed speech | Normal | Normal | Mild motor and speech delay | Normal | Normal social and motor development | Variable |
| Main features | Dolichocephaly, triangular face, prominent heels, pectus deformity, short thorax, winged scapulae, hyperlordosis, hip dysplasia, slender long bones and vertebral bodies | Delayed bone age, expressive language delay, triangular face, prominent nose, deep set eyes | Metaphyseal dysplasia, congenital adrenal hypoplasia, male genital anomalies | Prominent forehead, long thin philtrum, may have raised lactate, hypertrophic cardiomyopathy, Wolf–Parkinson–White syndrome | Triangular face, short broad neck, misshapen sternum, hepatomegaly, increased tumor risk (Wilms, ovarian stromal) | S—short stature H—hyperextensibility of joints/inguinal hernia O—ocular depression R—Rieger anomaly T—teething delay | Triangular face, hypotrichosis, onychodysplasia, short long bones, hypogonadism, high pitched voice | Macrocephaly, triangular face, downturned mouth, limb asymmetry, feeding difficulties, fifth finger clinodactyly, brachydactyly, syndactyly, hypospadias, cryptorchidism, café au lait spots |
| Inheritance | AR | AD | Paternally imprinted | AR | AR | AD | AR | Most de novo |
| Genetics | CUL7, OBSL1 (exons 1-9), CCDC8 mutations | SRCAP mutations | CDKN1C mutations | c.64T>C, p.Trp22Arg NDUFB3 mutation | TRIM37 mutations High prevalence in Finnish population | PIK3R1 mutations | POC1A mutations | 11p15 loss of methylation, maternal UPD chromosome 7 |
3-M syndrome has a wide differential diagnosis. Syndromes with macrocephaly and normocephaly have been included. Differential diagnoses include isolated complex I deficiency, caused by a specific NDUFB3 mutation, as demonstrated by Cases 3 and 4.
Abbreviations: AR, autosomal recessive; AD, autosomal dominant.
Short Stature With Endocrine Abnormalities in the Context of a Complex Multisystem Disorder
Case 5
A male infant had been under the care of pediatric endocrinology from early life with significant prenatal and postnatal growth failure associated with adrenal insufficiency, severe undervirilization, and an undefined immunodeficiency. He also had microcephaly and significant learning difficulties. By age 7 years, growth stasis (with height velocity 1 cm/year) and escalating weight velocity were evident, with central adiposity and a protuberant abdomen. There was associated fatty liver and elevated triglycerides. By this time, he had developed multiple vertebral fractures, warranting treatment with bisphosphonate infusions. Abdominal protuberance continued into the teenage years, alongside wasting of gluteal and limb muscles. He went on to develop a hemophagocytic lymphohistiocytosis–type illness triggered by Epstein–Barr virus, and at 15 years of age, he received an unrelated donor stem cell transplant which was successful. By age 19 years, he had ongoing adrenal insufficiency, severe short stature, hypertriglyceridemia, osteopenia, epilepsy, delayed puberty, and insulin resistance with acanthosis nigricans. His older sister had died in early childhood of overwhelming sepsis infection related to immunodeficiency. The family were keen for a precise diagnosis to be made.
During his early life, Sanger sequencing formed the mainstay of genetic analysis and specific genes potentially responsible for his phenotype were considered. In view of his adrenal deficiency, the nuclear receptor DAX1 and NR5A1 were sequenced, and no mutations were found. These were unlikely targets in view of the complexity of his clinical features. No mutations were identified in CDKN1C associated with IMAGe (intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genitourinary abnormalities in males) syndrome.
Reports of a syndrome associated with facial dysmorphism, immunodeficiency, livedo and short stature (FILS syndrome) (19), associated with a homozygous intronic variant (c.4444+3A>G) in POLE (polymerase epsilon), had previously been described in a single large consanguineous family. POLE encodes the catalytic subunit of human DNA POLE1, which is an enzyme involved in nuclear DNA replication and repair (20). Targeted genetic testing confirmed mutations in POLE in our case and DNA testing for both unrelated parents confirmed that they were carriers. An intronic variant (c.1686+32C>G) variant, which alters splicing, was identified. This same variant was found in a total of 15 individuals from 12 families (21). Our case also had a loss of function variant in trans (c.2091dupC, p.Phe699Valfs∗11), with these biallelic mutations leading to cellular deficiency in DNA polymerase epsilon (21). As expected, his deceased sister, on whom stored DNA was available, had the same mutations.
