





Abstract
Sporadic pseudohypoparathyroidism type 1B (sporPHP1B) is an imprinting disease without a defined genetic cause, characterized by broad methylation changes in differentially methylated regions (DMRs) of the GNAS gene.
This work aims to provide insights into the causative event leading to the GNAS methylation defects through comprehensive molecular genetic analyses of a pair of female monozygotic twins concordant for sporPHP1B who were conceived naturally, that is, without assisted reproductive techniques.
Using the leukocyte genome of the twins and family members, we performed targeted bisulfite sequencing, methylation-sensitive restriction enzyme (MSRE)-quantitative polymerase chain reaction (qPCR), whole-genome sequencing (WGS), high-density single-nucleotide polymorphism (SNP) array, and Sanger sequencing.
Methylation analyses by targeted bisulfite sequencing and MSRE-qPCR revealed almost complete losses of methylation at the GNAS AS, XL, and A/B DMRs and a gain of methylation at the NESP55 DMR in the twins, but not in other family members. Except for the GNAS locus, we did not find apparent methylation defects at other imprinted genome loci of the twins. WGS, SNP array, and Sanger sequencing did not detect the previously described genetic defects associated with familial PHP1B. Sanger sequencing also ruled out any novel genetic alterations in the entire NESP55/AS region. However, the analysis of 28 consecutive SNPs could not exclude the possibility of paternal heterodisomy in a span of 22 kb comprising exon NESP55 and AS exon 5.
Our comprehensive analysis of a pair of monozygotic twins with sporPHP1B ruled out all previously described genetic causes. Twin concordance indicates that the causative event was an imprinting error earlier than the timing of monozygotic twinning.
Pseudohypoparathyroidism (PHP) is the first described example of end-organ resistance to a hormone in mammals, which is primarily characterized by proximal renal tubular resistance to parathyroid hormone (PTH) (1). The molecular defects in the GNAS gene on chromosome 20, encoding the Gαs subunit of the heterotrimeric G protein, play a causal role in PHP (2, 3). The GNAS locus is a highly complex, imprinted locus. In addition to the messenger RNA encoding Gαs, the locus gives rise to several transcripts using alternative promoters and first exons: NESP55, XLαs, and A/B. There is also a noncoding antisense transcript called GNAS-AS1 (also known as NESPAS). The promoters of these 4 transcripts lie within differentially methylated regions (DMRs) and are active on the nonmethylated allele, leading to parent-of-origin specific expression. The AS, XLαs, and A/B DMRs are maternally methylated, whereas the NESP55 DMR is paternally methylated. Although the promoter of Gαs per se does not have a DMR, its expression from the paternal allele is silenced through an unknown mechanism in specific tissues, such as the proximal renal tubule, thyroid, and pituitary. Coding pathogenic variants of the maternal Gαs result in PHP1A, in which patients have Albright hereditary osteodystrophy (AHO), as well as multihormone resistance. Such Gαs coding variants are not typically found in PHP1B patients, who generally lack AHO features and display end-organ resistance only to PTH and, in some cases, thyrotropin. Instead, these patients exhibit methylation defects at the GNAS DMRs.
PHP1B manifests as autosomal dominant (AD-PHP1B) or sporadic (sporPHP1B) forms. AD-PHP1B patients have deletions in STX16 or NESP55 loci on the maternal allele and generally show a loss of methylation restricted to the A/B DMR (4-9). SporPHP1B patients have broad methylation changes at the GNAS DMRs with unknown causes, except in 8% to 10% of cases with paternal uniparental disomy involving the long arm of chromosome 20 (2, 3). A previous study suggested a postzygotic event caused somatic mosaicism in sporPHP1B patients with partial GNAS methylation defects (10). However, in sporPHP1B patients with complete GNAS methylation defects, the mechanistic basis of methylation defects, including the timing of the causative event, is still unclear.
