





Abstract
Genetic syndromes that affect the nervous system may also disrupt testicular function, and the mechanisms for these effects may be interrelated. Most often neurological signs and symptoms predominate and hypogonadism remains undetected and untreated, while in other cases, a thorough evaluation of a hypogonadal male reveals previously unrecognized ataxia, movement disorder, muscle weakness, tremor, or seizures, leading to a syndromic diagnosis. Androgen deficiency in patients with neurological diseases may aggravate muscle weakness and fatigue and predispose patients to osteoporosis and obesity. The purpose of this mini review is to provide a current understanding of the clinical, biochemical, histologic, and genetic features of syndromes in which male hypogonadism and neurological dysfunction may coexist and may be encountered by the clinical endocrinologist.
Systemic disorders are often associated with abnormal hypothalamic-pituitary-testicular function. While poor nutrition, weight loss, inflammatory cytokines, and medications contribute to testosterone deficiency and hypospermatogenesis, systemic disorders and the male reproductive system may be linked mechanistically. This is often the case with genetic and neurodevelopmental disorders, as advances in molecular genetics have led to the discovery of many genes that link nervous and male reproductive system functions. Hypogonadism may be present in patients with ataxia, mental retardation, seizures, blindness, hearing loss, and anosmia, among other symptoms. In some neurological disorders, hypogonadism is evident at birth with micropenis and/or cryptorchidism; others present as a failure to enter or complete puberty, and some are first recognized in adulthood because of clinical hypogonadism and infertility. The variation in phenotype among men with the same genetic neurodevelopmental disorder can be striking, and some disorders that result from ubiquitously expressed genes may cause both primary and secondary hypogonadism. Subjects without an established neurological diagnosis may consult with an endocrinologist because of hypogonadism, and a careful evaluation may progress to an understanding of their underlying disorder. Moreover, hypogonadism may aggravate neurologically related health issues including muscle weakness, obesity, osteoporosis, and fatigue. This mini review was developed to provide a current understanding of the clinical, biochemical, histologic, and genetic features of disorders in which male hypogonadism and neurological dysfunction may coexist. Due to space limitations, certain disorders are listed only in the tables. Acquired neurological conditions that may result in hypogonadism [eg, traumatic brain injury, brain tumors, X-irradiation, central nervous system (CNS) infection, vascular events] are also not discussed.
Search Strategy
I searched the PubMed database for articles written in the English language using the search terms “male hypogonadism,” “infertility,” “reproduction” “testis,” AND “neurological disorders,” “weakness,” and “ataxia.” Bibliographies of these citations were also reviewed. The criteria used to define male hypogonadism included clinical features consistent with androgen deficiency throughout the lifespan, evidence for hypospermatogenesis, and/or abnormal levels of testosterone, luteinizing hormone (LH), and follicle-stimulating hormone (FSH). Very few studies mentioned time of day of blood sampling or repeat sampling to confirm the results. Testosterone was generally measured by immunoassay. Case reports of 1 or 2 cases were generally not included.
Disorders Associated With Primary Testicular Failure
The biochemical hallmark of primary male hypogonadism is an elevated serum level of FSH (Table 1). Moreover, the testes are generally small, the sperm count is low, and inhibin-B levels are decreased. LH levels are usually elevated and testosterone may be reduced, but this is a less constant finding because Leydig cells may be affected less by testicular pathologies than is spermatogenesis and morbidities that suppress gonadotropin-releasing hormone (GnRH) secretion tend to reduce circulating LH more than FSH. In these disorders, abnormal gene expression, RNA splicing, protein trafficking, and intracellular metabolism disrupt spermatogenesis and adversely affect neuronal development and function. Several neurodevelopmental disorders in which primary testicular failure may occur are listed in Table 1.
Neurological disorders associated with primary testicular insufficiency
| Mutation | Functional effect | Clinical features | Reference(s) | |
|---|---|---|---|---|
| Adrenoleukodystrophy-adrenomyeloneuropathy | ACBD1 | Peroxisome long chain fatty acid oxidation | Ataxia, bladder/bowel dysfunction Adrenal insufficiency | (4) |
| Alstrom syndrome | ALMS1 | Cilia function and cell cycle regulation | Obesity, vision, and hearing loss; type 2 diabetes mellitus; cardiomyopathy. | (30, 178) |
| Bardet Biedl syndrome | BBS1-BBS21, MKKS, ARL6, TTC8, CEP290, NPHP1, SCAPER, LZTFL1, BB1P1. | Cilia development and function | Retinal dystrophy, polydactyly, cognitive Dysfunction, ataxia obesity, renal insufficiency, hearing loss, anosmia, micropenis, cryptorchidism. | (27) |
| Bulbospinal Muscle atrophy (Kennedy disease) | Increased CAG repeats in the androgen receptor | Toxic RNA and misfolded protein, impaired androgen receptor transactivation | Weakness, gynecomastia | (73, 74) |
| Down syndrome | Trisomy 21 | Overexpression of chromosome 21 genes | Cognitive impairment, dysmorphic features, hypotonia | (33) |
| Hereditary moyamoya syndrome | RNF213 | Ubiquitin ligase activity | Supraclinoid internal carotid artery occlusion, facial dysmorphism, cardiomyopathy, short stature | (179) |
| Klinefelter syndrome and polyploid variants | 47, XXY 48, XXYY | Testosterone deficiency, estrogen excess, overexpression of X chromosome genes | Essential tremor Seizure disorder | (44, 45) |
| Marinesco-Sjogren syndrome | SIL1 (suppressor of Ire1/Lhs1 double mutant) | Deficient cofactor for BiP (estrogen receptor chaperone-binding immunoglobulin protein) causing disrupted protein folding | Cerebellar ataxia, cataracts, mental retardation, skeletal abnormalities, myopathy | (180) |
| Methylmalonic aciduria-homocystinuria | MMACHC | Methionine synthesis defect | Ataxia, intellectual decline, seizures, leukoencephalopathy | (3) |
| Myotonic muscular dystrophy 1 | Myotonic dystrophy protein kinase | Myotonic dystrophy protein kinase toxic messenger RNA | Weakness, myotonia, cardiac arrhythmias and conduction defects, frontal balding | (176) |
| Noonan syndrome | PTPN11 | RAS/mitogen-activated protein kinase pathway | Short stature, dysmorphic facies, heart defects, cryptorchidism, infertility | (181) |
| Mutation | Functional effect | Clinical features | Reference(s) | |
|---|---|---|---|---|
| Adrenoleukodystrophy-adrenomyeloneuropathy | ACBD1 | Peroxisome long chain fatty acid oxidation | Ataxia, bladder/bowel dysfunction Adrenal insufficiency | (4) |
| Alstrom syndrome | ALMS1 | Cilia function and cell cycle regulation | Obesity, vision, and hearing loss; type 2 diabetes mellitus; cardiomyopathy. | (30, 178) |
| Bardet Biedl syndrome | BBS1-BBS21, MKKS, ARL6, TTC8, CEP290, NPHP1, SCAPER, LZTFL1, BB1P1. | Cilia development and function | Retinal dystrophy, polydactyly, cognitive Dysfunction, ataxia obesity, renal insufficiency, hearing loss, anosmia, micropenis, cryptorchidism. | (27) |
| Bulbospinal Muscle atrophy (Kennedy disease) | Increased CAG repeats in the androgen receptor | Toxic RNA and misfolded protein, impaired androgen receptor transactivation | Weakness, gynecomastia | (73, 74) |
| Down syndrome | Trisomy 21 | Overexpression of chromosome 21 genes | Cognitive impairment, dysmorphic features, hypotonia | (33) |
| Hereditary moyamoya syndrome | RNF213 | Ubiquitin ligase activity | Supraclinoid internal carotid artery occlusion, facial dysmorphism, cardiomyopathy, short stature | (179) |
| Klinefelter syndrome and polyploid variants | 47, XXY 48, XXYY | Testosterone deficiency, estrogen excess, overexpression of X chromosome genes | Essential tremor Seizure disorder | (44, 45) |
| Marinesco-Sjogren syndrome | SIL1 (suppressor of Ire1/Lhs1 double mutant) | Deficient cofactor for BiP (estrogen receptor chaperone-binding immunoglobulin protein) causing disrupted protein folding | Cerebellar ataxia, cataracts, mental retardation, skeletal abnormalities, myopathy | (180) |
| Methylmalonic aciduria-homocystinuria | MMACHC | Methionine synthesis defect | Ataxia, intellectual decline, seizures, leukoencephalopathy | (3) |
| Myotonic muscular dystrophy 1 | Myotonic dystrophy protein kinase | Myotonic dystrophy protein kinase toxic messenger RNA | Weakness, myotonia, cardiac arrhythmias and conduction defects, frontal balding | (176) |
| Noonan syndrome | PTPN11 | RAS/mitogen-activated protein kinase pathway | Short stature, dysmorphic facies, heart defects, cryptorchidism, infertility | (181) |
Neurological disorders associated with primary testicular insufficiency