This case illustrates that perseverance with genetic investigations ended the diagnostic odyssey for this family, explaining not only the proband's phenotype but the reason for his sister's death. The associated increased malignancy risk in this condition resulted in referral to a specific clinic for surveillance, demonstrating an alteration in his management. This case also contributed to POLE being established as the second gene in which mutations cause IMAGe syndrome (21). This child presented more than 20 years ago. Today early use of ES, indicated because of the complexity of the phenotype, would have led to an immediate diagnosis.
SGA With Persistent Short Stature and Dysmorphism
Case 6
An 11-month-old girl was referred with growth impairment. Routine antenatal imaging was unremarkable at 12 and 20 weeks but following a low PAPP-A level in the mother, indicating a greater IUGR risk, a further ultrasound at 30 weeks identified asymmetrical IUGR and oligohydramnios. Further imaging at 32 and 34 weeks demonstrated no further growth, and she was delivered at 35 weeks. Birth weight was 1.43 kg (−2.76 SDS), length 39 cm (−3.08 SDS), and head circumference 31 cm (−0.1 SDS). She was fed via nasogastric tube for 1 week and remained in the neonatal unit for 1 month due to slow weight gain.
At 11 months of age, weight was 6.3 kg (−2.5 SDS) and length 60.1 cm (−4.5 SDS) with body mass index of 16.6 (+0.4 SDS). Parental adjusted height SDS was −5.3. The mid face was noted to be small and the forehead protuberant. There was right sided hemihypertrophy with the left leg 1.5 cm shorter than the right. Development was normal. Baseline biochemical testing and chromosomal microarray were normal.
The clinical presentation is that of the child born SGA with evidence of continued growth impairment during the first year of life. The presence of mid-facial hypoplasia with a protuberant forehead and hemihypertrophy raise a strong possibility of Silver–Russell syndrome (SRS). Current consensus guidelines recommend that the Netchine–Harbison Clinical Scoring System (NH-CSS) should be used to identify patients for genetic testing (22). She did not at that time meet the criterion to score on the NH-CSS for postnatal growth impairment as she was too young, nor did she meet the criterion for feeding difficulties. Despite this, the clinical impression was a diagnosis of SRS. Therefore, 11p15 methylation studies and testing for maternal uniparental disomy of chromosome 7 were undertaken and found to be normal. Rarer causes of SRS include gain of function mutations in CDKN1C on the maternal allele (23) and loss of function mutations in IGF2 on the paternal allele (24). ES was undertaken in the proband, mother and father which revealed a maternally inherited heterozygous missense variant in CDKN1C c.842G>C p.(Arg281Thr) (25). This missense variant has not previously been identified in a child with SRS or IMAGe syndrome. It has not been detected in control datasets and is conserved across species. In silico analysis predicted the mutation to be damaging using multiple programs. In keeping with previous reports (23, 26), mother did not have any clinical features of SRS and further family segregation studies identified she had inherited the missense variant from her unaffected father (the proband's grandfather).
Given the finding of this mutation and the association between CDKN1C mutations and IMAGe syndrome, a skeletal survey was undertaken at 4.4 years of age which demonstrated gracile long bones and a delayed bone age (3 years) but no evidence of any skeletal dysplasia, while synacthen testing confirmed normal cortisol secretion.
Testing for methylation defects associated with SRS will remain guided by the NH-CSS — testing is recommended for a score of 4/6 or more, with consideration of testing for a score of 3/6. Uniparental disomy may be detected by a single nucleotide polymorphism (SNP) array (but not a comparative genomic hybridization array) and next-generation sequencing (27). For the rare variants like those in CDKN1C or IGF2, testing with targeted ES is required.
Idiopathic Short Stature
Cases 7 and 8
The proband presented to endocrinology for evaluation of short stature at age 10 years with a height of 122.2 cm (−2.6 SDS) and weight of 21.1 kg (−3.0 SDS). He had been born by cesarean section at 37 weeks due to maternal pre-eclampsia with a birth weight of 2 kg (−2.3 SDS). Weight gain in infancy was poor with a diagnosis of cow's milk allergy and gastroesophageal reflux. Through childhood, he had a persistent poor appetite and poor weight gain. He was noted to have hypermobile joints, a trait also recognized in his mother. He had anxiety, obsessive–compulsive traits, communication and social difficulties and a poor sleep pattern. His younger brother had a height of 111.4 cm (−2.1 SDS) at 7.2 years of age, and had joint hypermobility, constipation, and a poor sleep pattern. Mother's height was 150 cm (−2.1 SDS) and father's 165 cm (−1.7 SDS), with a distant paternal family history of short stature.
