





Abstract
Multiple endocrine neoplasia type 1 (MEN1)-related neuroendocrine tumors (NETs) of the lung are mostly indolent, with a good prognosis. Nevertheless, cases of aggressive lung NET do occur, and therefore the management of individual patients is challenging.
To assess tumor growth and the survival of patients with MEN1-related lung NETs at long-term follow-up.
The population-based Dutch MEN1 Study Group database (n = 446) was used to identify lung NETs by histopathological and radiological examinations. Tumor diameter was assessed. Linear mixed models and the Kaplan-Meier method were used for analyzing tumor growth and survival. Molecular analyses were performed on a lung NET showing particularly aggressive behavior.
In 102 patients (22.9% of the total MEN1 cohort), 164 lesions suspected of lung NETs were identified and followed for a median of 6.6 years. Tumor diameter increased 6.0% per year. The overall 15-year survival rate was 78.0% (95% confidence interval: 64.6–94.2%) without lung NET-related death. No prognostic factors for tumor growth or survival could be identified. A somatic c.3127A > G (p.Met1043Val) PIK3CA driver mutation was found in a case of rapid growing lung NET after 6 years of indolent disease, presumably explaining the sudden change in course.
MEN1-related lung NETs are slow growing and have a good prognosis. No accurate risk factors for tumor growth could be identified. Lung NET screening should therefore be based on well-informed, shared decision-making, balancing between the low absolute risk of an aggressive tumor in individuals and the potential harms of frequent thoracic imaging.
Multiple endocrine neoplasia type 1 (MEN1) is a rare autosomal dominant disorder caused by loss-of-function of the MEN1 gene, a tumor suppressor gene encoding the protein menin (1). Patients with MEN1 are predisposed to the development of various endocrine tumors at a young age, with primary hyperparathyroidism due to parathyroid adenomas, neuroendocrine tumors (NETs) of the pancreas and duodenum, and pituitary adenomas being the most common, so-called major manifestations. MEN1 patients are also at risk of adrenal tumors, lung NETs, thymic NETs and gastric NETs. Nonendocrine tumors such as angiofibromas, lipomas, leiomyomas, meningiomas, and probably breast cancer are also recognized as manifestations of the syndrome (2–5).
Lung NETs are reported in 4.7% to 31.3% of MEN1 patients, depending on whether the diagnosis was histopathologically proven or based on a combination of histopathological and radiological examinations, respectively (6–10). Clinical practice guidelines advise annual or biannual screening for lung and thymic NET by thoracic computed tomography (CT) scan or magnetic resonance imaging (MRI) scan, although the frequency of imaging is debated (11, 12).
Outcomes of previous studies suggest that MEN1-related lung NETs are associated with a relatively indolent course and a good prognosis. Growth analysis by our group showed a 17% tumor diameter growth per year (tumor doubling time, 4.5 years), with a median patient follow-up of 3.3 years. Tumor doubling time appeared to be shorter in males compared with females (2.5 vs 5.5 years) (7). Similar results were reported from other MEN1 cohorts, further confirming a benign natural course of disease (8, 9).
However, despite the indolent course of lung NETs in growth analyses, aggressive and fatal cases of lung NET do occur. Aggressive lung tumors, including large cell neuroendocrine carcinomas (LCNECs) and small cell neuroendocrine carcinomas with lethal consequences were described in 7 MEN1 patients in a recent French study of the Groupe d’étude des Tumeurs Endocrines (GTE). However, given the large cohort size of 1023 MEN1 patients, the long-term follow-up of median 48.7 years, high frequency of smokers, and lack of molecular analyses, a causal relationship with MEN1 syndrome was unclear (10).
The aggressive tumor behavior in some patients raises questions whether lung NETs truly remain indolent over the course of longer follow-up, and which factors associate with aggressive tumor biology. In this respect, of potential interest are additional somatic mutations that can drive accelerated tumor growth and smoking status, because high-grade NETs were more frequently diagnosed among smokers in the above-mentioned French GTE study.
The aims of this study were to assess growth patterns and survival of MEN1-related lung NETs during longer-term follow-up and to identify risk factors for tumor growth and survival. Moreover, we tried to elucidate the unexpected aggressive course of a lung NET in an individual patient with sudden accelerated growth and aggressive biological behavior at the molecular level.