In imprinting diseases with unknown etiology, like sporPHP1B, the findings from monozygotic twin cases provide critical information. In particular, to estimate the timing of the causative imprinting errors, monozygotic twin discordance for imprinting diseases is essential: Twins are clinically discordant if one sibling is (completely) affected and the other is unaffected (or partially affected); and twins are epigenetically discordant if they show differences in methylation levels. Previous studies have demonstrated that most monozygotic twin cases with imprinting diseases, such as Beckwith-Wiedemann syndrome and Silver-Russell syndrome, show both clinical and epigenetic discordance (11-16). In patients with Beckwith-Wiedemann syndrome, based on the discordance between monozygotic twins, it is speculated that the causative methylation defects can trigger monozygotic twinning (17). By contrast, only one set of monozygotic twins with sporPHP1B has been described to date (18).
In this study, we report a pair of monozygotic twins with sporPHP1B. The twins were concordant for clinical phenotype and the GNAS methylation defects. Our comprehensive molecular genetic analyses have ruled out the previously described genetic defects underlying PHP1B. The present findings provide insights into the causative event leading to the GNAS methylation defects in sporPHP1B.
Materials and Methods
Twin Patients and Their Family
The probands were a pair of 26-year-old Japanese female monozygotic twins. Twin 1 was referred to our department for further evaluation of incidentally found hypocalcemia at a local clinic. Twin 2 had noticed occasional hand numbness and facial spasm for 2 years, and she visited our department because of the diagnosis of hypocalcemia in twin 1. The parents were nonconsanguineous (Fig. 1A). The twins were conceived without assisted reproductive techniques and were born of an uncomplicated pregnancy and term delivery. We could not obtain information on amnionicity and chorionicity of the twins. Both twins had normal intellectual and physical development. On physical examination, each showed a positive Chvostek sign but no evidence of AHO. In both twins, the laboratory tests revealed hypocalcemia, hyperphosphatemia, hypocalciuria, and elevated serum intact PTH levels. The Ellsworth-Howard tests showed blunted urinary cAMP (3′,5′-cyclic adenosine 5′-monophosphate) excretion and phosphaturia in response to teriparatide. The twins were treated with alfacalcidol (with calcium lactate for twin 2) (Supplementary Fig. S1) (19). The calcium, phosphate, and intact PTH levels of other family members were within the normal range, except for the father’s slightly elevated PTH level, probably due to vitamin D deficiency. There was no other family history of endocrine or bone disease (Table 1). Genomic DNA of all family members was extracted from peripheral blood leukocytes using QIAamp DNA Mini Kit (Qiagen). Analyses were performed with informed consent from each family member using forms approved by the ethics committee of Tazuke Kofukai Medical Research Institute, Kitano Hospital.
Clinical features of all family members
| Twin 1 | Twin 2 | Father | Mother | Brother | |
|---|---|---|---|---|---|
| Sex | Female | Female | Male | Female | Male |