| Mutation | Functional effect | Clinical features | Reference(s) | |
|---|---|---|---|---|
| Adrenoleukodystrophy-adrenomyeloneuropathy | ACBD1 | Peroxisome long chain fatty acid oxidation | Ataxia, bladder/bowel dysfunction Adrenal insufficiency | (4) |
| Alstrom syndrome | ALMS1 | Cilia function and cell cycle regulation | Obesity, vision, and hearing loss; type 2 diabetes mellitus; cardiomyopathy. | (30, 178) |
| Bardet Biedl syndrome | BBS1-BBS21, MKKS, ARL6, TTC8, CEP290, NPHP1, SCAPER, LZTFL1, BB1P1. | Cilia development and function | Retinal dystrophy, polydactyly, cognitive Dysfunction, ataxia obesity, renal insufficiency, hearing loss, anosmia, micropenis, cryptorchidism. | (27) |
| Bulbospinal Muscle atrophy (Kennedy disease) | Increased CAG repeats in the androgen receptor | Toxic RNA and misfolded protein, impaired androgen receptor transactivation | Weakness, gynecomastia | (73, 74) |
| Down syndrome | Trisomy 21 | Overexpression of chromosome 21 genes | Cognitive impairment, dysmorphic features, hypotonia | (33) |
| Hereditary moyamoya syndrome | RNF213 | Ubiquitin ligase activity | Supraclinoid internal carotid artery occlusion, facial dysmorphism, cardiomyopathy, short stature | (179) |
| Klinefelter syndrome and polyploid variants | 47, XXY 48, XXYY | Testosterone deficiency, estrogen excess, overexpression of X chromosome genes | Essential tremor Seizure disorder | (44, 45) |
| Marinesco-Sjogren syndrome | SIL1 (suppressor of Ire1/Lhs1 double mutant) | Deficient cofactor for BiP (estrogen receptor chaperone-binding immunoglobulin protein) causing disrupted protein folding | Cerebellar ataxia, cataracts, mental retardation, skeletal abnormalities, myopathy | (180) |
| Methylmalonic aciduria-homocystinuria | MMACHC | Methionine synthesis defect | Ataxia, intellectual decline, seizures, leukoencephalopathy | (3) |
| Myotonic muscular dystrophy 1 | Myotonic dystrophy protein kinase | Myotonic dystrophy protein kinase toxic messenger RNA | Weakness, myotonia, cardiac arrhythmias and conduction defects, frontal balding | (176) |
| Noonan syndrome | PTPN11 | RAS/mitogen-activated protein kinase pathway | Short stature, dysmorphic facies, heart defects, cryptorchidism, infertility | (181) |
| Mutation | Functional effect | Clinical features | Reference(s) | |
|---|---|---|---|---|
| Adrenoleukodystrophy-adrenomyeloneuropathy | ACBD1 | Peroxisome long chain fatty acid oxidation | Ataxia, bladder/bowel dysfunction Adrenal insufficiency | (4) |
| Alstrom syndrome | ALMS1 | Cilia function and cell cycle regulation | Obesity, vision, and hearing loss; type 2 diabetes mellitus; cardiomyopathy. | (30, 178) |
| Bardet Biedl syndrome | BBS1-BBS21, MKKS, ARL6, TTC8, CEP290, NPHP1, SCAPER, LZTFL1, BB1P1. | Cilia development and function | Retinal dystrophy, polydactyly, cognitive Dysfunction, ataxia obesity, renal insufficiency, hearing loss, anosmia, micropenis, cryptorchidism. | (27) |
| Bulbospinal Muscle atrophy (Kennedy disease) | Increased CAG repeats in the androgen receptor | Toxic RNA and misfolded protein, impaired androgen receptor transactivation | Weakness, gynecomastia | (73, 74) |
| Down syndrome | Trisomy 21 | Overexpression of chromosome 21 genes | Cognitive impairment, dysmorphic features, hypotonia | (33) |
| Hereditary moyamoya syndrome | RNF213 | Ubiquitin ligase activity | Supraclinoid internal carotid artery occlusion, facial dysmorphism, cardiomyopathy, short stature | (179) |
| Klinefelter syndrome and polyploid variants | 47, XXY 48, XXYY | Testosterone deficiency, estrogen excess, overexpression of X chromosome genes | Essential tremor Seizure disorder | (44, 45) |
| Marinesco-Sjogren syndrome | SIL1 (suppressor of Ire1/Lhs1 double mutant) | Deficient cofactor for BiP (estrogen receptor chaperone-binding immunoglobulin protein) causing disrupted protein folding | Cerebellar ataxia, cataracts, mental retardation, skeletal abnormalities, myopathy | (180) |
| Methylmalonic aciduria-homocystinuria | MMACHC | Methionine synthesis defect | Ataxia, intellectual decline, seizures, leukoencephalopathy | (3) |
| Myotonic muscular dystrophy 1 | Myotonic dystrophy protein kinase | Myotonic dystrophy protein kinase toxic messenger RNA | Weakness, myotonia, cardiac arrhythmias and conduction defects, frontal balding | (176) |
| Noonan syndrome | PTPN11 | RAS/mitogen-activated protein kinase pathway | Short stature, dysmorphic facies, heart defects, cryptorchidism, infertility | (181) |
Leukodystrophies are genetically heterogeneous rare white matter neurodegenerative disorders characterized by symmetrical gait abnormalities and cognitive decline, and often early death (1), with increased white matter signal on T2-weighted magnetic resonance imaging (MRI) sequences (2). X-linked adrenoleukodystrophy-adrenomyeloneuropathy [adrenoleukodystrophy (ALD)/adrenomyeloneuropathy (AMN)] and methylmalonic aciduria-homocystinuria (3) are 2 leukodystrophies associated with primary testicular failure.
ALD/AMN is an X-linked disorder that disrupts peroxisomal oxidation. The disorder results from over 800 mutations in the ACBD1 [adenosine 5′-triphosphate (ATP)-binding cassette transporter subfamily D] gene and has a variable phenotype, with an incidence of 1:17 000 in all geographic areas and all racial/ethnic groups. Childhood ALD is a rapidly progressive disease that includes behavioral problems, strabismus, cognitive decline, gait disturbance, and quadriparesis, with death within 5 to 10 years of diagnosis. AMN is a slowly progressive spinal cord disease of adult men, with weakness, spasticity, bladder dysfunction, and behavioral changes (4) although neurological symptoms/signs may be subtle. Female heterozygotes may exhibit symptoms later in life. There is no recognized association between genotype and phenotype (5).
ACBD1 encodes the peroxisome membrane protein ATP-binding cassette subfamily D member 1, which functions in the transmembrane transport of very long chain fatty acids (VLCFA) into peroxisomes for degradation. In ALD/AMN, levels of VCLFA (hexacosanoic C26:0 and lignoceric C24:0) increase in nervous system tissues, resulting in white matter demyelination, and in plasma (6).
VLCFA also accumulate in the adrenal cortex and testes. Adrenal insufficiency develops gradually, and affected children often have increased adrenocorticotropic hormone (ACTH) levels and a subnormal cortisol response to cosyntropin stimulation even prior to the development of neurological symptoms and signs (7). Symptomatic adrenal failure may first manifest from early childhood through late adulthood, and Addison-only cases have been reported (8). Most adult men with AMN develop adrenal insufficiency, and AMN accounts for more than 15% of nonimmune primary adrenal failure in men (9). While the mechanism for steroid deficiency is not known, VLCFAs could interfere with tropic hormone receptor binding and activation (10) or with steroidogenesis directly.
Biochemical and clinical hypogonadism may also occur (11), most often together with adrenal insufficiency (12, 13). Cryptorchidism has been reported. Chronic adrenocorticotropic hormone elevation may lead to the development of testicular nodules (testicular adrenal rest tumors) as occurs in congenital adrenal hyperplasia (14). Many men report fertility prior to the onset of neurological disease (15) followed by the development of sexual dysfunction, decreased body hair, gynecomastia in mid adulthood, and premature male pattern baldness (16). FSH and/or LH levels are often increased, and while total testosterone levels are most often in the normal range, the testosterone response to human chorionic gonadotropin (hCG) stimulation is generally subnormal (13). High levels of sex hormone-binding globulin (SHBG) may increase total testosterone levels. An androgen receptor (AR) signaling defect (17, 18) is suggested by the elevated product of serum testosterone X LH levels. Men who are clinically and/or biochemically testosterone deficient should receive androgen replacement therapy.
Bardet-Biedl syndrome (BBS) is a pleiotropic autosomal recessive or oligogenic disorder characterized by progressive vision loss from retinal dystrophy, polydactyly, intellectual impairment, cystic kidneys, extreme obesity, and hypogonadism. Some patients also have ataxia, craniofacial abnormalities, cardiac anomalies, hearing loss, and anosmia (19). The prevalence of BBS averages 1:160 000 in North America and Europe but is 1:17 500 and 1:13 500 in isolated communities in Newfoundland and Kuwait, respectively (20).
BBS is a ciliopathy, a disorder of the microtubule-based organelles that extend from the surface of nearly all cells. BBS is associated with mutations of more than 20 genes on different chromosomes including BBS1-BBS19, MKS1, CEP290, LZTFL1, and FT27 (21) although mutations in BBS1 and BBS10 account for most cases. Together, the protein products form a complex known as the BBSome that regulates cilia development and intracellular transport by activating Wnt and other signaling pathways that function in cell growth and survival (22). BBS mutant cilia have loss or accumulation of abnormal proteins.