Methods
Study design and patient selection
Patients were selected from the Dutch national MEN1 database of the Dutch MEN1 Study Group (DMSG). This longitudinal database—which includes >90% of the Dutch MEN1 population—includes all MEN1 patients ≥16 years of age at the end of 2017 under treatment at 1 of the Dutch university medical centers (UMCs) between 1990 and 2017. MEN1 diagnosis was established following current international guidelines (11). Using a predefined protocol, clinical and demographic data were collected from 1990 to 2017 by a standardized medical record review. Detailed information on the DMSG database methods have been described previously (13). The study protocol was approved by the medical ethical committees of all UMCs.
As previously described, patients with lung NETs or lung lesions suspect of lung NETs were identified based upon histopathological and radiological findings (7). All pulmonary lesions on CT or MRI scan were reviewed to select potential lung NETs. Nodules were suspected of being a lung NET based on the report from a senior radiologist and confirmation in follow-up scans. In case of doubt, individual cases were discussed (M.B., J.L., G.V.). Potential lung metastases from other NETs were excluded on histological and/or radiological grounds. Contralateral lung NETs and ipsilateral recurrence of lung NETs after surgery were considered separate lung NETs for the growth analysis.
Outcome
The primary outcomes were the growth rate of lung NETs (measured in the percentage of increase of the largest tumor diameter) and all-cause mortality. The potential influence of gender, smoking status, age at lung NET diagnosis, and baseline tumor size on growth rate and survival was evaluated. Previously reported genotype–phenotype associations in other cohorts were also assessed: genotype was dichotomized according to the type of mutation (missense vs nonsense/frameshift), interacting domain (JunD, CHES1) and a combination of exon and type of mutation (nonsense and frameshift mutations in exons 2,9,10) (14–17). Furthermore, we studied the effect of tumor classification and stage—based on the Classification of Malignant Tumours (TNM) and the World Health Organization Classification of Tumours of the Lung, Pleura, Thymus and Heart (2015)—and the effect of lung surgery on survival (18, 19). Histopathological tumor characteristics (size, mitotic index, lymph node status), type of surgery, and follow-up status of histopathologically proven lung NETs were reported.
Statistical analysis
Tumor growth was studied using multilevel, linear mixed models analysis, accounting for clustering of observations within distinctive lung tumors (eg, left- and right-sided tumors) within patients. Follow-up time (years) started at the time of lung NET diagnosis. Due to the violation of model assumptions (ie, abnormal distribution of residuals), logarithmic transformed lung NET diameter was used as a dependent variable. Because current management recommendations advise surgical resection of lung NETs ≥ 20 mm upon discovery, tumors with a baseline size ≥20 mm in diameter were excluded from growth analysis (20). Possible effect modification was assessed for gender, genotype, smoking status, age at lung NET diagnosis, and baseline tumor size.
Survival analysis was performed using Kaplan-Meier plots. The time from diagnosis of lung NET until death, lost to follow-up, or the end of follow-up was included for analysis. The effect of gender, genotype, smoking status, baseline tumor size, surgery, World Health Organization classification, and lymph node involvement on survival was determined with log-rank tests.
Continuous variables are presented as mean value and standard deviation or median and interquartile range (IQR) when appropriate. Categorical variables are described as percentages. Comparisons between groups were performed using the chi-square test for categorical variables and a Student’s t-test (normal distribution) or Mann-Whitney U test (not normal distribution) for continuous data. Statistical significance was set at P < 0.05.
Investigations of the tumor showing accelerated growth
One patient with accelerated tumor growth and aggressive tumor behavior is described in more detail. Several genetic analyses were performed: next generation sequencing (NGS) was performed using Ion Ampliseq (Ion Torrent) with a custom-made panel used for analysis of lung tumors (genes specified in the Supplementary Material; all supplementary material and figures are located in a digital research materials repository) (21). Whole genome sequencing (WGS) on fresh-frozen tissue was performed at the Hartwig Medical Foundation according to all international standards (reference genome version GRCh37) (22). Copy number variation analysis was based on single nucleotide peptide data using the Infinium CytoSNP-850K BeadChip version 1.2 array and deoxyribonucleic acid (DNA) methylation data generated by the Illumina MethylationEPIC array platform, which was analyzed with R package “Conumee” (23). By using a purity ploidy estimator on the WGS data, the copy number profile of the tumor was assessed in more detail (24). Additionally, ribonucleic acid (RNA) was isolated and processed to investigate possible receptor tyrosine kinase mesenchymal-epithelial transition factor (cMET) exon 14 skipping. The possibility of a translocation in the rearranged translocation proto-oncogene (RET gene) was explored using fluorescence in situ hybridization (FISH). Furthermore, the presence of alternative lengthening of telomeres was studied by FISH, and loss of alpha-thalassemia or mental retardation syndrome X-linked protein (ATRX) and death domain-associated protein (DAXX) expression was immunohistochemically determined, as previously described (25). Likewise, menin immunohistochemistry was performed using recombinant antimenin antibody GeneTex EPR3986.