| Age at investigation, y | 26 | 26 | 58 | 53 | 22 |
| Symptoms | Asymptomatic | Hand numbness, facial spasm | Asymptomatic | Asymptomatic | Asymptomatic |
| Height, cm (percentile) | 152.3 (10-25) | 154 (10-25) | 168.4 (50-75) | 163.9 (95-99) | 179.5 (90-95) |
| Body weight, kg | 52 | 54 | 84.3 | 78.9 | 74 |
| Body mass index | 22.5 | 22.8 | 29.7 | 29.4 | 23 |
| Birth weight, g | 2072 | 2200 | 2700 | ||
| Physical and mental development | Normal | Normal | Normal | Normal | Normal |
| Physical examination | Chvostek sign + | Chvostek sign + | – | – | – |
| AHO feature | No | No | No | No | No |
| Past medical history | IDA, exotropia | IDA, AD | Colonic polyp | No | No |
| Calcium, 8.8-10.1 mg/dL | 7.6 | 5.5 | 9.2 | 9.4 | 9.5 |
| Phosphate, 2.7-4.6 mg/dL | 4.8 | 6.4 | 3.8 | 4 | 4 |
| PTH, 18.5-88.0 pg/mL | 509.2 | 384.9 | 96.6 | 69.2 | 42.6 |
| 25OH vitamin D, > 20 ng/mL | 4.5 | 12 | 12.7 | 8.9 | 19.5 |
| 1,25OH vitamin D, 20-60 pg/mL | 54 | 52.3 | |||
| Urinary Ca/creatinine | < 0.01 | < 0.01 | 0.083 | 0.092 | 0.14 |
| Bone ALP, 2.6-22.6 μg/L | 11.6 | 18.6 | |||
| TRACP-5b, 120-420 mIU/dL | 302 | 487 | |||
| TSH, 0.34-3.88 μIU/mL | 2.834 | 1.526 | |||
| FT4, 0.95-1.74 ng/dL | 0.942 | 0.82 | |||
| Ellsworth-Howard test | |||||
| Increase in urinary excretion of P (> 35 mg), cAMP (> 1 μmol) | P: 33.5 cAMP: 0.238 | P: 26.3 cAMP: 0.165 | |||
| T score of bone density, SD; lumbar/hip/radius | –1.3/–0.8/–0.5 | –0.8/–1.3/–0.9 |
| Twin 1 | Twin 2 | Father | Mother | Brother | |
|---|---|---|---|---|---|
| Sex | Female | Female | Male | Female | Male |
| Age at investigation, y | 26 | 26 | 58 | 53 | 22 |
| Symptoms | Asymptomatic | Hand numbness, facial spasm | Asymptomatic | Asymptomatic | Asymptomatic |
| Height, cm (percentile) | 152.3 (10-25) | 154 (10-25) | 168.4 (50-75) | 163.9 (95-99) | 179.5 (90-95) |
| Body weight, kg | 52 | 54 | 84.3 | 78.9 | 74 |
| Body mass index | 22.5 | 22.8 | 29.7 | 29.4 | 23 |
| Birth weight, g | 2072 | 2200 | 2700 | ||
| Physical and mental development | Normal | Normal | Normal | Normal | Normal |
| Physical examination | Chvostek sign + | Chvostek sign + | – | – | – |
| AHO feature | No | No | No | No | No |
| Past medical history | IDA, exotropia | IDA, AD | Colonic polyp | No | No |
| Calcium, 8.8-10.1 mg/dL | 7.6 | 5.5 | 9.2 | 9.4 | 9.5 |
| Phosphate, 2.7-4.6 mg/dL | 4.8 | 6.4 | 3.8 | 4 | 4 |
| PTH, 18.5-88.0 pg/mL | 509.2 | 384.9 | 96.6 | 69.2 | 42.6 |
| 25OH vitamin D, > 20 ng/mL | 4.5 | 12 | 12.7 | 8.9 | 19.5 |
| 1,25OH vitamin D, 20-60 pg/mL | 54 | 52.3 | |||
| Urinary Ca/creatinine | < 0.01 | < 0.01 | 0.083 | 0.092 | 0.14 |
| Bone ALP, 2.6-22.6 μg/L | 11.6 | 18.6 | |||
| TRACP-5b, 120-420 mIU/dL | 302 | 487 | |||
| TSH, 0.34-3.88 μIU/mL | 2.834 | 1.526 | |||
| FT4, 0.95-1.74 ng/dL | 0.942 | 0.82 | |||
| Ellsworth-Howard test | |||||
| Increase in urinary excretion of P (> 35 mg), cAMP (> 1 μmol) | P: 33.5 cAMP: 0.238 | P: 26.3 cAMP: 0.165 | |||
| T score of bone density, SD; lumbar/hip/radius | –1.3/–0.8/–0.5 | –0.8/–1.3/–0.9 |
Abbreviations: AD, atopic dermatitis; AHO, Albright hereditary osteodystrophy; ALP, alkaline phosphatase; Ca, calcium; cAMP, 3′,5′-cyclic adenosine 5′-monophosphate; FT4, free thyroxine; IDA, iron-deficiency anemia; PTH, parathyroid hormone; TSH, thyrotropin.