Both primary testicular failure and hypogonadotropic hypogonadism may occur in BBS. Most affected males have a small penis and scrotum, and about 10% have cryptorchidism (23). Puberty may be delayed (24), but clinical androgen deficiency is uncommon in adults. FSH and LH levels may be elevated while total testosterone is usually normal (20). In some cases, low total testosterone is explained by low SHBG linked to severe obesity, insulin resistance, and the metabolic syndrome (25). Desai et al (26) and Kocinski et al (27) have proposed that the undeveloped genitalia in BBS males is due to GnRH deficiency during fetal life that subsequently reverses. Desai et al (26) reported the case of an obese male with BBS with absent pubertal development and gonadotropin deficiency at age 16.5 years whose testosterone level was 3.44 ng/mL with normal LH and FSH levels and normal sized testes with active spermatogenesis when he was reexamined at age 30. Kocinski et al (27) noted the experiments by Koemeter-Cox et al (28) that demonstrated cilia uniquely on GnRH neurons that express the Kiss1 receptor, with multiciliated GnRH neurons increasing with development from birth until age 60 days in the mouse. They proposed that increased cilia during development might increase Kiss1 receptor signaling and allow for pubertal development in BBS.
Most adult men with BBS are infertile. Semen volume may be low, sperm morphology is abnormal, and motility is low while the sperm count is generally within the reference range (27). The cause for these abnormalities is uncertain. The sperm tail axoneme, a motile microtubule structure with 9 doublet arms surrounding 2 singlet arms that is essential for sperm motility, appears to be structurally normal, however (27). One case report that included a testicular biopsy described arrested spermatogenesis with focal germinal aplasia (29). Anosmia has been explained by ciliary defects in olfactory neurons.
Alstrom syndrome is a second but far less common ciliopathy that may likewise result in both primary and hypogonadotropic hypogonadism. The disorder is caused by recessive mutations of ALS1 and is characterized variably by childhood obesity, insulin resistance and type 2 diabetes mellitus, vision and hearing loss, and cardiomyopathy. Adult men have small testes and may have a small phallus and cryptorchidism. LH and FSH levels may be elevated or within the reference range (30).
Down syndrome (trisomy 21) is the most common genetic disease at birth with an incidence of 1:700 newborns that is increased with maternal age. There is selective overexpression of chromosome 21 genes with up to 40-fold upregulation in various tissues (31). Its main clinical characteristics are dysmorphic features; cognitive impairment; and cardiac, ophthalmological, and endocrine abnormalities (32). As many as 50% of boys with Down syndrome are born with cryptorchidism, and FSH levels during minipuberty are elevated with or without undescended testes (33), implying newborn Sertoli cell failure. Men with Down syndrome have small testes and impaired spermatogenesis and are almost always infertile (34). FSH and LH levels are increased while total testosterone levels are generally within the normal range (35). The extra chromosome may result in germ cell apoptosis with meiotic or postmeiotic failure perhaps through overexpression of the chromosome 21 gene DYRK1A (dual-specificity tyrosine phosphorylation-regulated kinase 1A), a dual specificity phosphorylation dependent tyrosine kinase (36).
Klinefelter syndrome (KS; 47, XXY) affects 1 in 500 to 600 men, including 10% to 12% of men with azoospermia (37). KS is characterized by small testes, reduced body hair, gynecomastia, and high levels of FSH and LH while testosterone levels are generally low (38). In addition to endocrine dysfunction, there is an increase in gene dosage due to lack of inactivation of specific genes on the short arm of the extra X (39, 40). Many of these genes are highly expressed in the brain (41), causing KS patients to express a range of brain-related phenotypes including behavioral, psychiatric, cognitive, and neurological abnormalities (42, 43). In 1 survey, 50% of KS cases admitted to essential tremor, primarily affecting the hands (44) while as many as 5% to 17 % may have a seizure disorder (45). The risk for intellectual disability increases with increasing polysomy (46, 47).
Myotonic dystrophy (DM) is the most common adult-onset muscular dystrophy, with a global prevalence between 1:3000 and 1:8000. It is an autosomal dominant disorder characterized by myotonia (prolonged muscle contraction), muscle atrophy with progressive weakness, and cognitive decline as well as frontal balding, cataracts, cardiac conduction defects and tachyarrhythmias, respiratory dysfunction, and endocrine disorders (48). DM1 results from increased CTG repeats on chromosome 19q 13.3 in the 3’ region of the DM protein kinase gene (49) while the related disorder DM2 results from a CCTG expansion in intron 1 of the cellular nucleic acid-binding protein on chromosome 3q21 (50). Mutant messenger RNAs (mRNAs) remain nuclear and form intranucleoplasmic hairpin loops that sequester RNA-binding proteins including muscle blind-like protein 1 and CUGBP Elav-like family member 1 that regulate mRNA splicing. The result is loss of function and the abnormal processing of multiple gene products.
DM protein kinase and cellular nucleic acid-binding protein mRNAs are expressed in testis (51, 52). Men with DM develop primary testicular failure (53), and a few with minimal-mild muscle symptoms have presented with infertility (54, 55). Testis size is generally reduced while testicular biopsy findings range from normal sized tubules with mild germ cell disorganization to germ cell maturation arrest and focal to diffuse seminiferous tubule fibrosis (53, 56-58). Inhibin-B and Müllerian inhibitory hormone levels are low (58, 59), and most men have elevated levels of FSH (58, 60). Although sperm production and quality are impaired, many men with DM report fertility earlier in life (53, 58). Leydig cell dysfunction in DM is more modest (58). Pubertal development is generally normal (58) while body hair may be reduced, and gynecomastia may occur in adulthood. Most men have normal total testosterone levels while LH levels may be normal or elevated (58, 60-63).
The mechanism for hypogonadism in DM is incompletely understood. Numerous RNA binding proteins function in spermatogenesis (64) to control spermatogonia renewal, mitotic and meiotic division, and spermiogenesis. As shown in Figure 1, dysregulated mRNA splicing may disrupt germ cell maturation and cause inhibin production to decline and FSH levels to increase (65). Testosterone production may decrease and LH may increase because less inhibin is available antagonize activin-suppression of testosterone biosynthesis (66) or because the mutant DM protein kinase in Leydig cells adversely affects steroidogenesis. Massive CTG expansion may occur in the sons of affected DM1 mothers (67), and genetic counseling, in vitro fertilization, and preimplantation genetic testing are suggested for couples planning to have children (68).
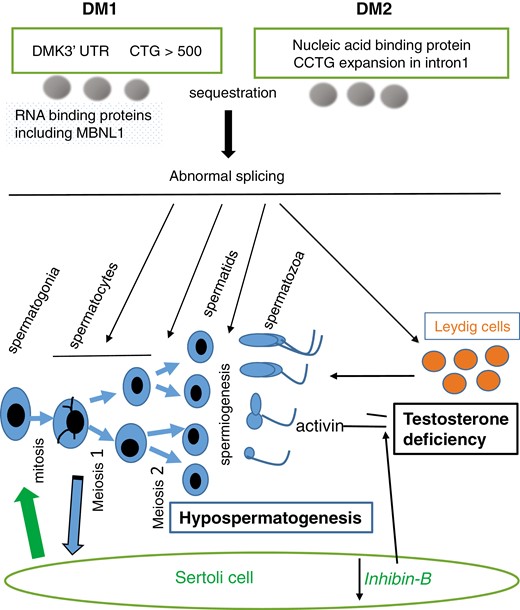
A schema for the impact of expanded 3’ CTG repeats in the myotonic dystrophy protein kinase gene in DM1 and a CCTG expansion in intron 1 of the nucleic acid binding protein in DM2 in testis. Resultant sequestration of RNA-binding proteins leads to abnormal splicing and thereby hypospermatogenesis and testosterone deficiency.
Spinal and bulbar muscular atrophy (Kennedy disease; KD) is an X-linked disorder in which there is motor neuron loss in the anterior horn of the spinal cord and the bulbar regions of the brain (69). KD patients develop progressive symmetrical weakness and wasting of the limbs (lower extremity > upper extremity) and facial muscles, fasciculation, and tremor, leading to bulbar weakness with dysphagia and dysarthria. Aspiration pneumonia and respiratory failure may lead to premature death. Nearly all men with KD have gynecomastia, and many have small testes, oligospermia, and clinical androgen deficiency (70, 71). They are also at increased risk for diabetes, hyperlipidemia, and nonalcoholic fatty liver disease (72-74).
KD is caused by an expanded CAG repeat (>38-68 + vs 11-32 in the normal population) in exon 1 of the AR gene encoding glutamine (75). KD is 1 of 9 recognized neurological disorders (polyglutamine diseases) that result from CAG repeat expansions (eg, Huntington disease). As summarized in Figure 2, expanded repeats produce a toxic mRNA that encodes a mutant AR protein, which is misfolded and aggregates following testosterone binding and translocation to the nucleus. The result is death of bulbospinal cord neurons (76), as well as injury to muscle cells (77) and various other tissues including testis. Altered AR structure also decreases AR binding capacity (78) and modifies coactivator protein interactions leading to reduced receptor transcriptional activity and androgen insensitivity (79). Adult men generally present at age 30 to 50 years, in inverse correlation with the CAG repeat length (80), and the diagnosis is confirmed by polymerase chain reaction analysis. Carrier females are usually asymptomatic.
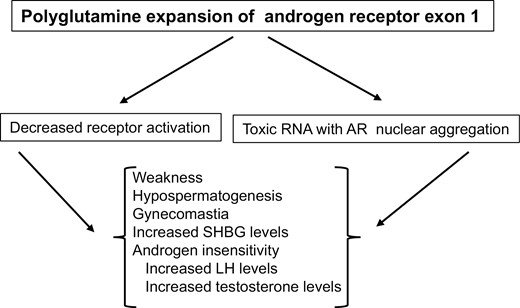
Clinical features in men with bulbospinal muscle atrophy (Kennedy disease). Expansion of CAG repeats in exon 1 of the androgen receptor gene produces a toxic messenger RNA that encodes a mutant AR protein that damages neurons and is insensitive to androgen activation.
