





Abstract
The elderly population is increasing faster than other segments of the population throughout the world. Age is the leading predictor for most chronic diseases and disorders, multimorbidity, geriatric syndromes, and impaired ability to recover from accidents or illnesses. Enhancing the duration of health and independence, termed healthspan, would be more desirable than extending lifespan merely by prolonging the period of morbidity toward the end of life. The geroscience hypothesis posits that healthspan can be extended by targeting fundamental aging mechanisms, rather than attempting to address each age-related disease one at a time, only so the afflicted individual survives disabled and dies shortly afterward of another age-related disease. These fundamental aging mechanisms include, among others, chronic inflammation, fibrosis, stem cell/ progenitor dysfunction, DNA damage, epigenetic changes, metabolic shifts, destructive metabolite generation, mitochondrial dysfunction, misfolded or aggregated protein accumulation, and cellular senescence. These processes appear to be tightly interlinked, as targeting any one appears to affect many of the rest, underlying our Unitary Theory of Fundamental Aging Mechanisms. Interventions targeting many fundamental aging processes are being developed, including dietary manipulations, metformin, mTOR (mechanistic target of rapamycin) inhibitors, and senolytics, which are in early human trials. These interventions could lead to greater healthspan benefits than treating age-related diseases one at a time. To illustrate these points, we focus on cellular senescence and therapies in development to target senescent cells. Combining interventions targeting aging mechanisms with disease-specific drugs could result in more than additive benefits for currently difficult-to-treat or intractable diseases. More research attention needs to be devoted to targeting fundamental aging processes.
The elderly comprise the fastest growing segment of the population across the developed and developing worlds. Aging is linked to an exponentially increasing risk of morbidity and mortality across species from fish to humans. Targeting fundamental aging mechanisms and delaying age-related declines in function and increases in chronic diseases, morbidity, and mortality is now possible, at least in experimental animals, by dietary interventions, genetic manipulations, and drugs. These interventions show early promise for extending healthspan in experimental animals, with coordinated delays in multiple age-related diseases. Because aging processes are generally conserved across the vertebrates, these interventions might be more readily translatable into humans than interventions for human diseases developed in animal models (eg, mice) artificially created to resemble a particular disease.
Aging is the leading predictor for most chronic diseases and disorders, metabolic dysfunction, multimorbidity, geriatric syndromes (eg, immobility, cognitive impairment, or incontinence), and decreased ability to recover from injuries or illnesses (decreased physical resilience; frailty). The prevalence of multimorbidity (simultaneous presence of 3 or more chronic disorders within an individual) increases exponentially during the latter phase of the lifespan (1). Perhaps aging is even a root-cause contributor to many or most of these disorders. Enhancing healthspan, the period of life free of disabilities, diseases, and dependencies, would for many people be more desirable than extending lifespan at all costs through prolonging the period of disability and morbidity toward the end of life. The geroscience hypothesis posits that by targeting fundamental aging mechanisms, rather than targeting age-related diseases one at a time, healthspan could be extended (Fig. 1). For example, preventing death from a heart attack only so the afflicted individual survives disabled and dies shortly afterward of another age-related disease such as cancer or dementia would add little to healthspan, quality of life, or lifespan.
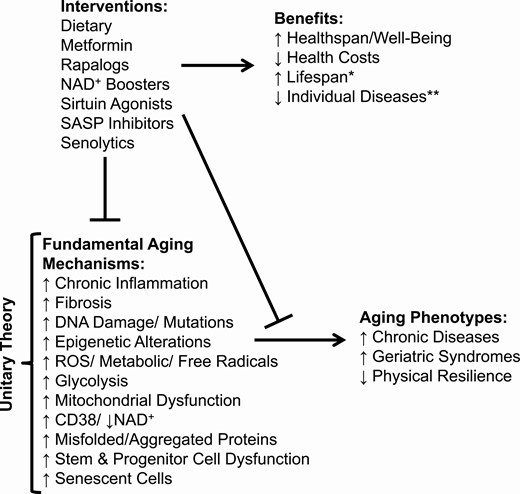
The Geroscience Hypothesis. *↑Lifespan is a possible by-product of ↑healthspan. **Interventions targeting fundamental aging mechanisms combined with disease-specific interventions can perhaps achieve an additive or even more than additive benefit.