Results
Longitudinal cohort study
A total of 446 patients (247 female, 55.4%) were included in the DMSG database by the end of 2017. The median age at MEN1 diagnosis was 37 years (range 4–82 years). The diagnosis of MEN1 was confirmed by a pathogenic MEN1 mutation in 355 cases (79.6%) and 38 patients (8.5%) were obligate carriers of the familial occurring pathogenic MEN1 mutation because they had at least 1 major MEN1-associated tumor in combination with a first-degree relative with confirmed MEN1 mutation. A total of 53 patients (11.9%) were diagnosed on clinical grounds (2/3 major MEN1-associated tumors). In 51 of those patients, genetic analysis showed no pathogenic MEN1 mutation (11.4%). A CDKN1B mutation was found in 3 of these 51 patients. In the 2 remaining patients diagnosed on clinical grounds, no genetic analysis was performed.
Periodic screening for lung NETs by means of interval thoracic CT scan was performed in 352 patients (78.9%). Patients who underwent CT examination did not differ from the rest of the MEN1 cohort in terms of gender, smoking status, and genotype. Pulmonary nodules were detected in 177 patients (50.3% of patients who were under periodic screening). A lung NET was excluded in 75 patients based on pathology results (n = 5), radiological evidence of metastatic origin of the lesion (n = 15), radiological evidence of another (benign) origin of the lesion (n = 19), or lack of confirmation on follow-up imaging (n = 36). See Fig. 1 for the full flowchart. A total of 164 lesions suspect of lung NET in 102 patients (22.9% of the entire cohort) were therefore included in the analysis.
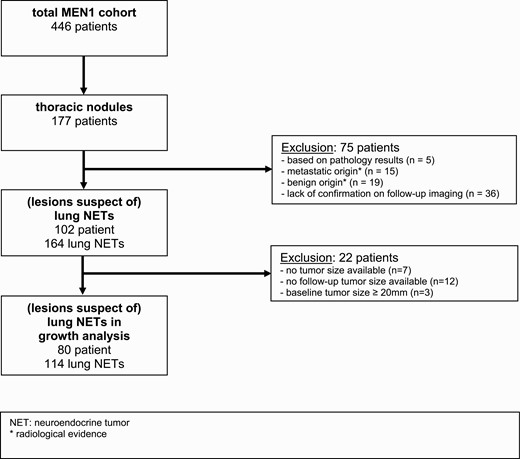
Flowchart of patient selection. Abbreviatons: MEN1, Multiple Endocrine Neoplasia type 1; NET, neuroendocrine tumor.
Histopathological and clinical characteristics
Lung NETs were diagnosed based on the combination of radiological and histopathological findings in 29 patients (6.5% of the entire cohort, 28.4% of patients included in the analysis) and were highly suspected of lung NET solely on radiological evidence in 73 patients (71.6% of patients included in the analysis). Lung NETs were diagnosed at a median age of 43 years (IQR 38–57 years). Patients with lung NETs were more frequently female (n = 61, 59.8%), reflecting the overall gender distribution within the cohort. There was no significant difference in smoking status (29.0% vs 37.3%, respectively) or genotype between patients with lung NETs and the other MEN1 patients. The prevalence of lesions suspect of lung NET was comparable between patients with a confirmed pathogenic MEN1 mutation (25.4%) and familial cases who were an obligate carrier (23.7%). In contrast, a lesion suspect of lung NET was found in only 3/53 (5.7%) clinically diagnosed MEN1 patients (2/3 major MEN1-associated tumors) without MEN1 mutation. In this patient group, 1 lesion was found in 1/3 patients with a CDKNB1 mutation, 1 lesion in 1/48 (4.8%) patients in whom genetic analysis showed neither a MEN1 nor a CDKN1B mutation, and 1 lesion in 1/2 patients in whom genetic analysis was not performed.