Examination of the proband revealed no dysmorphic features, no skeletal disproportion, nor any specific skeletal abnormalities (eg, Madelung deformity). There was no evidence of system or endocrine disease, confirmed on testing. Microarray analysis revealed 2 rare copy number gains upstream of the Short stature Homeobox-containing gene (SHOX) at Xp22.33 of 39 and 50 kb. One of these gains encompassed conserved noncoding regulatory element 2 with previous studies having associated such gains with short stature (Fig. 2) (28-31). Despite this, the variant was classified by the laboratory as “of unknown clinical significance”. These gains however were present in the younger brother and the mother, who both had heights <−2SDS. It is highly likely that these gains have disrupted the function of the SHOX gene.
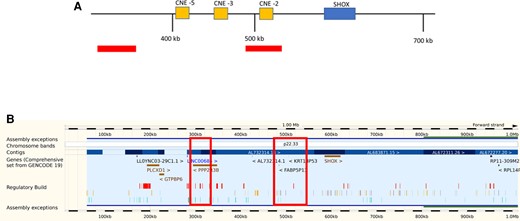
(A) Schematic representation of the genomic SHOX region with upstream flanking region and location of conserved noncoding elements (CNEs −2, −3, and −5). The distance from Xp/Yp telomere is indicated in kilobases, and areas where the patient's copy number gains were found are shown by the bars. One of the copy number gains encompasses CNE −2. (B) The same data presented using the Ensembl genome database for the SHOX region showing the genomic location for the copy number gains (61). These are indicated by the large boxes. (Other gene browsers eg, UCSC can be used).
This family illustrate that (1) there may be no clearly defined pointers to a specific growth diagnosis, with familial idiopathic short stature accepted as an explanation, (2) other health issues can be present, some of which may be related to the eventual growth diagnosis (in this case communication and social difficulties), and (3) first-line genetic testing with a microarray analysis can reveal a diagnosis but in the absence of a recognized phenotype may be classified as a variant of unknown significance.
The American College of Medical Genetics uses a quantitative framework to assign copy number and sequence variants into 1 of 5 categories (pathogenic, likely pathogenic, variant of unknown significance, likely benign and benign) (32, 33). To be classified as pathogenic the level of certainly is >99%, and for likely pathogenic >90%. The broadest level of certainty of any category is that for the variant of uncertain significance. With additional evidence, these variants can be later reclassified as pathogenic or benign. All sources of potential evidence including published literature, prediction of pathogenicity and cases reported in public databases such as ClinvarCNV should be explored. Importantly clinicians should carefully assess how the phenotype of a disorder associated with a variant of unknown significance matches the patient's presentation in the context of affected family members. Providing such information to the laboratory may aid in variant classification both for the index and future patients. In this case, the laboratory reassigned these variants to be of likely pathogenic significance. We recognize in some cases there will be a degree of uncertainty on the significance of a variant. Making a balanced judgement for the family may be helpful for therapeutic decisions—in this case the decision to treat both boys with r-hGH.
Heterozygous mutations in SHOX cause Leri–Weill dyschondrosteosis and Madelung deformity, while homozygous mutations cause Langer mesomelic dysplasia (34). The absence of SHOX, located on the short arm of the X (and Y) chromosomes, is also a major contributor to short stature in Turner syndrome. SHOX was one of the first genes to be recognized as carrying pathogenic mutations in short children without a defined etiology (35). It is estimated that 3% to 15% of children with idiopathic short stature carry a mutation, that disrupts the expression and/or action of SHOX, in the gene itself or its regulatory elements, which are located both up- and down-stream.
Other genes associated with skeletal abnormalities include Fibroblast growth factor receptor 3 (FGFR3) (hypochondroplasia and achondroplasia), Natriuretic peptide receptor 2 (NPR2) (acromesomelic dysplasia), and Aggrecan (ACAN) (spondyloepimetaphyseal dysplasia). Heterozygous mutations in these genes have been found in children with idiopathic short stature, with the yield dependent on phenotype. For instance, in short children with a degree of skeletal disproportion and/or a minor skeletal abnormality, insufficient to make a diagnosis, pathogenic variants in genes related to bone metabolism can be found in ∼20% (36).