Clinical features of all family members
| Twin 1 | Twin 2 | Father | Mother | Brother | |
|---|---|---|---|---|---|
| Sex | Female | Female | Male | Female | Male |
| Age at investigation, y | 26 | 26 | 58 | 53 | 22 |
| Symptoms | Asymptomatic | Hand numbness, facial spasm | Asymptomatic | Asymptomatic | Asymptomatic |
| Height, cm (percentile) | 152.3 (10-25) | 154 (10-25) | 168.4 (50-75) | 163.9 (95-99) | 179.5 (90-95) |
| Body weight, kg | 52 | 54 | 84.3 | 78.9 | 74 |
| Body mass index | 22.5 | 22.8 | 29.7 | 29.4 | 23 |
| Birth weight, g | 2072 | 2200 | 2700 | ||
| Physical and mental development | Normal | Normal | Normal | Normal | Normal |
| Physical examination | Chvostek sign + | Chvostek sign + | – | – | – |
| AHO feature | No | No | No | No | No |
| Past medical history | IDA, exotropia | IDA, AD | Colonic polyp | No | No |
| Calcium, 8.8-10.1 mg/dL | 7.6 | 5.5 | 9.2 | 9.4 | 9.5 |
| Phosphate, 2.7-4.6 mg/dL | 4.8 | 6.4 | 3.8 | 4 | 4 |
| PTH, 18.5-88.0 pg/mL | 509.2 | 384.9 | 96.6 | 69.2 | 42.6 |
| 25OH vitamin D, > 20 ng/mL | 4.5 | 12 | 12.7 | 8.9 | 19.5 |
| 1,25OH vitamin D, 20-60 pg/mL | 54 | 52.3 | |||
| Urinary Ca/creatinine | < 0.01 | < 0.01 | 0.083 | 0.092 | 0.14 |
| Bone ALP, 2.6-22.6 μg/L | 11.6 | 18.6 | |||
| TRACP-5b, 120-420 mIU/dL | 302 | 487 | |||
| TSH, 0.34-3.88 μIU/mL | 2.834 | 1.526 | |||
| FT4, 0.95-1.74 ng/dL | 0.942 | 0.82 | |||
| Ellsworth-Howard test | |||||
| Increase in urinary excretion of P (> 35 mg), cAMP (> 1 μmol) | P: 33.5 cAMP: 0.238 | P: 26.3 cAMP: 0.165 | |||
| T score of bone density, SD; lumbar/hip/radius | –1.3/–0.8/–0.5 | –0.8/–1.3/–0.9 |
| Twin 1 | Twin 2 | Father | Mother | Brother | |
|---|---|---|---|---|---|
| Sex | Female | Female | Male | Female | Male |
| Age at investigation, y | 26 | 26 | 58 | 53 | 22 |
| Symptoms | Asymptomatic | Hand numbness, facial spasm | Asymptomatic | Asymptomatic | Asymptomatic |
| Height, cm (percentile) | 152.3 (10-25) | 154 (10-25) | 168.4 (50-75) | 163.9 (95-99) | 179.5 (90-95) |
| Body weight, kg | 52 | 54 | 84.3 | 78.9 | 74 |
| Body mass index | 22.5 | 22.8 | 29.7 | 29.4 | 23 |
| Birth weight, g | 2072 | 2200 | 2700 | ||
| Physical and mental development | Normal | Normal | Normal | Normal | Normal |
| Physical examination | Chvostek sign + | Chvostek sign + | – | – | – |
| AHO feature | No | No | No | No | No |
| Past medical history | IDA, exotropia | IDA, AD | Colonic polyp | No | No |
| Calcium, 8.8-10.1 mg/dL | 7.6 | 5.5 | 9.2 | 9.4 | 9.5 |
| Phosphate, 2.7-4.6 mg/dL | 4.8 | 6.4 | 3.8 | 4 | 4 |
| PTH, 18.5-88.0 pg/mL | 509.2 | 384.9 | 96.6 | 69.2 | 42.6 |
| 25OH vitamin D, > 20 ng/mL | 4.5 | 12 | 12.7 | 8.9 | 19.5 |
| 1,25OH vitamin D, 20-60 pg/mL | 54 | 52.3 | |||
| Urinary Ca/creatinine | < 0.01 | < 0.01 | 0.083 | 0.092 | 0.14 |
| Bone ALP, 2.6-22.6 μg/L | 11.6 | 18.6 | |||
| TRACP-5b, 120-420 mIU/dL | 302 | 487 | |||
| TSH, 0.34-3.88 μIU/mL | 2.834 | 1.526 | |||
| FT4, 0.95-1.74 ng/dL | 0.942 | 0.82 | |||
| Ellsworth-Howard test | |||||
| Increase in urinary excretion of P (> 35 mg), cAMP (> 1 μmol) | P: 33.5 cAMP: 0.238 | P: 26.3 cAMP: 0.165 | |||
| T score of bone density, SD; lumbar/hip/radius | –1.3/–0.8/–0.5 | –0.8/–1.3/–0.9 |
Abbreviations: AD, atopic dermatitis; AHO, Albright hereditary osteodystrophy; ALP, alkaline phosphatase; Ca, calcium; cAMP, 3′,5′-cyclic adenosine 5′-monophosphate; FT4, free thyroxine; IDA, iron-deficiency anemia; PTH, parathyroid hormone; TSH, thyrotropin.