Fundamental Aging Mechanisms and the Unitary Theory
Fundamental aging mechanisms can be divided into categories, the “pillars of aging” (2) (see Fig. 1). These aging processes are operative across most vertebrate species. They include, among others, chronic low-grade inflammation, fibrosis, stem and progenitor cell dysfunction with reduced tissue regenerative capacity, DNA damage and mutations, epigenetic changes, metabolic shifts (a partial Warburg shift toward reliance on glycolysis), destructive metabolite generation (reactive oxygen species [ROS], saturated ceramides, etc), mitochondrial dysfunction, decreased nicotinamide adenine dinucleotide (NAD+; related to increased macrophage CD38 and decreased sirtuin activity (3)), increased misfolded or aggregated proteins (including α-synuclein, β-amyloid, tau), immune dysfunction, and senescent cell accumulation.
Fundamental aging processes may be interlinked. For example, cellular senescence leads to inflammation, fibrosis, DNA damage, mitochondrial dysfunction, ROS generation, NAD+ depletion, protein aggregation, failed autophagy, lipotoxicity, and stem/progenitor cell dysfunction. In turn, cellular senescence can be caused by other fundamental aging processes, including DNA damage, low NAD+, mitochondrial dysfunction, ROS, high glucose, protein aggregates, and lipotoxicity. These findings led us to formulate a Unitary Theory of Fundamental Aging Processes (see Fig. 1), positing that interventions, such as senolytics, that target any one of these processes, such as cellular senescence, could affect many or perhaps all the rest. Here, we will use cellular senescence, its deleterious effects, and strategies targeting senescent cells as examples to illustrate current efforts to develop interventions that extend healthspan by delaying age-related disorders and diseases as a group, instead of one at a time.
The Example of Cellular Senescence
Senescence entails cell cycle arrest with maintenance of metabolic activity, and is essentially a cell fate, like differentiation, proliferation, apoptosis, or necrosis (4-6). External and internal signals can contribute to driving a cell into senescence. These are generally cell or tissue damage-related, including DNA alterations (dysfunctional telomeres, strand breaks, etc), metabolic dysfunction (ROS, high glucose, bioactive lipids, decreased NAD+, mitochondrial dysfunction), protein alterations (aggregates, misfolded proteins, failed autophagy), inflammatory signals, mechanical/shear stress, pathogen-associated molecular pattern factors (viruses, bacterial/fungal proteins, lipopolysaccharides, etc), damage-associated molecular pattern factors (tissue damage signals [extracellular nucleotides, etc]), intracellular damage signals (DNA damage, telomeric dysfunction, mitochondrial dysfunction), oncogenes, and mitogens (insulin-like growth factor-1 [IGF-1], etc). Once initiated, senescence takes 10 days to 6 weeks to become established through transcription factor cascades that may, but do not always, include p16INK4a/ retinoblastoma protein and/or p53/p21CIP1. This causes extensive changes in gene expression, histone modifications, altered organelle function (eg, mitochondria, endoplasmic reticulum, nucleolus, nuclear envelope), elevated protein production due to increased mTOR (mechanistic target of rapamycin) and decreased autophagy, and profound morphological and metabolic shifts (6). After drug-induced removal, senescent cells take from 10 days to 6 weeks to reoccur if cellular senescence inducers remain present, at least in cell culture.