Genetic Studies in Those With Short Stature of Undefined Etiology
To date the most accessible whole genome assessment has been that for copy number variation using either array comparative genomic hybridization or SNP arrays. The diagnostic yield in published studies utilizing these technologies in children with short stature of undefined etiology has been between 4% and 16% (see Table 2). Diagnostic yield is increased with increasing severity of short stature and where children have additional features such as dysmorphism or intellectual disability.
Diagnostic yield in copy number variant analysis studies of short stature patients
| References | n | Selection criteria | Diagnostic yield | Short stature genes/microdeletion/duplication region |
|---|---|---|---|---|
| Zahnleiter et al (54) | 200 | Idiopathic short stature—sporadic in 92 and familial in 108 | 10% | PAX3, POMC, GCKR, 22q11, 3q29, 1q21, 2q33, 1p36, and 1q21 |
| Canton et al (55) | 51 | Born SGA with short stature present at ≥ 4 years. One of intellectual disability, developmental delay, or dysmorphic features | 16% | 22q11, 10q26, GAB1, IGFBP2, CSNK2A1, CHD8, GALNS, MIR421 |
| Wit et al (56) | 49 | Born SGA with short stature present at ≥ 3 years | 16% | mUPD7, mUPD14, SHOX, 15q26, 22q11, 2q31, 7p21, 8q24 |
| Van Duyvenvoorde et al (57) | 162 | Short stature of unknown origin both normal size and small at birth included | 12% | SHOX, IGF1R, 15q26, 7q3, 1q44 |
| Hu et al (58) | 119 | Idiopathic short stature. SGA children excluded. | 4% | 22q11, 4q11, 4q12, Yp11 |
| Homma et al (59) | 229 | Short stature with dysmorphic features and developmental delay | 14% | 1p36, 2p15, 2q31.1, Wolf–Hirschhorn syndrome, 12q14, Temple syndrome, 15q11, Smith–Magenis syndrome, NF1 deletion syndrome, Miller–Dieker syndrome, 22q11 deletion, SHOX |
| Hars-Isono et al (17) | 86 | Short stature aged ≥ 2 years. Born SGA | 9% | 1q21, 22q11, Xp22.33, 19p13.12, 5q35, 10q36 |
| References | n | Selection criteria | Diagnostic yield | Short stature genes/microdeletion/duplication region |
|---|---|---|---|---|
| Zahnleiter et al (54) | 200 | Idiopathic short stature—sporadic in 92 and familial in 108 | 10% | PAX3, POMC, GCKR, 22q11, 3q29, 1q21, 2q33, 1p36, and 1q21 |
| Canton et al (55) | 51 | Born SGA with short stature present at ≥ 4 years. One of intellectual disability, developmental delay, or dysmorphic features | 16% | 22q11, 10q26, GAB1, IGFBP2, CSNK2A1, CHD8, GALNS, MIR421 |
| Wit et al (56) | 49 | Born SGA with short stature present at ≥ 3 years | 16% | mUPD7, mUPD14, SHOX, 15q26, 22q11, 2q31, 7p21, 8q24 |
| Van Duyvenvoorde et al (57) | 162 | Short stature of unknown origin both normal size and small at birth included | 12% | SHOX, IGF1R, 15q26, 7q3, 1q44 |
| Hu et al (58) | 119 | Idiopathic short stature. SGA children excluded. | 4% | 22q11, 4q11, 4q12, Yp11 |
| Homma et al (59) | 229 | Short stature with dysmorphic features and developmental delay | 14% | 1p36, 2p15, 2q31.1, Wolf–Hirschhorn syndrome, 12q14, Temple syndrome, 15q11, Smith–Magenis syndrome, NF1 deletion syndrome, Miller–Dieker syndrome, 22q11 deletion, SHOX |
| Hars-Isono et al (17) | 86 | Short stature aged ≥ 2 years. Born SGA | 9% | 1q21, 22q11, Xp22.33, 19p13.12, 5q35, 10q36 |
Diagnostic yield in copy number variant analysis studies of short stature patients
| References | n | Selection criteria | Diagnostic yield | Short stature genes/microdeletion/duplication region |
|---|---|---|---|---|
| Zahnleiter et al (54) | 200 | Idiopathic short stature—sporadic in 92 and familial in 108 | 10% | PAX3, POMC, GCKR, 22q11, 3q29, 1q21, 2q33, 1p36, and 1q21 |