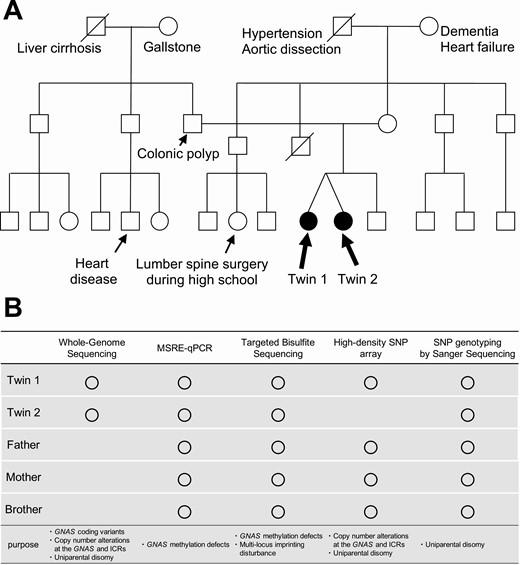
Pedigree and comprehensive genetic analysis for family members. A, Pedigree of the family. There were no consanguineous marriages in this family. Only the twins showed the biochemical features of pseudohypoparathyroidism type 1B (PHP1B). B, Methods used for genetic analysis of the family. ICR; imprinting control region; MSRE-qPCR, methylation-sensitive restriction enzyme–quantitative polymerase chain reaction; SNP, single-nucleotide polymorphism.
Targeted Bisulfite Sequencing
Targeted bisulfite sequencing was performed on all family members using SureSelect Human Methyl-Seq (Agilent). Target epigenetic regions on fragmented DNA were hybridized with biotinylated RNA probes and captured on beads. The selected fragments were bisulfite treated, polymerase chain reaction (PCR) amplified, and sequenced on the HiSeq platform. After sequencing, the BSMAP 2.90 package was used to align reads and calculate the methylation ratio of every single cytosine location.
Methylation-Sensitive Restriction Enzyme–Quantitative Polymerase Chain Reaction
Genomic DNA was digested with methylation-sensitive restriction enzyme (MSRE) HpaII or methylation-insensitive restriction enzyme MspI. Quantitative PCR (qPCR) reactions were performed using KOD SYBR qPCR mix (TOYOBO) and primers flanking each CpG island. In every qPCR experiment, we confirmed a single melt curve peak in each sample. The calibration curve was generated using Ct values of serially diluted undigested genome samples. The relative methylated ratios were calculated based on the Ct values of the HpaII-digested samples. Primer sequences are available on request.
Whole-Genome Sequencing Analysis
Whole-genome sequencing (WGS) was performed on the twins using the TruSeq DNA PCR-Free Kit (Illumina) and the Illumina Novaseq6000 (Illumina) in a 2 × 150-bp paired-end mode. The average coverage depths of twin 1 and twin 2 were 34.95 × and 29.59 ×, respectively. Structural variants were detected using Manta structural variant caller.