Accumulation of senescent cells can cause local and systemic inflammation, tissue destruction, immune system inhibition, and stem and progenitor cell dysfunction due to their senescence-associated secretory phenotype (SASP) (6-9). Generally 30% to 70% of senescent cells develop an SASP that entails release of proinflammatory, proapoptotic cytokines (tumor necrosis factor α [TNFα], interleukin-1α [IL-1α], IL-6, IL-7, IL-8, interferon γ, and many more), chemokines (eg, monocyte chemoattractant protein‐1 [MCP-1], macrophage inflammatory protein 1α [MIP1-α], RANTES, RARRES2) that attract, activate, and anchor immune cells (including macrophages, dendritic cells, T-lymphocyte subsets, and neutrophils), tissue-destroying proteases (matrix metalloproteinase [MMP]-3, -9, -12, etc), procoagulant factors (plasminogen activator inhibitor-1 [PAI-1], etc), factors that impede stem cell/progenitor function and drive fibrosis (activin A, transforming growth factor-β–related proteins), ferritin, growth factors (eg, granulocyte colony-stimulating factor), bioactive lipids (bradykinins, saturated ceramides, prostanoids), microRNAs, tissue-damaging noncoding nucleotides (including mitochondrial DNA, which attracts, activates, and anchors dendritic cells), microvesicles including exosomes, and other factors that cause tissue necrosis, systemic inflammation, stem cell/progenitor dysfunction, fibrosis, and spread of senescence to nonsenescent cells.
Likely owing to their SASP, a small number of senescent cells can cause considerable dysfunction. In preclinical experiments, transplanting senescent cells around the knee joints of young mice was sufficient to induce an osteoarthritis-like phenotype, whereas transplanting nonsenescent cells did not (10). Transplanting 106 radiation- or chemotherapy-induced senescent autologous ear fibroblasts or syngeneic adipocyte progenitors intraperitoneally into lean, adult mice, so that fewer than 1/10 000 of all cells in the transplanted mice were senescent, induced physical dysfunction (frailty) and premature death due to early onset of the same age-related diseases that cause death in naturally aged mice (10, 11). This also occurred after human senescent (vs nonsenescent) cells were transplanted into immune-deficient mice. When labeled senescent cells were transplanted intraperitoneally, they remained within the peritoneum, yet the transplant recipients developed nonlabeled senescent cells in their limbs, demonstrating that senescence can spread from cell to cell, even at a distance in an “endocrine” fashion (10). An example of this might be preeclampsia, in which senescent cells or their products may escape from the placenta and seed or drive senescence in the mother’s vascular-wall endothelial cells (12). This spread of senescent cells to distant sites lends support to the view that systemic senolytics, if safe, could lead to better overall healthspan outcomes than administering senolytics locally, for example, injection into a joint, particularly if that joint is already sufficiently damaged that the injected senolytic escapes into the circulation anyway.
Cellular senescence contributes to age-related dysfunction and multiple diseases throughout the lifespan (5). Senescent cell burden is low in young individuals but increases with aging in several tissues, including adipose tissue, lung, skeletal muscle, heart, kidney, brain, bone, and skin (5, 6, 13-15). Senescent cells accumulate at sites of pathology in multiple chronic diseases, such as the lungs in chronic obstructive pulmonary disease, asthma, idiopathic pulmonary fibrosis, and smoking; adipose and other tissues in obesity and type 2 diabetes; the brain in Alzheimer and other dementias; and blood vessels and the heart in cardiovascular disease.
Senescent cells can also have beneficial effects, for example, in clearing dead tissue during wound healing or after injury, in decreasing the spread of cancer (cells with cancerous mutations or expressing oncogenes can become senescent, slowing cancer development and spread), for remodeling tissues during prenatal and postnatal development, and, in the placenta during childbirth, in releasing cytokines that drive parturition (16). Thus, unlike clearing already generated senescent cells with a proinflammatory SASP, interfering with senescent cell generation would not likely be beneficial. Indeed, mutations of p16INK4A or p53, which can be involved in the generation of senescent cells, predisposes to cancers, whereas removing already generated senescent cells from mice delays multiple forms of cancers with aging, in tandem with delaying a range of other age-related diseases (10).
Interventions Targeting Fundamental Aging Processes
Interventions targeting fundamental aging processes are being developed, including some in early human trials. These include dietary manipulations (eg, caloric restriction, intermittent fasting), exercise, metformin, mTOR inhibitors (“Rapalogs” such as rapamycin), NAD+ precursors (eg, nicotinamide adenine dinucleotide, nicotinamide riboside), sirtuin agonists (eg, resveratrol, pterostilbene), SASP inhibitors, and senolytics. Some of these interventions may act to target several fundamental aging mechanisms. For example, metformin, which is under investigation as an agent to improve healthspan, seems to affect the SASP, mTOR activity, as well as mitochondrial function (17). Exercise has been shown to prevent obesity-induced senescent cell formation in mice (18); however, it reduced inflammation without affecting senescent cell burden in a human study of aged cyclists (19). In terms of targeting senescent cells, the effects of SASP inhibition would be expected to be less durable than those of senolytics, which trigger apoptosis of senescent cells leading to their overt removal from tissues. Because of this, senolytics have been the focus of most efforts toward clinical translation of strategies to target senescent cells. Therefore we will review the development and possible promise of senolytics as an example here.