Median follow-up time from lung NET diagnosis until the end of follow-up (death, lost to follow-up, or the end of the study) was 6.6 years (IQR 3.4–9.1 years, range: 0.5–38.0 years). The clinical and histopathological characteristics of all patients with a pathological diagnosis of lung NET are shown in Table 1. Tumor size was <15 mm without accelerating growth in only 4 patients who underwent surgery. Histopathological examination showed a typical carcinoid in 20 patients and an atypical carcinoid in 8 patients. The mitotic index was >5 in only 2 cases. In addition, there was 1 case with high-grade neuroendocrine neoplasm (patient 20), which was difficult to classify as either atypical carcinoid or LCNEC (see results, description of the case with an exeptional tumor course).
Clinical, genetic, and histopathological characteristics of patients with pathological diagnosis of lung NET
| Patient | Sex | Genetic Mutation | Smoking | Age at Diagnosis (yr) | Type of Surgery | Lymfadenectomy (Positive/Total Number of Lymph Nodes) | Tumor Size on PA (mm) | Mitotic Index (per 10 hpf) | WHO Classification | TNM Classification | Length of Follow-up (yr) | Follow-up Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | FS | 44 | Wedge resection lower left lobe | N | 7 | 1 | Typical carcinoid | pT1cN0M0 | 9.2 | Nodule ≥10 mm IL |
| 2 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 62 | Wedge resection lower left lobe | Y (0/ND) | 23 | 0 | Typical carcinoid | pT1N0cM0 | 13.2 | No lung lesions |
| 3 | F | Frameshift exon 10: c.1430dupG (p.Glu478fs) | NS | 42 | Wedge resection upper left lobe | N | 5 | ND | Typical carcinoid | pT1c(m)N0M0 | 14.0 | Nodule <10 mm IL, nodule ≥10 mm CLd |
| 4 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | NS | 42 | Wedge resection upper left lobe | N | 14 | 0 | Typical carcinoid | pT1N0M0 | 8.8 | Nodules <10 mm IL and CL |
| 5 | M | Deletion exon 1 to 3: c.-110-?_669+?del(p.?) | CS | 45 | Lobectomy upper right lobe | Y (0/11) | 18 | 1 | Typical carcinoid | pT1N0cM0 | 5.5 | Nodules ≥10 mm IL and CL; diede |
| 6 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 62 | Lobectomy upper left lobe | Y (0/11) | 30 | <1 | Typical carcinoid | pT1N0cM0 | 5.0 | No lung lesions; diedf |
| 7 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | CS | 38 | Wedge resection right middle lobe | N | 7 | <2 | Typical carcinoid | pT1cN0M0 | 5.6 | No lung lesions; I diedg |
| 8 | F | Missense exon 4: c.683TC (p.Leu228Pro) | NS | 54 | Lobectomy upper right lobe | Y (1/>6) | 15 | 5 | Atypical carcinoid | pT1N1cM0 | 23.3 | Nodule <10 mm CL |
| 9 | M | Nonsense exon 6: c.819TG (p.Y273X) or c.819TA(p.Tyr273X) | NS | 41 | Wedge resection in upper and lower left lobe | N | ND | <5 | Atypical carcinoid | pT1cN0pM1 | 12.8 | Nodule <10 mm CL |
| 10 | F | Frameshift exon 3: c.653_660del (p.Ala218fs) | FS | 24 | Segmentectomy lower left lobe | N | 13 | ND | Typical carcinoid | pT1cN0M0 | 36.2 | Nodules ≥10 mm CLj |
| 11 | F | Nonsense exon 8: c.1074CG (p.Tyr358X) | NS | 23 | Bilobectomy of middle and lower right lobes | Y (0/6) | 25 | 2 | Atypical carcinoid | pT1N0cM0 | 11.3 | No lung lesions |
| 12 | M | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | ND | 37 | Lobectomy right middle lobe | ND | ND | ND | Typical carcinoid | ND | 36.2 | Nodule <10 mm IL, nodule ≥10 mm CL; diedh |