Gynecomastia occurs in most men with KD, sometimes as early as young adulthood (81), and testicular atrophy and oligo/azoospermia may follow or precede neurological symptoms. Nuclear inclusions are found in the testis, and hypospermatogenesis may result from aggregation and dysfunction of the mutant AR in Sertoli cells. Hormone levels are variable suggesting multiple pathological effects. Total testosterone levels in adults are variably (0-68%) elevated (74, 81-83) while LH levels are generally normal, although the LH response to GnRH stimulation may be exaggerated (81). Androgen resistance is implied by the combination of elevated LH and testosterone levels, which follows from disrupted testosterone negative feedback inhibition of GnRH secretion. FSH levels may be elevated selectively (78, 81) while no results for inhibin B were found in the publications reviewed. Estradiol levels are increased in some men presumably because testicular aromatase is stimulated by increased LH, and increased estradiol and androgen insensitivity may elevate SHBG (84), favoring the bioactivity of estradiol over testosterone. The absence of elevated testosterone and LH levels in some KD patients may be partly explained by an older age at diagnosis (83) and chronic illness, including dysphagia, which may cause testosterone and LH levels to decline. Muscle atrophy and weakness, as well as chronic illness and androgen insensitivity, increase the risk for osteoporosis.
Given the notion that cell death is caused by a testosterone-activated toxic nuclear AR, suppression of testosterone signaling is a logical potential therapeutic approach and was supported by experiments in a transgenic mouse model in which androgen deprivation delayed the onset and slowed neurological disease progression (85). In a placebo-controlled multicenter clinical trial, however, the GnRH analog leuprorelin failed to benefit barium swallowing by videofluorography (86), and in a second study, the testosterone 5 alpha-reductase inhibitor dutasteride failed to increase muscle strength (87) in men with KD. Although men may be clinically hypogonadal, there is also no evidence that androgen treatment is beneficial. Gynecomastia may require surgical management.
Hypogonadism With Developmental Disorders of the Hypothalamus and Pituitary
Kallmann syndrome is the association of congenital isolated hypogonadotropic hypogonadism (CIHH) with anosmia (and other developmental defects). The syndrome results from failure of GnRH neurons to migrate from the cribriform plate to the hypothalamus during fetal life where connections to the median eminence are normally established. More than 20 genes are known play a role in this migration of neurons (Table 2). Deleterious mutations are associated with neuronal developmental abnormalities leading to hearing loss and mirror movements as well as anosmia. CIHH may also occur without developmental defects when causative mutations involve GnRH function rather than neuronal migration. CIHH and Kallmann syndrome are the subject of several excellent reviews (88-91) and are not discussed in detail due to space limitations, nor are complex disorders of embryonic development of the CNS and face with congenital hypopituitarism including septo-optic dysplasia (92), CHARGE syndrome (93, 94), and SOX 2 disorder (95), which exceed the scope of this review.
Genes associated with neurological symptoms in Kallmann syndrome patients
| Gene | Protein defect | Inheritance | Neuro-phenotype | Reference |
|---|---|---|---|---|
| KAL-1 (ANOS1) | Anosmin-1 | X-linked recessive | Anosmia, synkinesias | (182) |
| FEZF1 | Transcriptional regulator | Autosomal recessive | Anosmia | (183) |
| FGF-R1 | Anosmin-1 binding protein; RAS/MAPK activation | Autosomal dominant | Anosmia craniofacial abnormalities | (184) |
| FGF8 | FGFR tyrosine kinase phosphorylation | Autosomal dominant | anosmia craniofacial abnormalities | (185) |
| PROK2 | G-protein coupled receptor activation in CNS and testis | Oligogenic, recessive, autosomal dominant | Anosmia | (186) |
| PROKR2 | Gq pathways | Oligogenic, recessive, autosomal dominant | Anosmia, septo-optic dysplasia | (187) |
| WDR11 | Hedgehog signaling in cilium | Autosomal dominant | Anosmia | (188) |
| HS6ST1 | HS-6-O-sulfotransferase; Sulfation of anosmin and FGFR1 | Autosomal dominant oligogenic | Anosmia | (189) |
| SEMA3A | Semaphorin-3A | Oligogenic | Anosmia | (190) |
| SOX10 | Transcriptional regulator | Autosomal dominant oligogenic | Anosmia, sensorineural deafness, Waardenburg syndrome | (191) |
| TCF12 | Transcription factor regulating STUB1 | Autosomal dominant | Anosmia, craniosynostosis, autism spectrum disorder | (192) |
| NELF | Transcriptional regulator | Autosomal recessive oligogenic | Anosmia | (193) |
| PRDM13 | GABAergic control of cerebellum and kisspeptin neurons | Autosomal recessive | Ataxia, hypotonia, intellectual disability, scoliosis | (194) |
| Gene | Protein defect | Inheritance | Neuro-phenotype | Reference |
|---|---|---|---|---|
| KAL-1 (ANOS1) | Anosmin-1 | X-linked recessive | Anosmia, synkinesias | (182) |
| FEZF1 | Transcriptional regulator | Autosomal recessive | Anosmia | (183) |
| FGF-R1 | Anosmin-1 binding protein; RAS/MAPK activation | Autosomal dominant | Anosmia craniofacial abnormalities | (184) |
| FGF8 | FGFR tyrosine kinase phosphorylation | Autosomal dominant | anosmia craniofacial abnormalities | (185) |
| PROK2 | G-protein coupled receptor activation in CNS and testis | Oligogenic, recessive, autosomal dominant | Anosmia | (186) |
| PROKR2 | Gq pathways | Oligogenic, recessive, autosomal dominant | Anosmia, septo-optic dysplasia | (187) |
| WDR11 | Hedgehog signaling in cilium | Autosomal dominant | Anosmia | (188) |
| HS6ST1 | HS-6-O-sulfotransferase; Sulfation of anosmin and FGFR1 | Autosomal dominant oligogenic | Anosmia | (189) |
| SEMA3A | Semaphorin-3A | Oligogenic | Anosmia | (190) |
| SOX10 | Transcriptional regulator | Autosomal dominant oligogenic | Anosmia, sensorineural deafness, Waardenburg syndrome | (191) |
| TCF12 | Transcription factor regulating STUB1 | Autosomal dominant | Anosmia, craniosynostosis, autism spectrum disorder | (192) |
| NELF | Transcriptional regulator | Autosomal recessive oligogenic | Anosmia | (193) |
| PRDM13 | GABAergic control of cerebellum and kisspeptin neurons | Autosomal recessive | Ataxia, hypotonia, intellectual disability, scoliosis | (194) |
Genes associated with neurological symptoms in Kallmann syndrome patients
| Gene | Protein defect | Inheritance | Neuro-phenotype | Reference |
|---|---|---|---|---|
| KAL-1 (ANOS1) | Anosmin-1 | X-linked recessive | Anosmia, synkinesias | (182) |
| FEZF1 | Transcriptional regulator | Autosomal recessive | Anosmia | (183) |
| FGF-R1 | Anosmin-1 binding protein; RAS/MAPK activation | Autosomal dominant | Anosmia craniofacial abnormalities | (184) |
| FGF8 | FGFR tyrosine kinase phosphorylation | Autosomal dominant | anosmia craniofacial abnormalities | (185) |
| PROK2 | G-protein coupled receptor activation in CNS and testis | Oligogenic, recessive, autosomal dominant | Anosmia | (186) |
| PROKR2 | Gq pathways | Oligogenic, recessive, autosomal dominant | Anosmia, septo-optic dysplasia | (187) |
| WDR11 | Hedgehog signaling in cilium | Autosomal dominant | Anosmia | (188) |
| HS6ST1 | HS-6-O-sulfotransferase; Sulfation of anosmin and FGFR1 | Autosomal dominant oligogenic | Anosmia | (189) |
| SEMA3A | Semaphorin-3A | Oligogenic | Anosmia | (190) |
| SOX10 | Transcriptional regulator | Autosomal dominant oligogenic | Anosmia, sensorineural deafness, Waardenburg syndrome | (191) |
| TCF12 | Transcription factor regulating STUB1 | Autosomal dominant | Anosmia, craniosynostosis, autism spectrum disorder | (192) |
| NELF | Transcriptional regulator | Autosomal recessive oligogenic | Anosmia | (193) |
| PRDM13 | GABAergic control of cerebellum and kisspeptin neurons | Autosomal recessive | Ataxia, hypotonia, intellectual disability, scoliosis | (194) |
| Gene | Protein defect | Inheritance | Neuro-phenotype | Reference |
|---|---|---|---|---|
| KAL-1 (ANOS1) | Anosmin-1 | X-linked recessive | Anosmia, synkinesias | (182) |
| FEZF1 | Transcriptional regulator | Autosomal recessive | Anosmia | (183) |
| FGF-R1 | Anosmin-1 binding protein; RAS/MAPK activation | Autosomal dominant | Anosmia craniofacial abnormalities | (184) |
| FGF8 | FGFR tyrosine kinase phosphorylation | Autosomal dominant | anosmia craniofacial abnormalities | (185) |
| PROK2 | G-protein coupled receptor activation in CNS and testis | Oligogenic, recessive, autosomal dominant | Anosmia | (186) |
| PROKR2 | Gq pathways | Oligogenic, recessive, autosomal dominant | Anosmia, septo-optic dysplasia | (187) |
| WDR11 | Hedgehog signaling in cilium | Autosomal dominant | Anosmia | (188) |
| HS6ST1 | HS-6-O-sulfotransferase; Sulfation of anosmin and FGFR1 | Autosomal dominant oligogenic | Anosmia | (189) |
| SEMA3A | Semaphorin-3A | Oligogenic | Anosmia | (190) |
| SOX10 | Transcriptional regulator | Autosomal dominant oligogenic | Anosmia, sensorineural deafness, Waardenburg syndrome | (191) |
| TCF12 | Transcription factor regulating STUB1 | Autosomal dominant | Anosmia, craniosynostosis, autism spectrum disorder | (192) |
| NELF | Transcriptional regulator | Autosomal recessive oligogenic | Anosmia | (193) |
| PRDM13 | GABAergic control of cerebellum and kisspeptin neurons | Autosomal recessive | Ataxia, hypotonia, intellectual disability, scoliosis | (194) |
Multisystem Disorders With Neurological Manifestations and Hypogonadotropic Hypogonadism
The signaling pathways that regulate the function and survival of GnRH neurons and gonadotrophs are often shared by other nervous system tissues such that hypogonadotropic hypogonadism is often found in individuals with developmental delay, ataxia, and cognitive decline (Table 3). In most syndromes, although pathogenic mutations have been identified, there remains a limited understanding of the mechanistic details leading to hypogonadism, and extreme obesity, medications (eg, glucocorticoids), and confounding disorders (eg, chronic kidney disease) may be contributing factors in some individuals. In some cases, most of the data are in children.