In 2004 Krishnamurthy et al(20) showed senescent cell accumulation is delayed in Ames dwarf mice (with pituitary hormone deficiencies, including low growth hormone [GH] and, in turn, low IGF-1) and calorically restricted mice, models with increased healthspan and lifespan. This prompted us to test the hypothesis that targeting senescent cells may alleviate multiple age-related disorders. Efforts to discover senolytics, drugs that selectively eliminate senescent cells, began in 2004 to 2005 employing traditional approaches, including creating fusion proteins comprising a senescent cell–binding domain coupled to a toxin and high-throughput compound library screens. These approaches were not initially successful, so a hypothesis-driven discovery paradigm was turned to for discovering senolytics (6, 21, 22). Since senescent cells kill cells around them and damage tissues because of their inflammatory, apoptotic SASP, yet senescent cells do not die, the hypothesis was tested that prosurvival networks defend senescent cells against their own SASP: senescent cell antiapoptotic pathways (SCAPs) (15, 21-23).
From proteomic/transcriptomic databases and using bioinformatics methods, the SCAP network was discovered (21). RNA interference analyses demonstrated that transiently disabling this SCAP network leads to apoptosis of senescent cells, whereas normal cells without a SASP were unaffected, the “Achilles heel” of senescent cells. Bioinformatics approaches were used to identify agents that act on critical SCAP nodes. Among these were fisetin, quercetin (Q), dasatinib (D), and now more than 20 others that selectively eliminate senescent cells in mice and humans (15).
The target of senolytics is senescent cells, not a single receptor, enzyme, or biochemical pathway. By targeting prosurvival networks instead of single molecules, specificity for senescent cells can be increased, side-effect profiles flattened, and off-target effects on nonsenescent cells reduced. Successful strategies for developing senolytics appear to have more in common with approaches for developing antibiotics than the conventional one-drug/one-target/one-disease drug development approach. Senolytic drugs can be administered via intermittent dosing and do not interfere with generation of senescent cells when they are needed, such as to mitigate cancer development. Intermittent dosing reduces the risk of side and off-target effects and also allows the avoidance of clearing senescent cells during wound healing, tissue remodeling, or pregnancy, when they can have beneficial effects.
The Immune Threshold Theory of Senescent Cell Burden
Generally, senescent cells are cleared by the immune system (9). They can attract, activate, and anchor immune cells, including macrophages, dendritic cells, T lymphocytes, and neutrophils through SASP chemokines (eg, MCP-1, MIP1-α, RANTES, and RARRES2), cytokines such as IL-6, and TNFα, extracellular mitochondrial DNA, possibly microRNAs, and other factors (9, 10, 24). However, above a threshold burden, senescent cells might interfere with the immune system and its ability to remove senescent cells. For example, IL-6, an SASP component, interferes with macrophage migration, MMPs can cleave FAS ligand and other immune cell surface proteins, senescent cells cause fibrosis that could impede immune cell infiltration and trap immune cells within inflammatory foci, and senescent cells can express “don’t eat me” signals (9). Based on these findings, we formulated a threshold theory of senescent cell burden: Once senescent cell abundance is sufficient to cause spread of senescence that exceeds capacity of the immune system to keep up with clearing these cells, an accelerated aging-like state ensues (9-11). Consistent with this are the following: 1) the number of senescent cells that needs to be transplanted to cause frailty, limit healthspan, and cause premature death, including from cancer, is higher in young than old mice and in young lean than young obese mice. Old mice or young, obese mice have more preexisting senescent cells than young, lean mice (10). 2) The abundance of presenescent and senescent adipose progenitors with limited replicative remains low until early old age in mice, followed by an upward inflection in older mice (25). 3) The lag between induction of senescence by chemotherapy and development of age-related morbidities is longer in childhood cancer survivors than adults who received higher chemotherapy doses in preparation for bone marrow transplantation (26). These findings support the hypothesis that there is a threshold above which senescent cell burden due to spread of cellular senescence becomes self-amplifying. This self-amplification could contribute to an increased risk of senescence- and age-related phenotypes, physical dysfunction/frailty, hyperinflammation, and diseases, perhaps contributing to age-related multimorbidity (1). Therefore, reducing systemic senescent cell burden to below a threshold might allow the immune system to clear more senescent cells, including those remaining senescent cells that are resistant to the senolytic drug administered, and to extend healthspan.