| Canton et al (55) | 51 | Born SGA with short stature present at ≥ 4 years. One of intellectual disability, developmental delay, or dysmorphic features | 16% | 22q11, 10q26, GAB1, IGFBP2, CSNK2A1, CHD8, GALNS, MIR421 |
| Wit et al (56) | 49 | Born SGA with short stature present at ≥ 3 years | 16% | mUPD7, mUPD14, SHOX, 15q26, 22q11, 2q31, 7p21, 8q24 |
| Van Duyvenvoorde et al (57) | 162 | Short stature of unknown origin both normal size and small at birth included | 12% | SHOX, IGF1R, 15q26, 7q3, 1q44 |
| Hu et al (58) | 119 | Idiopathic short stature. SGA children excluded. | 4% | 22q11, 4q11, 4q12, Yp11 |
| Homma et al (59) | 229 | Short stature with dysmorphic features and developmental delay | 14% | 1p36, 2p15, 2q31.1, Wolf–Hirschhorn syndrome, 12q14, Temple syndrome, 15q11, Smith–Magenis syndrome, NF1 deletion syndrome, Miller–Dieker syndrome, 22q11 deletion, SHOX |
| Hars-Isono et al (17) | 86 | Short stature aged ≥ 2 years. Born SGA | 9% | 1q21, 22q11, Xp22.33, 19p13.12, 5q35, 10q36 |
| References | n | Selection criteria | Diagnostic yield | Short stature genes/microdeletion/duplication region |
|---|---|---|---|---|
| Zahnleiter et al (54) | 200 | Idiopathic short stature—sporadic in 92 and familial in 108 | 10% | PAX3, POMC, GCKR, 22q11, 3q29, 1q21, 2q33, 1p36, and 1q21 |
| Canton et al (55) | 51 | Born SGA with short stature present at ≥ 4 years. One of intellectual disability, developmental delay, or dysmorphic features | 16% | 22q11, 10q26, GAB1, IGFBP2, CSNK2A1, CHD8, GALNS, MIR421 |
| Wit et al (56) | 49 | Born SGA with short stature present at ≥ 3 years | 16% | mUPD7, mUPD14, SHOX, 15q26, 22q11, 2q31, 7p21, 8q24 |
| Van Duyvenvoorde et al (57) | 162 | Short stature of unknown origin both normal size and small at birth included | 12% | SHOX, IGF1R, 15q26, 7q3, 1q44 |
| Hu et al (58) | 119 | Idiopathic short stature. SGA children excluded. | 4% | 22q11, 4q11, 4q12, Yp11 |
| Homma et al (59) | 229 | Short stature with dysmorphic features and developmental delay | 14% | 1p36, 2p15, 2q31.1, Wolf–Hirschhorn syndrome, 12q14, Temple syndrome, 15q11, Smith–Magenis syndrome, NF1 deletion syndrome, Miller–Dieker syndrome, 22q11 deletion, SHOX |
| Hars-Isono et al (17) | 86 | Short stature aged ≥ 2 years. Born SGA | 9% | 1q21, 22q11, Xp22.33, 19p13.12, 5q35, 10q36 |
In addition to copy number variant studies, both targeted and exome sequencing approaches have been used in children with short stature of undefined etiology with variable diagnostic yield (Table 3). In a targeted gene approach using >1000 genes, of which a third were associated with growth disorders, in 192 short children without a diagnosis, only 2% had a pathogenic mutation that matched the phenotype (37). In another targeted study using 232 genes in a cohort of 263 children with idiopathic short stature (with 263 ethnically matched controls), 10% had pathogenic or likely pathogenic mutations in known growth genes (38). Using ES in a larger group of children with either isolated short stature (n = 257) or apparently syndromic short stature (n = 304), causal variants were found in 11% of the former group and 35% of the latter group (39). Genes involved in fundamental cellular processes were prominent. In an even bigger study of 814 children with short stature and at least one factor suggestive of a monogenic disorder, pathogenic/likely pathogenic variants, copy number variants or chromosomal abnormalities were found in 44%, with RASopathy genes the most commonly identified (40).