High-density Single-Nucleotide Polymorphism Array
The genome-wide single-nucleotide polymorphism (SNP) typing was performed on twin 1, the parents, and the brother, using the InfiniumOmni5-4v1-2_A1 (Illumina). Genomic DNA was amplified, fragmented, and hybridized on BeadChip. Array data were extracted, processed, and analyzed using Illumina GenomeStudio v.2 with copy number variation (cnvPartition v3.2.0).
Sanger Sequencing and Fragment Analysis
The genomic region from upstream of AS exon 5 to the XL DMR of the twins was PCR-amplified with 46 amplicons and Sanger-sequenced. PCR amplicons were designed to overlap each other. Especially, the region from upstream of AS exon 5 to exon NESP55 was completely Sanger-sequenced in all family members. In other regions, all amplicons containing the twins’ informative heterozygous SNPs were sequenced in other family members. PCR reactions were performed using KOD ONE (TOYOBO), a high-fidelity DNA polymerase. Sequencing reactions were performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific), and the samples were analyzed by a 3730xl DNA Analyzer (Thermo Fisher Scientific). For fragment analysis, PCR reactions were performed using 6FAM-labeled primers, and the products were analyzed by a 3730xl DNA Analyzer using GeneMapper Software v5 (Thermo Fisher Scientific).
Results
Both monozygotic twin patients showed concordant clinical phenotypes consistent with PHP1B: hypocalcemia, hyperphosphatemia, and resistance to PTH without AHO. To investigate the cause of PHP1B in the twins, we performed comprehensive molecular genetic analyses: targeted bisulfite sequencing, MSRE-qPCR, WGS, high-density SNP array, and Sanger sequencing (Fig. 1B). Methylation analysis with targeted bisulfite sequencing of all family members revealed almost complete losses of methylation at the AS, XL, and A/B DMRs and a gain of methylation at the NESP55 DMR in the twins but not in other family members (Fig. 2A and 2B). We further confirmed the GNAS methylation defects by MSRE-qPCR (Supplementary Fig. S2) (19). Consistent with the clinical presentation, the results indicated that the twins had concordant, broad, and almost complete methylation defects at the GNAS locus. In targeted bisulfite sequencing, except for the GNAS locus, we did not find apparent methylation defects at multiple other representative imprinted genome loci of the twins (Fig. 2C). Therefore, it was unlikely that the twins had multilocus imprinting disturbances (MLIDs), which has been reported in some patients with PHP1B (20).
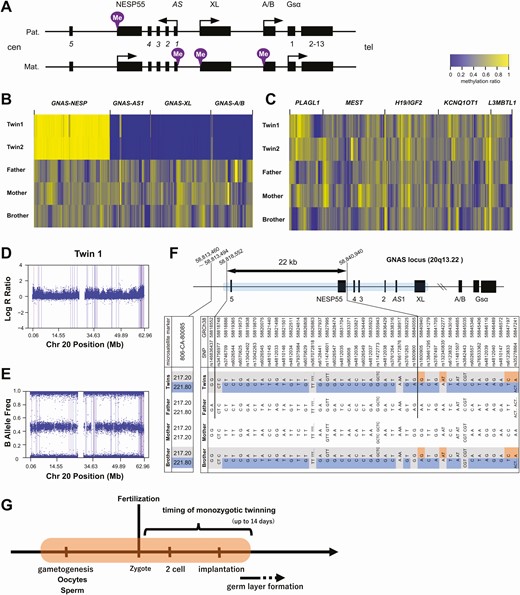
Genetic analyses of the twins and family members. A, Diagram showing the GNAS locus. The methylated allele is indicated by the purple mark at each differentially methylated region (DMR). Arrows indicate transcription start sites and the direction of transcription. B, Heat map visualization of methylation ratio at the GNAS DMRs and C, other representative imprinted loci. The methylation ratio at each CpG site was measured by target bisulfite sequencing. D and E, single nucleotide polymorphism (SNP) array results of twin 1. Plots of D, log R ratio, and E, B allele frequency in chromosome 20 are shown. F, Sanger-sequencing results of the region from AS exon 5 to the XL DMR. The blue box in the schema represents the Sanger-sequenced area. Informative SNPs and fragment length results are listed. The result of fragment analysis of a nearby short tandem repeat (806CA-80085) is also shown. The double arrow indicates the 22-kb region of possible paternal (Pat.) uniparental heterodisomy in the twins. The base positions at both ends are shown, referred to GRCh38. In the table, the blue and the orange boxes indicate paternally and maternally (Mat.) derived alleles, respectively. The gray boxes indicate alleles of undetermined parental origin. G, Schematic representation of the timing of the causative methylation error. The orange-shaded area represents the possible window for the error during development.