| 13 | F | Splice mutation intron 4: c.799-9GA(p.?) | NS | 38 | Lobectomy upper left lobe | Y (1/3) | 20 | ND | Typical carcinoid | pT1N1cM0 | 8.2 | Nodules <10 mm IL and CL |
| 14 | F | Nonsense exon 2: c.377GA (p.Trp126X) | FS | 54 | Lobectomy upper right lobe | Y (0/2) | 12 | <2 | Typical carcinoid | pT1N0cM0 | 7.3 | Nodule <10 mm IL |
| 15 | F | Nonsense exon 10: c.1594CT (p.Arg532X) | NS | 41 | Lobectomy right middle lobe | N | 10 | 0 | Typical carcinoid | pT1cN0M0 | 10.7 | Nodules <10 mm IL and CL |
| 16 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del) | FS | 44 | Segmentectomy lower left lobe | Y (0/3) | 35 | 2 | Atypical carcinoid | pT2N0cM0 | 7.5 | No lung lesions |
| 17 | F | Nonsense exon 8: c.1192C > T (p.Gln398X) | NS | 46 | Wedge resection upper left lobe | N | 10 | 2 | Atypical carcinoid | pT1cN0M0 | 0.1 | ND |
| 18 | M | Frameshift exon 10: c.1430dupG (p.Glu478fs) | FS | 43 | Wedge resection lower left lobe | N | 5 | 1 | Typical carcinoid | pT1cN0M0 | 6.0 | No lung lesions |
| 19 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | NS | 43 | Wedge resection right middle lobe | Y (1/1) | 6 | ND | Typical carcinoid | pT1N1cM0 | 6.8 | Nodule ≥10 mm IL, nodule <10 mm CL |
| 20 | M | Frameshift exon 2: c.249_252del (p.Ile85fs) | FS | 38 | Lobectomy upper left lobe | Y (15/16) | 15 | 10 | Atypical carcinoidc | pT3N2cM0 | 0.7 | Liver metastasesk |
| 21 | M | Frameshift exon 2: c.249_252del (p.Ile85fs) | ND | 66 | Lobectomy upper left lobe | Y (1/6) | 15 | 6 | Atypical carcinoid | pT1N1cM0 | 5.8 | Nodules <10 mm IL and CL |
| 22 | M | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 56 | Lobectomy lower right lobe | Y (2/7) | 37 | 4 | Atypical carcinoid | pT4N2cM0 | 4.3 | Nodule <10 mm CL |
| 23 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 57 | Lobectomy right middle lobe | Y (2/?) | 19 | ND | Typical carcinoid | pT3N2cM0 | 2.9 | ND |
| 24 | F | Deletion whole gene: c.-110-?_1848+?del (p.?) | NS | 64 | Wedge resection lower left lobe | N | 9 | 2 | Atypical carcinoid | pT1cN0M0 | 0.8 | ND |
| 25 | F | Missense exon 10: c.1489C > T (p.Pro497Ser)b | NS | 27 | Partial resection upper left lobe | ND | ND | ND | Typical carcinoid | ND | 38.0 | Nodule <10 mm IL |
| 26 | F | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 54 | Lobectomy lower right lobe | Y (1/12) | 12 | 0 | Typical carcinoid | pT3N1cM0 | 6.0 | No lung lesions |
| 27 | F | Nonsense exon 2: c.270T > G (p.Tyr90*) | NS | 44 | Lobectomy upper left lobe | Y (4/4) | 14 | <1 | Typical carcinoid | pT3N2M0 | 2.2 | Nodules <10 mm IL and CL |
| 28 | F | Frameshift exon 10: c.1677_1684dup8 (p.Lys562fs)a | NS | 57 | CT-guided biopsy upper left lobe | ND | ND | ND | Typical carcinoid | cT1N0M0 | 5.0 | No lung lesions l |
| 29 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 43 | Wedge resection lower left lobe | ND | 10 | <2 | Typical carcinoid | pT1cN0M0 | 6.3 | Nodule <10 mm IL, nodule ≥10 mm CLm |
| Patient | Sex | Genetic Mutation | Smoking | Age at Diagnosis (yr) | Type of Surgery | Lymfadenectomy (Positive/Total Number of Lymph Nodes) | Tumor Size on PA (mm) | Mitotic Index (per 10 hpf) | WHO Classification | TNM Classification | Length of Follow-up (yr) | Follow-up Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | FS | 44 | Wedge resection lower left lobe | N | 7 | 1 | Typical carcinoid | pT1cN0M0 | 9.2 | Nodule ≥10 mm IL |