Genes identified in exome screens in those with short stature of undefined etiology
| References | Targeted or ES | N | Selection criteria | Diagnostic yield | Genes | |||
|---|---|---|---|---|---|---|---|---|
| Fundamental cellular processes | Extracellular matrix maintenance | Paracrine signaling | Hormonal signaling | |||||
| Li et al (40) | ES Targeted | 330 484 | Height < −2 SDS, plus 1 or more of: height < −3 SDS Severe IGHD, MPHD, GHI, SGA no catch-up growth, additional congenital anomalies/dysmorphic features, evidence of skeletal dysplasia, developmental delay, microcephaly, mother with recurrent miscarriage | 11% (those with height < −3SDS alone) to 70% (those with developmental delay) | PTPN11 NF1 KMT2A ANKRD11 SHOX NIPBL SLCA12A1 | PHEX ACAN COL2A1 COL1A1 RUNX2 | FGFR3 FGFR2 FGFR1 GALNS NPR2 | GH1 GLI2 IGF1R DUOX2 |
| Fan et al (39)a | ES | 561 | Height < −2 SDS Age <18 years Excluded those with pituitary tumor, chronic disease, iatrogenic SS | 11% (24%a) | PTPN11 BLM NF1 | ACAN COL2A1 | FGFR3 NPR2 | GH1 GLI2 STAT5B IGF1R POU1F1 |
| Perchard et al (38) | Targeted | 263 | Age ≥ 2 years SS (height ≤ −2.5 SDS) without a defined etiology ≥1 GH peak ≥7µg/L | 10% | PTPN11 HRAS TP63 OBSL1 FANCB GDF5 IKBKG | ACAN RUNX2 MMP13 | NPR2 GNAS | GH1 IGF1R LHX4 |
| Hauer et al (60) | Targeted ES | 200 | Short stature with no identified cause (common nongenetic causes excluded) | 17% | KRAS MAP2K1 PTPN11 NF1 KDM6A TRIM37 | ACAN COL2A1 | NPR2 FGFR3 PDE4D | GHSR IGF1R |
| Wang et al (37) | Targeted, 1077 genes | 192 | Height < −2 SDS, with no defined genetic etiology Could have comorbidities, dysmorphic features or other hormone deficiencies | 2% | PTPN11 TRPV4 | IGF1R | ||
| References | Targeted or ES | N | Selection criteria | Diagnostic yield | Genes | |||
|---|---|---|---|---|---|---|---|---|
| Fundamental cellular processes | Extracellular matrix maintenance | Paracrine signaling | Hormonal signaling | |||||
| Li et al (40) | ES Targeted | 330 484 | Height < −2 SDS, plus 1 or more of: height < −3 SDS Severe IGHD, MPHD, GHI, SGA no catch-up growth, additional congenital anomalies/dysmorphic features, evidence of skeletal dysplasia, developmental delay, microcephaly, mother with recurrent miscarriage | 11% (those with height < −3SDS alone) to 70% (those with developmental delay) | PTPN11 NF1 KMT2A ANKRD11 SHOX NIPBL SLCA12A1 | PHEX ACAN COL2A1 COL1A1 RUNX2 | FGFR3 FGFR2 FGFR1 GALNS NPR2 | GH1 GLI2 IGF1R DUOX2 |
| Fan et al (39)a | ES | 561 | Height < −2 SDS Age <18 years Excluded those with pituitary tumor, chronic disease, iatrogenic SS | 11% (24%a) | PTPN11 BLM NF1 | ACAN COL2A1 | FGFR3 NPR2 | GH1 GLI2 STAT5B IGF1R POU1F1 |
| Perchard et al (38) | Targeted | 263 | Age ≥ 2 years SS (height ≤ −2.5 SDS) without a defined etiology ≥1 GH peak ≥7µg/L | 10% | PTPN11 HRAS TP63 OBSL1 FANCB GDF5 IKBKG | ACAN RUNX2 MMP13 | NPR2 GNAS | GH1 IGF1R LHX4 |
| Hauer et al (60) | Targeted ES | 200 | Short stature with no identified cause (common nongenetic causes excluded) | 17% | KRAS MAP2K1 PTPN11 NF1 KDM6A TRIM37 | ACAN COL2A1 | NPR2 FGFR3 PDE4D | GHSR IGF1R |
| Wang et al (37) | Targeted, 1077 genes | 192 | Height < −2 SDS, with no defined genetic etiology Could have comorbidities, dysmorphic features or other hormone deficiencies | 2% | PTPN11 TRPV4 | IGF1R | ||
Example genes from these studies were included based on likelihood of pathogenicity and the frequency of variants in a gene.
Abbreviations: ES, exome sequencing; SS, short stature.