Multisystem syndromes with hypogonadotropic hypogonadism and neurological abnormalities
| Mutation | Inheritance | Functional effect | Neurological/clinical features | Reference(s) | |
|---|---|---|---|---|---|
| Boucher Neuhauser syndrome | PNPLA6 | Autosomal recessive | Deficient or abnormal neuropathy target esterase | Ataxia, chorioretinal dystrophy | (195) |
| CHARGE syndrome | CHD7 (chromodomain helicase DNA-binding protein) | De novo haploinsufficiency | Transcriptional regulator | Coloboma, heart defects, choanal atresia, retardation of growth, gonadal defects (due to CHH) and ear abnormalities, anosmia, micropenis, cryptorchidism | (93, 94) |
| Gordon-Holmes syndrome | STUB1, CHIP, PNPLA6 | Autosomal recessive | Ubiquitin ligase activity | Ataxia | (96) |
| Hartsfield syndrome | FGFR1 | Sporadic, Autosomal dominant | Receptor tyrosine-kinase signaling | Holoprosencephaly, ectrodactyly (split hand/foot malformation) | (196) |
| Kearns-Sayre syndrome | Mitochondrial genome deletions | Sporadic | Oxidative phosphorylation | Pigmentary retinopathy, external ophthalmoplegia, weakness, ataxia, deafness, diabetes | (109) |
| Moebius syndrome | ? | Sporadic | ? | Anosmia, cranial nerves VI/VII and other facial palsies, limb defects | (197) |
| PMM2-congenital disorder of glycosylation (CDG-type 1a) | PMM2 (Phosphomannomutase 2) | Autosomal recessive | Defective N-linked protein glycosylation | Intellectual disability, developmental delay, seizures, ataxia, strabismus, growth failure, cortisol deficiency, cryptorchidism | (198, 199) |
| Pol-III (4H) leukodystrophy | POLR3A/POLR3B/ BPOL3C POLR3K | Autosomal recessive | RNA polymerase III | Hypomyelination, hypodontia, myopia, cerebellar and cognitive dysfunction | (171) |
| POEMS syndrome | Paraneoplastic syndrome | Sporadic | IgG, IgA monoclonal gammopathy | Sensorimotor peripheral neuropathy, organomegaly, gynecomastia | (119, 120) |
| Prader Willi | Chromosome 15q deletion, maternal disomy 15 | Sporadic | Lack of paternal imprinted gene expression | Infantile hypotonia and failure to thrive, hyperphagia and obesity, dysmorphic features, short stature, learning disability and behavioral problems, and pituitary dysfunction | (134) |
| Proprotein convertase deficiency | PCSK1 | Autosomal recessive | Impaired conversion of inactive precursor to active peptides | Intestinal malabsorption, early onset, obesity, diabetes insipidus, ACTH, TSH, GH, and GnRH deficiency | (149, 200) |
| Septo-optic dysplasia | HESX1, SOX2, SOX3, OTX2 | Autosomal recessive, autosomal dominant | Transcription factors in pituitary development | Optic nerve hypoplasia, absent septum pellucidum, seizures, hypopituitarism | (92) |
| Sifrim Hitz Weiss syndrome | CHD4 | “Sporadic” | ATP hydrolysis, transcriptional repression | Developmental delay, intellectual disability, heart defects, micropenis, cryptorchidism | (201, 202) |
| SOX 2 disorder [sex-determining region Y (SRY) box2] | SOX2 | Autosomal recessive | Transcription factor | Anophthalmia, hearing loss, seizures, corpus collosum and pituitary hypoplasia | (203) |
| Waardenburg syndrome | SOX10 | Haploinsufficiency | SRY-box 10 | Anosmia, congenital sensorineural hearing loss, abnormal pigmentation of the hair, skin, and iris. | (191, 204, 205) |
| Warburg micro syndrome (Martsolf syndrome) | RAB18 RAB3GAP1, RAB3GAP2, TBC1D20 | Autosomal recessive | RAB18 deficiency (a GTPase) | Microphthalmia, congenital cataracts, microcephaly, mental retardation, hypoplasia of corpus callosum, cryptorchidism, micropenis | (166, 169, 206, 207) |
| Wolfram syndrome | WFS1, WFS2 | Autosomal recessive | Protein folding and processing in the endoplasmic reticulum | Diabetes mellitus, diabetes insipidus, optic atrophy, sensorineural deafness, ataxia, neurogenic bladder and bowel dysfunction | (208, 209) |
| Woodhouse Sakati syndrome | DCAF17 | Autosomal recessive | Unknown | Alopecia, ataxia, dystonia, intellectual disability, hearing loss, small pituitary, diabetes mellitus | (210) |
| Mutation | Inheritance | Functional effect | Neurological/clinical features | Reference(s) | |
|---|---|---|---|---|---|
| Boucher Neuhauser syndrome | PNPLA6 | Autosomal recessive | Deficient or abnormal neuropathy target esterase | Ataxia, chorioretinal dystrophy | (195) |
| CHARGE syndrome | CHD7 (chromodomain helicase DNA-binding protein) | De novo haploinsufficiency | Transcriptional regulator | Coloboma, heart defects, choanal atresia, retardation of growth, gonadal defects (due to CHH) and ear abnormalities, anosmia, micropenis, cryptorchidism | (93, 94) |
| Gordon-Holmes syndrome | STUB1, CHIP, PNPLA6 | Autosomal recessive | Ubiquitin ligase activity | Ataxia | (96) |
| Hartsfield syndrome | FGFR1 | Sporadic, Autosomal dominant | Receptor tyrosine-kinase signaling | Holoprosencephaly, ectrodactyly (split hand/foot malformation) | (196) |
| Kearns-Sayre syndrome | Mitochondrial genome deletions | Sporadic | Oxidative phosphorylation | Pigmentary retinopathy, external ophthalmoplegia, weakness, ataxia, deafness, diabetes | (109) |
| Moebius syndrome | ? | Sporadic | ? | Anosmia, cranial nerves VI/VII and other facial palsies, limb defects | (197) |
| PMM2-congenital disorder of glycosylation (CDG-type 1a) | PMM2 (Phosphomannomutase 2) | Autosomal recessive | Defective N-linked protein glycosylation | Intellectual disability, developmental delay, seizures, ataxia, strabismus, growth failure, cortisol deficiency, cryptorchidism | (198, 199) |
| Pol-III (4H) leukodystrophy | POLR3A/POLR3B/ BPOL3C POLR3K | Autosomal recessive | RNA polymerase III | Hypomyelination, hypodontia, myopia, cerebellar and cognitive dysfunction | (171) |
| POEMS syndrome | Paraneoplastic syndrome | Sporadic | IgG, IgA monoclonal gammopathy | Sensorimotor peripheral neuropathy, organomegaly, gynecomastia | (119, 120) |
| Prader Willi | Chromosome 15q deletion, maternal disomy 15 | Sporadic | Lack of paternal imprinted gene expression | Infantile hypotonia and failure to thrive, hyperphagia and obesity, dysmorphic features, short stature, learning disability and behavioral problems, and pituitary dysfunction | (134) |
| Proprotein convertase deficiency | PCSK1 | Autosomal recessive | Impaired conversion of inactive precursor to active peptides | Intestinal malabsorption, early onset, obesity, diabetes insipidus, ACTH, TSH, GH, and GnRH deficiency | (149, 200) |
| Septo-optic dysplasia | HESX1, SOX2, SOX3, OTX2 | Autosomal recessive, autosomal dominant | Transcription factors in pituitary development | Optic nerve hypoplasia, absent septum pellucidum, seizures, hypopituitarism | (92) |
| Sifrim Hitz Weiss syndrome | CHD4 | “Sporadic” | ATP hydrolysis, transcriptional repression | Developmental delay, intellectual disability, heart defects, micropenis, cryptorchidism | (201, 202) |
| SOX 2 disorder [sex-determining region Y (SRY) box2] | SOX2 | Autosomal recessive | Transcription factor | Anophthalmia, hearing loss, seizures, corpus collosum and pituitary hypoplasia | (203) |
| Waardenburg syndrome | SOX10 | Haploinsufficiency | SRY-box 10 | Anosmia, congenital sensorineural hearing loss, abnormal pigmentation of the hair, skin, and iris. | (191, 204, 205) |
| Warburg micro syndrome (Martsolf syndrome) | RAB18 RAB3GAP1, RAB3GAP2, TBC1D20 | Autosomal recessive | RAB18 deficiency (a GTPase) | Microphthalmia, congenital cataracts, microcephaly, mental retardation, hypoplasia of corpus callosum, cryptorchidism, micropenis | (166, 169, 206, 207) |
| Wolfram syndrome | WFS1, WFS2 | Autosomal recessive | Protein folding and processing in the endoplasmic reticulum | Diabetes mellitus, diabetes insipidus, optic atrophy, sensorineural deafness, ataxia, neurogenic bladder and bowel dysfunction | (208, 209) |
| Woodhouse Sakati syndrome | DCAF17 | Autosomal recessive | Unknown | Alopecia, ataxia, dystonia, intellectual disability, hearing loss, small pituitary, diabetes mellitus | (210) |
Abbreviations: ACTH, adrenocorticotropic hormone; ATP, adenosine 5′-triphosphates; CHH, congenital hypogonadotrophic hypogonadism; GH, growth hormone; GnRH, gonadotropin-releasing hormone; IgA, immunoglobulin A; IgG, immunoglobulin G; TSH, thyroid-stimulating hormone.