Potential of Senolytics for Endocrine Disorders
Senescent cells, particularly senescent adipocyte progenitors, accumulate in the adipose tissue of obese and diabetic mice and humans, and removal of senescent cells via the senolytic drug combination D + Q has been shown to improve metabolic function in obese mice (24). Among other senescent cell types, D + Q targets senescent human adipocyte progenitors, unlike most other senolytics to date. Age-related metabolic dysfunction is also associated with increased adipose tissue senescent cell burden that can be reduced by senolytic administration in mice (14). These improvements are mediated at least in part by decreased adipose tissue inflammation, increased adipose tissue and peripheral insulin sensitivity, and enhanced ability of progenitor cells to differentiate into insulin-responsive adipocytes (14, 24). Senescent cells also accumulate in the pancreas both in type 1 and type 2 diabetes, as well as with aging. This β-cell senescence has been proposed to promote insulin secretion, and aged human β cells have increased glucose-induced insulin secretion (27). However, when the burden of senescent pancreatic β cells in mice is reduced by treatment with navitoclax (which we discovered is senolytic for some, but not all cells types; it does not target senescent adipocyte progenitors [28]), insulin production is increased.
Clinical trials are needed to determine the overall effect of senolytics on type 2 diabetes in humans, given that impacts on insulin secretion as well as insulin sensitivity in peripheral tissues are expected based on preclinical studies. It is possible that senolytic therapies will need to be used or combined for each disease indication according to their cell type specificity, to optimize their therapeutic effects. Similarly, human studies are needed to determine the impact of preclinical findings in nonobese diabetic mice, which suggest that targeting senescent β cells may prevent type 1 diabetes progression by interfering with the immune destruction of β cells (29). In a small, proof-of-concept clinical trial, we showed that D + Q reduces p16INK4A- and senescence-associated β-galactosidase–expressing cells, indicating decreased senescent cell abundance, in abdominal subcutaneous adipose tissue of obese, diabetic individuals with renal dysfunction 11 days after the last of 3 daily doses of D + Q compared to biopsies before senolytics were administered (30, 31). Adipose tissue inflammatory cell infiltration (macrophages) and fibrosis (crown-like structures) were reduced by senolytics, as was a composite of circulating inflammatory SASP factors.
Regarding age-related osteoporosis, senescent cells accumulate in bone with aging both in mice and humans (32). Elimination of senescent cells from p16Ink4a-INK-ATTAC mice led to reduced bone resorption, enhanced differentiation of progenitor cells toward the osteoblast lineage, and increased bone strength. Based on these studies, a clinical trial is under way (NCT04313634) to investigate the effect of the senolytics, D + Q or fisetin vs placebo, on bone resorption and bone formation in elderly women.
GH-resistant or -deficient mice, which have increased longevity, have reduced senescent cell burden. Transgenic mice with increased IGF-1 levels due to expression of bovine growth hormone, conversely, exhibit premature aging and have increased cellular senescence. Prolonged IGF-1 exposure in vitro was also shown to induce cellular senescence in a p53-dependent manner. We found that long-term increased GH exposure in GH-overexpressing mice or due to prolonged GH treatment results in increased senescent cell burden, and the converse happens with decreased GH signaling (33). Whether increased senescent cell burden occurs in humans with acromegaly, which is associated with early death and age-related disease onset (34), or whether decreased senescent cell burden occurs in humans with Laron dwarfism, who have decreased IGF-1 signaling and delayed age-related disease onset (35), remains to be seen.