To identify the causative genetic defect, we performed WGS of the twins. There were no coding variants at the GNAS locus. We found no deletions, insertions, or inversions at the GNAS and STX16 loci, which are known to be associated with AD-PHP1B (4-9). A high-density SNP array also did not detect any copy number alteration on chromosome 20 of twin 1 (Fig. 2D) and family members (Supplementary Fig. S3) (19). In addition, Sanger sequencing of the region between exon NESP55 and AS exon 3 in the twins ruled out microdeletions that have been reported in AD-PHP1B patients with losses of methylation at all 3 maternal DMRs (7). We found at least 1 heterozygous SNP in every 2 consecutive amplicons of this region, which excluded the possibility of monoallelic PCR amplification. We further screened coding variants in candidate genes involved in imprinting control, including the known causative genes of MLIDs (Supplementary Table S1) (19, 21), but no coding variants were found. From WGS and SNP array, we found no regions of suspected uniparental isodisomy on chromosome 20 of the twins (Fig. 2E). Taken together, these results ruled out the previously described genetic defects in AD-PHP1B or sporPHP1B patients.
Based on the results of SNP array and Sanger sequencing of the NESP55/AS region of all family members, we could not exclude the possibility of paternal heterodisomy around exon NESP55. We further Sanger-sequenced an approximately 30-kb region and identified a 22-kb region, including 28 consecutive informative heterozygous SNPs, in which segmental paternal heterodisomy could not be excluded (Fig. 2F). The region extended from upstream of AS exon 5 to exon NESP55 and partially included the NESP55 DMR. The unaffected brother also had SNP genotypes similar to the twins in this region, except for 1 SNP located within the NESP55 DMR. No informative microsatellite markers were found within this region.
Discussion
We describe a pair of monozygotic twins with sporPHP1B, who were concordant for the clinical phenotype: hypocalcemia, hyperphosphatemia, elevated PTH levels, and no AHO features. No other family members showed these clinical features. In addition, the twins were concordant for methylation defects at the GNAS locus: almost complete losses of methylation at the AS, XL, and A/B DMRs and a gain of methylation at the NESP55 DMR. These methylation defects were not observed in other family members. From the aforementioned results, the twins were considered to be sporadic or de novo AD-PHP1B.
Comprehensive genetic analyses with WGS, SNP array, and Sanger sequencing ruled out previously described genetic causes of PHP1B, including deletion of STX16, deletion of the NESP55/AS region, or large paternal uniparental disomy (2, 3). In addition, targeted bisulfite sequencing excluded MLIDs, and WGS confirmed the absence of pathogenic variants in the known causative genes for MLIDs and other methylation regulatory genes. Collectively, our analyses excluded all known causes of PHP1B, suggesting that the cause is a previously uncharacterized event that leads to an epimutation limited to the GNAS locus.