Multisystem syndromes with hypogonadotropic hypogonadism and neurological abnormalities
| Mutation | Inheritance | Functional effect | Neurological/clinical features | Reference(s) | |
|---|---|---|---|---|---|
| Boucher Neuhauser syndrome | PNPLA6 | Autosomal recessive | Deficient or abnormal neuropathy target esterase | Ataxia, chorioretinal dystrophy | (195) |
| CHARGE syndrome | CHD7 (chromodomain helicase DNA-binding protein) | De novo haploinsufficiency | Transcriptional regulator | Coloboma, heart defects, choanal atresia, retardation of growth, gonadal defects (due to CHH) and ear abnormalities, anosmia, micropenis, cryptorchidism | (93, 94) |
| Gordon-Holmes syndrome | STUB1, CHIP, PNPLA6 | Autosomal recessive | Ubiquitin ligase activity | Ataxia | (96) |
| Hartsfield syndrome | FGFR1 | Sporadic, Autosomal dominant | Receptor tyrosine-kinase signaling | Holoprosencephaly, ectrodactyly (split hand/foot malformation) | (196) |
| Kearns-Sayre syndrome | Mitochondrial genome deletions | Sporadic | Oxidative phosphorylation | Pigmentary retinopathy, external ophthalmoplegia, weakness, ataxia, deafness, diabetes | (109) |
| Moebius syndrome | ? | Sporadic | ? | Anosmia, cranial nerves VI/VII and other facial palsies, limb defects | (197) |
| PMM2-congenital disorder of glycosylation (CDG-type 1a) | PMM2 (Phosphomannomutase 2) | Autosomal recessive | Defective N-linked protein glycosylation | Intellectual disability, developmental delay, seizures, ataxia, strabismus, growth failure, cortisol deficiency, cryptorchidism | (198, 199) |
| Pol-III (4H) leukodystrophy | POLR3A/POLR3B/ BPOL3C POLR3K | Autosomal recessive | RNA polymerase III | Hypomyelination, hypodontia, myopia, cerebellar and cognitive dysfunction | (171) |
| POEMS syndrome | Paraneoplastic syndrome | Sporadic | IgG, IgA monoclonal gammopathy | Sensorimotor peripheral neuropathy, organomegaly, gynecomastia | (119, 120) |
| Prader Willi | Chromosome 15q deletion, maternal disomy 15 | Sporadic | Lack of paternal imprinted gene expression | Infantile hypotonia and failure to thrive, hyperphagia and obesity, dysmorphic features, short stature, learning disability and behavioral problems, and pituitary dysfunction | (134) |
| Proprotein convertase deficiency | PCSK1 | Autosomal recessive | Impaired conversion of inactive precursor to active peptides | Intestinal malabsorption, early onset, obesity, diabetes insipidus, ACTH, TSH, GH, and GnRH deficiency | (149, 200) |
| Septo-optic dysplasia | HESX1, SOX2, SOX3, OTX2 | Autosomal recessive, autosomal dominant | Transcription factors in pituitary development | Optic nerve hypoplasia, absent septum pellucidum, seizures, hypopituitarism | (92) |
| Sifrim Hitz Weiss syndrome | CHD4 | “Sporadic” | ATP hydrolysis, transcriptional repression | Developmental delay, intellectual disability, heart defects, micropenis, cryptorchidism | (201, 202) |
| SOX 2 disorder [sex-determining region Y (SRY) box2] | SOX2 | Autosomal recessive | Transcription factor | Anophthalmia, hearing loss, seizures, corpus collosum and pituitary hypoplasia | (203) |
| Waardenburg syndrome | SOX10 | Haploinsufficiency | SRY-box 10 | Anosmia, congenital sensorineural hearing loss, abnormal pigmentation of the hair, skin, and iris. | (191, 204, 205) |
| Warburg micro syndrome (Martsolf syndrome) | RAB18 RAB3GAP1, RAB3GAP2, TBC1D20 | Autosomal recessive | RAB18 deficiency (a GTPase) | Microphthalmia, congenital cataracts, microcephaly, mental retardation, hypoplasia of corpus callosum, cryptorchidism, micropenis | (166, 169, 206, 207) |
| Wolfram syndrome | WFS1, WFS2 | Autosomal recessive | Protein folding and processing in the endoplasmic reticulum | Diabetes mellitus, diabetes insipidus, optic atrophy, sensorineural deafness, ataxia, neurogenic bladder and bowel dysfunction | (208, 209) |
| Woodhouse Sakati syndrome | DCAF17 | Autosomal recessive | Unknown | Alopecia, ataxia, dystonia, intellectual disability, hearing loss, small pituitary, diabetes mellitus | (210) |
| Mutation | Inheritance | Functional effect | Neurological/clinical features | Reference(s) | |
|---|---|---|---|---|---|
| Boucher Neuhauser syndrome | PNPLA6 | Autosomal recessive | Deficient or abnormal neuropathy target esterase | Ataxia, chorioretinal dystrophy | (195) |
| CHARGE syndrome | CHD7 (chromodomain helicase DNA-binding protein) | De novo haploinsufficiency | Transcriptional regulator | Coloboma, heart defects, choanal atresia, retardation of growth, gonadal defects (due to CHH) and ear abnormalities, anosmia, micropenis, cryptorchidism | (93, 94) |
| Gordon-Holmes syndrome | STUB1, CHIP, PNPLA6 | Autosomal recessive | Ubiquitin ligase activity | Ataxia | (96) |
| Hartsfield syndrome | FGFR1 | Sporadic, Autosomal dominant | Receptor tyrosine-kinase signaling | Holoprosencephaly, ectrodactyly (split hand/foot malformation) | (196) |
| Kearns-Sayre syndrome | Mitochondrial genome deletions | Sporadic | Oxidative phosphorylation | Pigmentary retinopathy, external ophthalmoplegia, weakness, ataxia, deafness, diabetes | (109) |
| Moebius syndrome | ? | Sporadic | ? | Anosmia, cranial nerves VI/VII and other facial palsies, limb defects | (197) |
| PMM2-congenital disorder of glycosylation (CDG-type 1a) | PMM2 (Phosphomannomutase 2) | Autosomal recessive | Defective N-linked protein glycosylation | Intellectual disability, developmental delay, seizures, ataxia, strabismus, growth failure, cortisol deficiency, cryptorchidism | (198, 199) |
| Pol-III (4H) leukodystrophy | POLR3A/POLR3B/ BPOL3C POLR3K | Autosomal recessive | RNA polymerase III | Hypomyelination, hypodontia, myopia, cerebellar and cognitive dysfunction | (171) |
| POEMS syndrome | Paraneoplastic syndrome | Sporadic | IgG, IgA monoclonal gammopathy | Sensorimotor peripheral neuropathy, organomegaly, gynecomastia | (119, 120) |
| Prader Willi | Chromosome 15q deletion, maternal disomy 15 | Sporadic | Lack of paternal imprinted gene expression | Infantile hypotonia and failure to thrive, hyperphagia and obesity, dysmorphic features, short stature, learning disability and behavioral problems, and pituitary dysfunction | (134) |
| Proprotein convertase deficiency | PCSK1 | Autosomal recessive | Impaired conversion of inactive precursor to active peptides | Intestinal malabsorption, early onset, obesity, diabetes insipidus, ACTH, TSH, GH, and GnRH deficiency | (149, 200) |
| Septo-optic dysplasia | HESX1, SOX2, SOX3, OTX2 | Autosomal recessive, autosomal dominant | Transcription factors in pituitary development | Optic nerve hypoplasia, absent septum pellucidum, seizures, hypopituitarism | (92) |
| Sifrim Hitz Weiss syndrome | CHD4 | “Sporadic” | ATP hydrolysis, transcriptional repression | Developmental delay, intellectual disability, heart defects, micropenis, cryptorchidism | (201, 202) |
| SOX 2 disorder [sex-determining region Y (SRY) box2] | SOX2 | Autosomal recessive | Transcription factor | Anophthalmia, hearing loss, seizures, corpus collosum and pituitary hypoplasia | (203) |
| Waardenburg syndrome | SOX10 | Haploinsufficiency | SRY-box 10 | Anosmia, congenital sensorineural hearing loss, abnormal pigmentation of the hair, skin, and iris. | (191, 204, 205) |
| Warburg micro syndrome (Martsolf syndrome) | RAB18 RAB3GAP1, RAB3GAP2, TBC1D20 | Autosomal recessive | RAB18 deficiency (a GTPase) | Microphthalmia, congenital cataracts, microcephaly, mental retardation, hypoplasia of corpus callosum, cryptorchidism, micropenis | (166, 169, 206, 207) |
| Wolfram syndrome | WFS1, WFS2 | Autosomal recessive | Protein folding and processing in the endoplasmic reticulum | Diabetes mellitus, diabetes insipidus, optic atrophy, sensorineural deafness, ataxia, neurogenic bladder and bowel dysfunction | (208, 209) |
| Woodhouse Sakati syndrome | DCAF17 | Autosomal recessive | Unknown | Alopecia, ataxia, dystonia, intellectual disability, hearing loss, small pituitary, diabetes mellitus | (210) |
Abbreviations: ACTH, adrenocorticotropic hormone; ATP, adenosine 5′-triphosphates; CHH, congenital hypogonadotrophic hypogonadism; GH, growth hormone; GnRH, gonadotropin-releasing hormone; IgA, immunoglobulin A; IgG, immunoglobulin G; TSH, thyroid-stimulating hormone.