Although the role of senescent cells in autoimmune thyroid disease has not been widely explored, it has been postulated that cellular senescence may play a role in the development of radiation-induced hypothyroidism, given that radiation is a potent and well-known trigger of cellular senescence. More study is needed to determine whether senolytics could be given safely to prevent the development of postradiation hypothyroidism.
Different senolytic drugs may have vastly different effects within the same disease model, depending on their cell type specificity. For example, senolytic drugs that have single or limited targets, such as navitoclax (ABT-263) (28, 36) or Nutlin3a, tend to have multiple off-target apoptotic effects on multiple nonsenescent cell types, making them “panolytic.” Navitoclax, a BCL-2/BCL-w/BCL-xL inhibitor, eliminates only a restricted range of senescent cells, for example, navitoclax is not senolytic against human adipocyte progenitors, one of the most abundant senescent cell types in aged humans or people with diabetes and obesity (24). Also, whereas Navitoclax decreases bone-senescent cells, it is toxic to osteoblasts, making osteoporosis worse, unlike D + Q, which alleviates osteoporosis (37).
In addition to the conditions listed earlier, senolytics have shown promise in preclinical studies of multiple other conditions in mice, including cardiovascular disorders, cognitive impairment, pulmonary dysfunction, frailty, kidney and liver dysfunction, and osteoarthritis, and delay cancer and extend healthspan and lifespan (5, 6, 15) (see Fig. 1).
Preclinical studies have laid the groundwork for early clinical trials in which senolytic drugs have been administered to humans. As noted previously, in patients with diabetic kidney disease, D + Q reduced adipose tissue–senescent cell abundance (31). A different study showed that D + Q reduces frailty in individuals with idiopathic pulmonary fibrosis, a fatal condition related to lung–senescent cell accumulation (31, 38). Additional senolytic clinical trials are under way or beginning for preventing COVID-19 complications, sequelae of bone marrow transplantation and childhood cancer treatment, Alzheimer disease, and osteoarthritis, among other conditions.
Conclusions
Targeting age-related diseases one at a time may add only modestly to healthspan. Targeting fundamental aging mechanisms, such as with senolytics to remove senescent cells, could lead to a far greater extension of healthspan. Because fundamental aging processes may contribute to earlier onset of multiple diseases, perhaps more than additive benefits for currently difficult-to-treat or intractable disorders can be achieved by combining interventions targeting the underlying fundamental aging processes with disease-specific drugs. Could, for example, a life-threatening condition such as diastolic dysfunction causing heart failure, for which disease-specific drugs are so far modestly effective at best, be alleviated by combining these disease-specific agents with senolytics? Could diabetes and its complications be prevented by slowing the accumulation of senescent cells in obesity and aging such as with a combination of exercise and senolytics? Based on the increasing burden of age-related, intractable, chronic disorders and diseases, more research needs to be devoted to developing interventions that target fundamental aging processes and translating these interventions, which are beginning to show early signs of promise, into treatments to reduce morbidity and increase healthspan.
Abbreviations
- D
dasatinib
- GH
growth hormone
- IGF-1
insulin-like growth factor-1
- mTOR
mechanistic target of rapamycin
- NAD+
nicotinamide adenine dinucleotide
- ROS
reactive oxygen species
- SASP
senescence-associated secretory phenotype
- SCAP
senescent cell antiapoptotic pathway
- Q
quercetin
Acknowledgments
Financial Support: This work was supported by the National Institutes of Health (Grants R37AG013925 and P01AG062413), the Translational Geroscience Network (Grant R33AG061456), the Alzheimer’s Association Part the Cloud Program, Robert and Arlene Kogod, the Connor Group, Robert J. and Theresa W. Ryan, and the Noaber Foundation.
Additional Information
Disclosure Summary: T.T., A.K.P., and J.L.K. have a financial interest related to this research. Patents on senolytic drugs are held by Mayo Clinic. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic Conflict of Interest policies.
Data Availability
All data generated or analyzed during this study are included in this published article or in the data repositories listed in “References.”
References

{kind=link}