| 2 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 62 | Wedge resection lower left lobe | Y (0/ND) | 23 | 0 | Typical carcinoid | pT1N0cM0 | 13.2 | No lung lesions |
| 3 | F | Frameshift exon 10: c.1430dupG (p.Glu478fs) | NS | 42 | Wedge resection upper left lobe | N | 5 | ND | Typical carcinoid | pT1c(m)N0M0 | 14.0 | Nodule <10 mm IL, nodule ≥10 mm CLd |
| 4 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | NS | 42 | Wedge resection upper left lobe | N | 14 | 0 | Typical carcinoid | pT1N0M0 | 8.8 | Nodules <10 mm IL and CL |
| 5 | M | Deletion exon 1 to 3: c.-110-?_669+?del(p.?) | CS | 45 | Lobectomy upper right lobe | Y (0/11) | 18 | 1 | Typical carcinoid | pT1N0cM0 | 5.5 | Nodules ≥10 mm IL and CL; diede |
| 6 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 62 | Lobectomy upper left lobe | Y (0/11) | 30 | <1 | Typical carcinoid | pT1N0cM0 | 5.0 | No lung lesions; diedf |
| 7 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | CS | 38 | Wedge resection right middle lobe | N | 7 | <2 | Typical carcinoid | pT1cN0M0 | 5.6 | No lung lesions; I diedg |
| 8 | F | Missense exon 4: c.683TC (p.Leu228Pro) | NS | 54 | Lobectomy upper right lobe | Y (1/>6) | 15 | 5 | Atypical carcinoid | pT1N1cM0 | 23.3 | Nodule <10 mm CL |
| 9 | M | Nonsense exon 6: c.819TG (p.Y273X) or c.819TA(p.Tyr273X) | NS | 41 | Wedge resection in upper and lower left lobe | N | ND | <5 | Atypical carcinoid | pT1cN0pM1 | 12.8 | Nodule <10 mm CL |
| 10 | F | Frameshift exon 3: c.653_660del (p.Ala218fs) | FS | 24 | Segmentectomy lower left lobe | N | 13 | ND | Typical carcinoid | pT1cN0M0 | 36.2 | Nodules ≥10 mm CLj |
| 11 | F | Nonsense exon 8: c.1074CG (p.Tyr358X) | NS | 23 | Bilobectomy of middle and lower right lobes | Y (0/6) | 25 | 2 | Atypical carcinoid | pT1N0cM0 | 11.3 | No lung lesions |
| 12 | M | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | ND | 37 | Lobectomy right middle lobe | ND | ND | ND | Typical carcinoid | ND | 36.2 | Nodule <10 mm IL, nodule ≥10 mm CL; diedh |
| 13 | F | Splice mutation intron 4: c.799-9GA(p.?) | NS | 38 | Lobectomy upper left lobe | Y (1/3) | 20 | ND | Typical carcinoid | pT1N1cM0 | 8.2 | Nodules <10 mm IL and CL |
| 14 | F | Nonsense exon 2: c.377GA (p.Trp126X) | FS | 54 | Lobectomy upper right lobe | Y (0/2) | 12 | <2 | Typical carcinoid | pT1N0cM0 | 7.3 | Nodule <10 mm IL |
| 15 | F | Nonsense exon 10: c.1594CT (p.Arg532X) | NS | 41 | Lobectomy right middle lobe | N | 10 | 0 | Typical carcinoid | pT1cN0M0 | 10.7 | Nodules <10 mm IL and CL |
| 16 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del) | FS | 44 | Segmentectomy lower left lobe | Y (0/3) | 35 | 2 | Atypical carcinoid | pT2N0cM0 | 7.5 | No lung lesions |
| 17 | F | Nonsense exon 8: c.1192C > T (p.Gln398X) | NS | 46 | Wedge resection upper left lobe | N | 10 | 2 | Atypical carcinoid | pT1cN0M0 | 0.1 | ND |
| 18 | M | Frameshift exon 10: c.1430dupG (p.Glu478fs) | FS | 43 | Wedge resection lower left lobe | N | 5 | 1 | Typical carcinoid | pT1cN0M0 | 6.0 | No lung lesions |
| 19 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | NS | 43 | Wedge resection right middle lobe | Y (1/1) | 6 | ND | Typical carcinoid | pT1N1cM0 | 6.8 | Nodule ≥10 mm IL, nodule <10 mm CL |