Gordon Holmes syndrome is a neurodegenerative disorder with autosomal recessive inheritance that is characterized by ataxia, cognitive decline, and congenital hypogonadism (96). Neurological symptoms generally begin in adolescence through young adulthood, and may include peripheral neuropathy, movement disorders, dysarthria, and dysphagia (97). Neuroimaging reveals cortical and cerebellar atrophy with focal areas of white matter hyperintensity.
The disorder is caused by mutations in proteins that regulate ubiquitination. The E1-E2-E3 enzyme cascade normally identifies misfolded proteins and targets them for ubiquitination and degradation in the proteasome (98). Toxic aggregates and/or the failure of this system to degrade proteins in neurons results in neurotoxicity. More than 600 E3 ligases have been identified. Gordon Holmes syndrome can result from biallelic inactivating mutations of the E3 ligase ring finger protein 216 (RNF216) or the combination of mutations of RNF216 and the deubiquitinase OTU domain containing 4 (99). The latter results from mutation of STIP1 homology and U-box containing protein 1 (97, 98), which encodes the protein C-terminus of HSC70-interating protein that functions as an E3 ligase. The syndrome may also be explained by recessive mutations of palatin-like phospholipase domain containing 6) (100), which encodes neuropathy targeted esterase, a lysophospholipase with potential effects on biological membranes and acetylcholine synthesis.
Affected men may have a small phallus and testes and cryptorchidism, as well as delayed and incomplete puberty (101), and there is endocrine evidence for hypogonadotropic hypogonadism (102). Small genitals imply a defect during fetal life (well before neurological symptoms). While the mechanism for gonadotropin insufficiency is unknown, the pituitary is normal on MRI, and other pituitary hormones most often function normally. The failure of long-term pulsatile GnRH to stimulate LH and FSH normally, together with a robust testosterone response to hCG stimulation, suggested a pituitary defect (99, 102). RNF216 is highly expressed in testis. Null mice developed germ cell degeneration and were infertile (103); however, the endocrine disturbance in this model is unknown as gonadotropins were not measured.
Palatin-like phospholipase domain containing 6 recessive homozygous or compound heterozygous mutations are associated with Boucher Neuhauser syndrome which is characterized by impaired vision due to chorioretinal dystrophy, ataxia with progressive cerebellar degeneration, and hypogonadotropic hypogonadism (100, 104, 105).
Mitochondrial diseases result from mutations of the 37 genes of the mitochondrial genome. Mitochondrial genes function primarily in oxidative phosphorylation to produce ATP, and regulate multiple systems including CNS, neuromuscular, cardiac, renal, hepatic, and endocrine function. Deletions or point mutations of these genes occur most often de novo although some cases are inherited as autosomal recessives or dominants. While the cause for most mitochondrial mutations is uncertain, mutations in several nuclear genes have been identified as driving mitochondrial DNA deletions (106).
Endocrine dysfunction in patients with multiorgan disease, especially individuals with deafness and short stature, may indicate a mitochondrial disorder (107, 108). Mitochondrial diseases are heterogeneous clinically even with the same mutation, phenotypes range from mild to severe, and symptoms may begin from childhood to mid-adulthood. Several syndromes are recognized. Kearns-Sayre syndrome describes children to young adults who present with weakness and ataxia associated with progressive external ophthalmoplegia, often with ptosis, salt and pepper retinal changes, and cardiomyopathy and/or cardiac conduction defects. It is caused by a large single deletion or rearrangement of mitochondrial DNA. Diabetes occurs in more than 10% of cases, and occasional patients have hypothyroidism, hypoparathyroidism, adrenal insufficiency, or hypogonadism (109-111). Case reports describe males with short stature and delayed puberty, cryptorchidism, small phallus and testes, and clinical hypogonadism. In the few case studies with endocrine testing, gonadotropin levels and the testosterone response to hCG suggest coexistent primary and secondary hypogonadism (112, 113). Hypogonadotropic hypogonadism may occasionally occur with MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes) (114) and MERRF (myoclonic epilepsy with ragged-red fibers) (115) syndromes.
The initial step in steroidogenesis, the conversion of cholesterol to pregnenolone, occurs within mitochondria. It is catalyzed by the cytochrome P450 side chain cleavage enzyme, P450scc, which is encoded by the nuclear gene CYP11A1 (116). In this very rare disorder, boys with inactivating mutations present with variably ambiguous genitalia and adrenal insufficiency. They are clinically and hormonally similar to boys with mutation of steroidogenic acute regulatory protein (lipoid congenital adrenal hyperplasia), other than the absence of large adrenal glands.
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M-protein, skin lesions), also known as Crow-Fukase syndrome, is a paraneoplastic disorder associated with a plasma cell neoplasm and immunoglobulin G or A monoclonal gammopathy (117), usually in middle-aged adults. Tumors have a heterogeneous genetic profile (118). Nearly all patients have a symmetrical sensorimotor neuropathy with numbness in the feet and hands, and pain. Stroke may occur. Sclerotic bone lesions, polycythemia, and thrombocytosis are common diagnostic findings, and vascular endothelial growth factor levels are very high and correlate with disease activity.
Endocrine abnormalities are present in nearly all cases, although they are often initially overlooked. Patients may develop primary hypothyroidism (36%), primary adrenal insufficiency (5%), mild hyperprolactinemia (24%), elevated insulin-like growth factor 1 levels, vitamin D deficiency, hypocalcemia with secondary hyperparathyroidism and increased bone turnover markers, abnormal glucose metabolism including diabetes (7%), and gonadal dysfunction (36%) (119, 120). In some studies, weight loss, renal dysfunction, or glucocorticoid treatment may have influenced endocrine test results. Nevertheless, male sexual dysfunction is common, and gynecomastia is often present (121, 122). Total and free testosterone levels are generally low while gonadotropin levels vary with evidence for both primary (30%) and secondary hypogonadism (70%). Estradiol levels are sometimes elevated, and increased bioconversion of dehydroepiandrosterone sulfate to estrogens has been reported (123). While the cause of the hypogonadism is unknown, a role for elevated cytokines including vascular endothelial growth factor as well as interleukin 1β, interleukin 6, and tumor necrosis factor α (124, 125) has been proposed. One idea is that cytokines activate the aromatase distal promoter 1.4 in adipose tissue (126) to increase estrogen production (127). Increased circulating estradiol suppresses testosterone production, and prolactin and IGF1 levels may increase. Testosterone treatment should be individualized and may increase the risk for polycythemia (119).
Prader-Willi syndrome (PWS) is a multisystem disorder characterized by infantile hypotonia and failure to thrive, progressive hyperphagia and obesity, dysmorphic features, short stature, learning disability and behavioral problems, and pituitary dysfunction, most notably hypogonadism. Hyperphagia and extreme obesity increase the risk for type 2 diabetes, sleep apnea, and right heart failure, shortening the life span (128).
PWS results from a lack of expression of genes in the region of 15q11.2-11.3. Many genes in this region are subject to genomic imprinting with expression of only the paternally derived allele while expression of the maternal allele is normally silenced by methylation. Over 60% of PWS subjects have a deletion in the paternally inherited allele while most others have inherited 2 copies of the entire, or segments of the maternal chromosome (129). PWS affects males and females equally with a worldwide prevalence of 1:10 000 to 1:30 000. Nearly all cases represent sporadic mutations, and the risk is increased with advanced maternal age.
Endocrine dysfunction is very common in PWS patients and stems in part from hypothalamic dysfunction and pituitary hypoplasia. Nearly all adults have obesity, growth hormone deficiency, and hypogonadism. Diabetes, central hypothyroidism, central adrenal insufficiency, and premature adrenarche are also relatively common (130).