Fan et al (39): Genes identified in participants with isolated short stature have been included. Further variants were found in those with syndromic short stature.
Genes identified in exome screens in those with short stature of undefined etiology
| References | Targeted or ES | N | Selection criteria | Diagnostic yield | Genes | |||
|---|---|---|---|---|---|---|---|---|
| Fundamental cellular processes | Extracellular matrix maintenance | Paracrine signaling | Hormonal signaling | |||||
| Li et al (40) | ES Targeted | 330 484 | Height < −2 SDS, plus 1 or more of: height < −3 SDS Severe IGHD, MPHD, GHI, SGA no catch-up growth, additional congenital anomalies/dysmorphic features, evidence of skeletal dysplasia, developmental delay, microcephaly, mother with recurrent miscarriage | 11% (those with height < −3SDS alone) to 70% (those with developmental delay) | PTPN11 NF1 KMT2A ANKRD11 SHOX NIPBL SLCA12A1 | PHEX ACAN COL2A1 COL1A1 RUNX2 | FGFR3 FGFR2 FGFR1 GALNS NPR2 | GH1 GLI2 IGF1R DUOX2 |
| Fan et al (39)a | ES | 561 | Height < −2 SDS Age <18 years Excluded those with pituitary tumor, chronic disease, iatrogenic SS | 11% (24%a) | PTPN11 BLM NF1 | ACAN COL2A1 | FGFR3 NPR2 | GH1 GLI2 STAT5B IGF1R POU1F1 |
| Perchard et al (38) | Targeted | 263 | Age ≥ 2 years SS (height ≤ −2.5 SDS) without a defined etiology ≥1 GH peak ≥7µg/L | 10% | PTPN11 HRAS TP63 OBSL1 FANCB GDF5 IKBKG | ACAN RUNX2 MMP13 | NPR2 GNAS | GH1 IGF1R LHX4 |
| Hauer et al (60) | Targeted ES | 200 | Short stature with no identified cause (common nongenetic causes excluded) | 17% | KRAS MAP2K1 PTPN11 NF1 KDM6A TRIM37 | ACAN COL2A1 | NPR2 FGFR3 PDE4D | GHSR IGF1R |
| Wang et al (37) | Targeted, 1077 genes | 192 | Height < −2 SDS, with no defined genetic etiology Could have comorbidities, dysmorphic features or other hormone deficiencies | 2% | PTPN11 TRPV4 | IGF1R | ||
| References | Targeted or ES | N | Selection criteria | Diagnostic yield | Genes | |||
|---|---|---|---|---|---|---|---|---|
| Fundamental cellular processes | Extracellular matrix maintenance | Paracrine signaling | Hormonal signaling | |||||
| Li et al (40) | ES Targeted | 330 484 | Height < −2 SDS, plus 1 or more of: height < −3 SDS Severe IGHD, MPHD, GHI, SGA no catch-up growth, additional congenital anomalies/dysmorphic features, evidence of skeletal dysplasia, developmental delay, microcephaly, mother with recurrent miscarriage | 11% (those with height < −3SDS alone) to 70% (those with developmental delay) | PTPN11 NF1 KMT2A ANKRD11 SHOX NIPBL SLCA12A1 | PHEX ACAN COL2A1 COL1A1 RUNX2 | FGFR3 FGFR2 FGFR1 GALNS NPR2 | GH1 GLI2 IGF1R DUOX2 |
| Fan et al (39)a | ES | 561 | Height < −2 SDS Age <18 years Excluded those with pituitary tumor, chronic disease, iatrogenic SS | 11% (24%a) | PTPN11 BLM NF1 | ACAN COL2A1 | FGFR3 NPR2 | GH1 GLI2 STAT5B IGF1R POU1F1 |
| Perchard et al (38) | Targeted | 263 | Age ≥ 2 years SS (height ≤ −2.5 SDS) without a defined etiology ≥1 GH peak ≥7µg/L | 10% | PTPN11 HRAS TP63 OBSL1 FANCB GDF5 IKBKG | ACAN RUNX2 MMP13 | NPR2 GNAS | GH1 IGF1R LHX4 |
| Hauer et al (60) | Targeted ES | 200 | Short stature with no identified cause (common nongenetic causes excluded) | 17% | KRAS MAP2K1 PTPN11 NF1 KDM6A TRIM37 | ACAN COL2A1 | NPR2 FGFR3 PDE4D | GHSR IGF1R |
| Wang et al (37) | Targeted, 1077 genes | 192 | Height < −2 SDS, with no defined genetic etiology Could have comorbidities, dysmorphic features or other hormone deficiencies | 2% | PTPN11 TRPV4 | IGF1R | ||
Example genes from these studies were included based on likelihood of pathogenicity and the frequency of variants in a gene.