| 20 | M | Frameshift exon 2: c.249_252del (p.Ile85fs) | FS | 38 | Lobectomy upper left lobe | Y (15/16) | 15 | 10 | Atypical carcinoidc | pT3N2cM0 | 0.7 | Liver metastasesk |
| 21 | M | Frameshift exon 2: c.249_252del (p.Ile85fs) | ND | 66 | Lobectomy upper left lobe | Y (1/6) | 15 | 6 | Atypical carcinoid | pT1N1cM0 | 5.8 | Nodules <10 mm IL and CL |
| 22 | M | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 56 | Lobectomy lower right lobe | Y (2/7) | 37 | 4 | Atypical carcinoid | pT4N2cM0 | 4.3 | Nodule <10 mm CL |
| 23 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 57 | Lobectomy right middle lobe | Y (2/?) | 19 | ND | Typical carcinoid | pT3N2cM0 | 2.9 | ND |
| 24 | F | Deletion whole gene: c.-110-?_1848+?del (p.?) | NS | 64 | Wedge resection lower left lobe | N | 9 | 2 | Atypical carcinoid | pT1cN0M0 | 0.8 | ND |
| 25 | F | Missense exon 10: c.1489C > T (p.Pro497Ser)b | NS | 27 | Partial resection upper left lobe | ND | ND | ND | Typical carcinoid | ND | 38.0 | Nodule <10 mm IL |
| 26 | F | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 54 | Lobectomy lower right lobe | Y (1/12) | 12 | 0 | Typical carcinoid | pT3N1cM0 | 6.0 | No lung lesions |
| 27 | F | Nonsense exon 2: c.270T > G (p.Tyr90*) | NS | 44 | Lobectomy upper left lobe | Y (4/4) | 14 | <1 | Typical carcinoid | pT3N2M0 | 2.2 | Nodules <10 mm IL and CL |
| 28 | F | Frameshift exon 10: c.1677_1684dup8 (p.Lys562fs)a | NS | 57 | CT-guided biopsy upper left lobe | ND | ND | ND | Typical carcinoid | cT1N0M0 | 5.0 | No lung lesions l |
| 29 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 43 | Wedge resection lower left lobe | ND | 10 | <2 | Typical carcinoid | pT1cN0M0 | 6.3 | Nodule <10 mm IL, nodule ≥10 mm CLm |
Patients 1–16 have already been reported in our earlier study on neuroendocrine tumors of thymus and lung (7).
Abbreviations: CL, contralateral lung; CS, current smoker; CT, computed tomography; F, female; FS, former smoker; hpf, high-power field; IL, ipsilateral lung; LCNEC, large cell neuroendocrine carcinoma; (m), multiple tumors; M, male; N, no; ND, not determined; NS, never smoked; PA, pathology; RTx, radiotherapy; TNM, TNM Classification of Malignant Tumors; WHO, World Health Organization; Y, yes; yr, year.
aBased on genetic analysis of family members.
bVariant of uncertain significance.
cHigh-grade tumor difficult to classify as either atypical carcinoid or LCNEC. Based on histological, immunohistochemical, and molecular findings, it was concluded that this tumor was best classified as a high-grade atypical carcinoid (see the Results section).
dThe contralateral nodule was removed by a lobectomy of the middle right lobe. Histopathological examination revealed a typical carcinoid (diameter 34 mm) with a mitotic index <1 and ipsilateral positive hilar lymph nodes (TNM classification: pT2N1Mx).
eCause of death: adenocarcinoma of unknown origin.
fCause of death: prostate carcinoma.
gCause of death: metastatic thymic NET.
hCause of death: complicated surgery (not MEN1-related).
iPatient received additional radiotherapy (unknown dose).
jThe contralateral nodules were removed by a lobectomy of the middle right lobe and segment resection of the right upper lobe. Histopathological examination showed a typical carcinoid (largest lesion: diameter 14 mm) with a mitotic index <1 (TNM classification: pT1N0M1). Follow-up imaging afterwards revealed a nodule <10 mm in the right lung.