The hypogonadism in PWS is complex and incompletely understood, and is schematized in Figure 3. Hypogonadism appears to reflect both testicular failure and gonadotropin insufficiency (131-135). More than 75% of boys are born with unilateral or bilateral cryptorchidism, an underdeveloped scrotum and/or microphallus. Testosterone and inhibin B levels in minipuberty are at the lower end of the reference range (136) while LH and FSH are high normal, suggesting a functional defect in Leydig and Sertoli cells. Findings on testicular biopsy at orchidopexy (137) varied from nearly normal spermatogenesis to germinal cell aplasia (Sertoli-cell only). Inhibin B levels remain lower than normal in prepubertal boys (138) at which time LH, FSH, and testosterone levels are low, as in normal boys (139). The testosterone response following hCG administration among boys age 1 to 2 years was normal (139). Pubertal development may be absent, delayed, or arrested. PWS adults typically have a small phallus and testes, little body hair, and a high-pitched voice. Testosterone levels are low (134, 140) in part because SHBG levels are low (141). Inhibin B levels in adults are generally below the fifth percentile while FSH levels are often elevated (141-143). LH levels are most often in the reference range but may be suppressed or elevated (140).
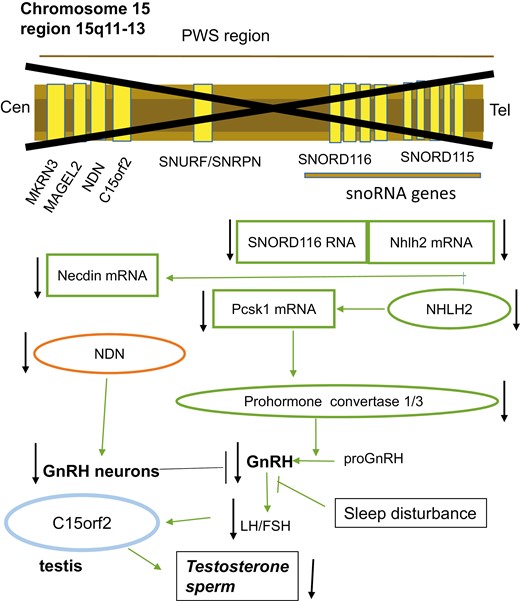
A schema for the hypogonadism in Prader-Willi syndrome (PWS). Deletion in the imprinted region of chromosome 15q11-q13 (marked with an X) may result in loss of function of the nectin gene NDN, C15orf2, and SNORD 116. The transcription factor necdin is abundantly expressed in many developing cells, including neuronal progenitor cells in the hypothalamus where it regulates neuronal development. Mice deficient in necdin fail to develop a full complement of gonadotropin-releasing hormone (GnRH) neurons. C15orf2 is expressed in testis and may be linked to testicular dysfunction. Absence of the paternally derived SNORD116 Sno RNAs is a major determinant of PWS. Normally, the noncoding Sno RNA interacts with targets including Nhlh2 messenger RNA. The enzyme prohormone convertase, encoded by PCSK1, is regulated by NHLH2, and both are reduced in PWS cells, as recently reviewed (177). Deficiency of the PCSK1 product, prohormone convertase 1/3, which processes peptide hormones including GnRH, results in gonadotropin deficiency. Finally, the sleep disturbance of PWS patients may also contribute to hypogonadism.
The cause for primary testicular dysfunction in PWS is uncertain. One candidate gene, C15orf2, located in the PWS region of chromosome 15, is expressed in testis (144). It encodes a predicted 1156-amino-acid protein whose function remains unknown. GnRH deficiency, as well as impaired secretion of other pituitary hormones, appears to result in part from deletion of SNORD116 (C/D box sno RNA; small nucleolar RNA) (145, 146). Downstream consequences of this deleted segment include reduced expression in the hypothalamus and pituitary of the transcription factor NHLH2 (147). One of the NHLH2 targets, proenzyme convertase 1, functions in the posttranslational processing of prohormones and neuropeptides including GnRH and GHRH (148, 149). Decreased expression of proprotein convertase subtilisin/Kexin type 1 and NHLH2 was found in PWS patient-derived stem cells (145). A second proposed mechanism for GnRH deficiency involves necdin, a neuronally expressed gene located on chromosome 15 that is necessary for the full complement of GnRH neurons (150). Finally, sleep disturbance in PWS patients may contribute to low testosterone (151, 152).
Male and female prepubertal children with PWS have higher dehydroepiandrosterone sulfate levels than age-matched normal weight controls, and a few children have levels that exceed the reference range, with premature adrenarche (140, 153, 154). A few cases of precocious puberty in PWS males as well as females have also been reported (155). Loss of function mutations of Makorin ring finger protein 3, a maternally imprinted gene located on the PWS region of chromosome 15 that functions as an E3 ubiquitin ligase are associated with precocious puberty in nonsyndromic patients (156-158). Loss of this gene may be silent in PWS, however, because other genes on chromosome 15 that are required for GnRH production (eg, SNORD116) are also nonfunctional (157).
Orchidopexy is recommended in early life for PWS boys with undescended testes although spermatogenesis and testosterone outcomes in adulthood are below those of nonsyndromic cryptorchidism (159), and PWS men are uniformly infertile. The American Academy of Pediatrics has recommended a trial of hCG before surgical intervention because of surgical risk (160) and because pretreatment with hCG may improve the success of orchidopexy (161). Testosterone replacement is recommended beginning in the teenage years for masculinization and possible beneficial effects on body composition and bone mass (162) although long-term outcome data are lacking. Low doses of testosterone are used initially with cautious dose escalation due to the possibility of stimulating adverse behaviors (133, 134, 162, 163).
Warburg micro and Martsolf syndromes are clinically and genetically overlapping disorders characterized by cataracts, hypotonia, intellectual disability, and progressive spasticity, short stature and hypogonadism, with the latter syndrome designating more mildly affected cases. These disorders are caused by mutations in RAB18, RAB3GAP1, RAB3GAP2, and TBC1D20. RAB3GAP1/2 form a heterodimeric complex that activates RAB18, a GTPase involved in intracellular membrane functions, whereas TBC1D20 shows modest RAB18 activity in vitro (164). RAB18 deficiency may lead to defective autophagic degradation (165). Micropenis and cryptorchidism are common in affected boys (166), suggesting hypogonadotropic hypogonadism. Endocrine test results are limited, however, with results for 2 adult male siblings with small testes and elevated gonadotropins who were the subjects of the initial report by Martsolf (167) and a 12-year-old male and 2 women with hypogonadotropic hypogonadism (168, 169). RAB18 dysfunction may, therefore, cause hypogonadism by affecting both the testis and hypothalamus-pituitary.
RNA polymerase III-related leukodystrophy (4H syndrome) refers to the triad of hypomyelination, hypodontia, and hypogonadotropic hypogonadism (170, 171). Symptoms usually begin with developmental delay in early childhood, with subsequent ataxia and nystagmus, and cognitive impairment. 4H syndrome is an autosomal recessive disorder most often due to biallelic inactivating mutation of POLR3A, POLR3B, BPOL3C, or POLR3K, which encode subunits of RNA polymerase III (172). This enzyme plays a role in the transcription of small noncoding RNAs that function in both transcription and translation. MRI findings include diffuse hypomyelination, cerebellar atrophy, and a thin corpus collosum (171). In an international survey of 150 patients with Pol-III mutations (173), puberty was viewed as delayed or absent in 64% of males and 89% of affected females. Testosterone levels were low in males as were LH levels following GnRH stimulation, beyond which the mechanism for gonadotropin deficiency is unknown. Short stature is also common, with evidence for growth hormone deficiency in a few patients while other pituitary function is generally normal (173). Hypogonadotropic hypogonadism may occur alone with few or no neurological symptoms (174, 175).
Summary and Future Research
This mini review summarizes studies demonstrating the occurrence of hypogonadism in males with a variety of genetic neurological disorders from infancy through adulthood. Many of the disorders discussed herein are very uncommon, even for experts, but are of importance beyond their prevalence because unraveling the disease mechanism provides insight into male reproductive biology. Many of these syndromes are also difficult to distinguish clinically, and others demonstrate a spectrum of phenotypes sometimes associated with mutations in the same or associated genes. Genetic sequencing has changed patient care and is continuously growing in importance as a diagnostic tool for the endocrinologist. An accurate diagnosis is essential to sound genetic advice and counseling, for patient and family education and support, and for treatment initiatives. The molecular mechanisms for hypogonadism in most of the disorders discussed are only partly understood and remain the subject of intense research. Furthermore, information on treatment approaches specific to most disorders is limited, so that coordinated studies to determine benefits and risks of androgen replacement are also needed.
Abbreviations
- ALD
adrenoleukodystrophy
- AMN
adrenomyeloneuropathy
- AR
androgen receptor
- BBS
Bardet-Biedl syndrome
- CIHH
congenital isolated hypogonadotropic hypogonadism
- DM
myotonic dystrophy
- FSH
follicle-stimulating hormone
- GnRH
gonadotropin-releasing hormone
- hCG
human chorionic gonadotropin
- KD
Kennedy disease
- LH
luteinizing hormone
- MRI
magnetic resonance imaging
- mRNA
messenger RNA
- SHBG
sex hormone-binding globulin
- VLCFA
very long chain fatty acids
Acknowledgments
Thank you to Drs. Banu Ayden and Arshpreet Kaur for helpful comments about the manuscript.
Funding
This research was supported in part by the Walter F. and Avis Jacobs Foundation.
Disclosures
The author has no conflict of interest to report.
Data Availability
Data sharing is not applicable to this article as no data sets were generated or analyzed during the present study.

{kind=link}
{kind=link}
{kind=link}