Monozygotic twin cases affected by imprinting disorders have important implications for estimating the timing of the causative event of methylation defects (11). Traditionally, monozygotic twinning has been explained by the splitting theory, which assumes postzygotic splitting events before 13 days postfertilization (22, 23). Another recent hypothesis is that monozygotic twinning occurs earlier, at the first division of the zygote, although this hypothesis is controversial (23-25). In any case, monozygotic twinning must precede the onset of germ layer formation 14 days postfertilization (22). Notably, several lines of evidence support monozygotic twin discordance in imprinting disorders. In Beckwith-Wiedemann syndrome, the frequency of monozygotic twins is much higher than the general population, and most twin pairs are discordant for the clinical phenotype (11-14). These twins also show discordant methylation defects at an imprinted region of 11p15.5 in certain tissues, suggesting mosaicism (11-14). These observations support the hypothesis that the monozygotic twinning is triggered, at least partly, by the failure of methylation maintenance close to the timing of twinning and the resultant growth asymmetry (17). Similar predominant clinical and epigenetic discordances are also reported for monozygotic twins with Silver-Russell syndrome or transient neonatal diabetes (11, 15, 16). By contrast, our twins with sporPHP1B were concordant for the clinical phenotype and GNAS methylation defects. To date, only 1 more pair of monozygotic twins was included in a previous study of 22 cases with sporPHP1B, and they were also concordant (18). These findings suggest that the timing of the causative event of sporPHP1B is earlier than those of other imprinting diseases mentioned earlier and must be before monozygotic twinning (Fig. 2G). This argument is apparently in line with a previous study speculating that an epimutation before the formation of the germ layer causes sporPHP1B (10). The present findings can further narrow the window for the causative event because monozygotic twinning is earlier than triple germ layer formation (22).
Paternal isodisomies or heterodisomies have been identified in a few patients with sporPHP1B (26). Intriguingly, based on the result of SNP genotyping, we could not exclude the possibility of paternal heterodisomy in the upstream region of the GNAS locus, maximum 22 kb in length, partially including the NESP55 DMR. It is unclear whether such a small region of paternal heterodisomy in the presently studied twins could play a causal role in the observed GNAS methylation defects. Note that very small uniparental heterodisomies have been identified in other diseases and suggested to play a causative role (27-33). Based on human (9) and mouse (34, 35) studies, NESP55 transcription and/or the genomic region comprising exon NESP55 is crucial for the control of GNAS imprinting. While one study suggested a role for NESP55 transcription during oogenesis (34), 2 other studies indicated that this region may also be required for methylation maintenance at downstream GNAS DMRs in other cells after fertilization (6, 36). It is thus tempting to speculate that the paternal heterodisomy led to the suppression of NESP55 transcription on both alleles very early in the zygote, leading to broad methylation defects at the GNAS locus. However, except for one in the NESP55 DMR, the brother shared SNP genotypes with the twins in the same region. Therefore, it is difficult to draw a definitive conclusion on the causal role of this region of possible paternal heterodisomy.
In this study, we performed a comprehensive analysis on a pair of monozygotic twins with sporPHP1B, ruling out all previously described genetic causes. Twin concordance indicates that the causative event was an imprinting error earlier than the timing of the monozygotic twinning.
Abbreviations
- AD-PHP1B
autosomal dominant pseudohypoparathyroidism type 1B
- AHO
Albright hereditary osteodystrophy
- DMR
differentially methylated region
- IDA
iron-deficiency anemia
- MLID
multilocus imprinting disturbance
- MSRE
methylation-sensitive restriction enzyme
- PHP
pseudohypoparathyroidism
- PTH
parathyroid hormone
- qPCR
quantitative polymerase chain reaction
- SNP
single-nucleotide polymorphism
- sporPHP1B
sporadic pseudohypoparathyroidism type 1B
- WGS
whole-genome sequencing
Acknowledgments
The study was supported in part by a grant from NIH/NIDDK (R01 DK073911 to M.B.). We thank the patients and other family members for their participation. We are grateful to Dr Harald Jüppner for helpful suggestions. We also thank the managing staff of the medical research institute, Kitano Hospital, for their support of our research.
Additional Information
Disclosures: The authors have nothing to disclose.
Data Availability
Restrictions apply to the availability of some or all data generated or analyzed during this study to preserve patient confidentiality or because they were used under license. The corresponding author will on request detail the restrictions and any conditions under which access to some data may be provided.
References
Author notes
Y.K. and Y.I. contributed equally to this work.

{kind=link}
{kind=link}