Abbreviations: ES, exome sequencing; SS, short stature.
Fan et al (39): Genes identified in participants with isolated short stature have been included. Further variants were found in those with syndromic short stature.
Overall, certain genes (Table 3) including PTPN11, ACAN, NPR2, FGFR3, IGF1R, and GH1, appear to be the most frequently identified in short children with undefined etiologies.
Ancestry and Defining a Pathogenic Variant
Short stature may result from a single rare monogenic variant. Therefore, an essential consideration when determining pathogenicity of a variant is its frequency within the relevant ancestry (eg, African, European, South Asian). Online resources such as the Genome Aggregation Database (gnomaD) (41) enable this, and the current dataset version (2.1.1) includes over 76 000 genomes from diverse ancestries.
Associations between SNPs and growth characteristics have been demonstrated. For instance in the European EPIGROW study (38), relationships between individual SNPs and shorter sitting height and smaller head circumference as well as lower IGF-I levels were found. Such relationships may also vary with ancestry. A reanalysis was undertaken only in those with Caucasian ancestry (42), with confirmation of the findings.
Despite this freely available online resource, and recognition of the value in studying diverse populations (43), the majority of height genome wide association studies include data from participants of European ancestry alone (44-46). This may reflect a wider issue, whereby certain ancestral groups are underrepresented in research studies (47, 48).
A number of short stature genes have been identified in particular populations. Amongst Europeans, Sardinians are the shortest (49) and genetic variations in KCNQ1 and GHR have been identified (50). Worldwide, one of the shortest populations are African Pygmies with a mean adult male height <150 cm, in whom variants in HESX1, APPL1, ASB14, DNAH12, and HYAL2 are associated with their short stature (51). This indicates that polygenic effects can combine to result in marked short stature.
The WHO have produced a report on medical genetic services in developing countries (52). Since resources to combat disease are scarce, the WHO recommend that developing countries should assess their disease burden and allocate budget accordingly, ensuring that there is capacity to care for genetic conditions. In the context of short stature, knowledge of population-specific short stature mutations could lead to potentially affordable, targeted candidate gene panels.
A 10-Year Vision
Over a relatively short period of ∼20 years, we have moved from checking karyotypes in short children to looking for abnormalities in multiple genes associated with a particular short stature category or exploring the whole exome for novel mutations. The latter approaches are producing a diagnosis in 10% to 40% of short children with diagnostic yield being higher in those with very severe short stature and/or suggestive clinical features (eg, skeletal disproportion). New genetic causes of short stature are also being recognized (53): it will be helpful if there could be an international repository for new findings to disseminate knowledge but also help to define the clinical spectrum of new conditions. Whole genome testing has not been applied systematically to growth disorders, but its wider use will inevitably increase diagnostic yield. In addition, developments in identifying epigenetic abnormalities are likely to be highly relevant to growth disorders, in particular in those with a history of IUGR.
We have presented scenarios where these genetic tests have provided vital insight into the cause of the condition and contributed to management decisions. It is likely that such testing will become much more widely used and applied earlier in the diagnostic process. It will be important that rigor in the attribution of a diagnosis to a particular gene mutation is applied with due attention to the clinical context.
Disclosures
R.P., P.M., and P.E.C. have nothing to disclose and have no conflicts of interest in relation to this manuscript.
Data Availability
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.
References
Abbreviations
- ACAN
Aggrecan
- FGFR3
Fibroblast growth factor receptor 3
- GH
growth hormone
- ES
exome sequencing
- IGF
insulin-like growth factor
- IMAGe
intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genitourinary abnormalities in males
- IUGR
intrauterine growth restriction
- MRI
magnetic resonance imaging
- NH-CSS
Netchine–Harbison Clinical Scoring System
- NPR2
Natriuretic peptide receptor 2
- NR
normal range
- r-h
recombinant human
- POLE
DNA polymerase epsilon catalytic subunit
- SDS
Standard deviation score
- SGA
small for gestational age
- SHOX
Short stature Homeobox-containing
- SNP
single nucleotide polymorphism
- SRS
Silver–Russell syndrome
- WHO
World Health Organization

{kind=link}
{kind=link}