kPatient received additional radiotherapy (60 Gy). After 12 months, new liver metastases were found, which were histopathologically proven, showing an atypical carcinoid with a Ki67 of 16%.
lPatient received radiotherapy on the lesion in the upper left lobe (55 Gy) and on another—not biopsied—lesion in the lower left lobe (60 Gy).
mThe contralateral nodule was removed by a wedge resection of the lower right lobe. Histopathological examination revealed a typical carcinoid (diameter 10 mm) with a mitotic index <1. Two lymph nodes were removed without tumor localization (TNM classification: pT1N0Mx)
Clinical, genetic, and histopathological characteristics of patients with pathological diagnosis of lung NET
| Patient | Sex | Genetic Mutation | Smoking | Age at Diagnosis (yr) | Type of Surgery | Lymfadenectomy (Positive/Total Number of Lymph Nodes) | Tumor Size on PA (mm) | Mitotic Index (per 10 hpf) | WHO Classification | TNM Classification | Length of Follow-up (yr) | Follow-up Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | FS | 44 | Wedge resection lower left lobe | N | 7 | 1 | Typical carcinoid | pT1cN0M0 | 9.2 | Nodule ≥10 mm IL |
| 2 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 62 | Wedge resection lower left lobe | Y (0/ND) | 23 | 0 | Typical carcinoid | pT1N0cM0 | 13.2 | No lung lesions |
| 3 | F | Frameshift exon 10: c.1430dupG (p.Glu478fs) | NS | 42 | Wedge resection upper left lobe | N | 5 | ND | Typical carcinoid | pT1c(m)N0M0 | 14.0 | Nodule <10 mm IL, nodule ≥10 mm CLd |
| 4 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | NS | 42 | Wedge resection upper left lobe | N | 14 | 0 | Typical carcinoid | pT1N0M0 | 8.8 | Nodules <10 mm IL and CL |
| 5 | M | Deletion exon 1 to 3: c.-110-?_669+?del(p.?) | CS | 45 | Lobectomy upper right lobe | Y (0/11) | 18 | 1 | Typical carcinoid | pT1N0cM0 | 5.5 | Nodules ≥10 mm IL and CL; diede |
| 6 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 62 | Lobectomy upper left lobe | Y (0/11) | 30 | <1 | Typical carcinoid | pT1N0cM0 | 5.0 | No lung lesions; diedf |
| 7 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | CS | 38 | Wedge resection right middle lobe | N | 7 | <2 | Typical carcinoid | pT1cN0M0 | 5.6 | No lung lesions; I diedg |
| 8 | F | Missense exon 4: c.683TC (p.Leu228Pro) | NS | 54 | Lobectomy upper right lobe | Y (1/>6) | 15 | 5 | Atypical carcinoid | pT1N1cM0 | 23.3 | Nodule <10 mm CL |
| 9 | M | Nonsense exon 6: c.819TG (p.Y273X) or c.819TA(p.Tyr273X) | NS | 41 | Wedge resection in upper and lower left lobe | N | ND | <5 | Atypical carcinoid | pT1cN0pM1 | 12.8 | Nodule <10 mm CL |
| 10 | F | Frameshift exon 3: c.653_660del (p.Ala218fs) | FS | 24 | Segmentectomy lower left lobe | N | 13 | ND | Typical carcinoid | pT1cN0M0 | 36.2 | Nodules ≥10 mm CLj |
| 11 | F | Nonsense exon 8: c.1074CG (p.Tyr358X) | NS | 23 | Bilobectomy of middle and lower right lobes | Y (0/6) | 25 | 2 | Atypical carcinoid | pT1N0cM0 | 11.3 | No lung lesions |
| 12 | M | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | ND | 37 | Lobectomy right middle lobe | ND | ND | ND | Typical carcinoid | ND | 36.2 | Nodule <10 mm IL, nodule ≥10 mm CL; diedh |
| 13 | F | Splice mutation intron 4: c.799-9GA(p.?) | NS | 38 | Lobectomy upper left lobe | Y (1/3) | 20 | ND | Typical carcinoid | pT1N1cM0 | 8.2 | Nodules <10 mm IL and CL |
| 14 | F | Nonsense exon 2: c.377GA (p.Trp126X) | FS | 54 | Lobectomy upper right lobe | Y (0/2) | 12 | <2 | Typical carcinoid | pT1N0cM0 | 7.3 | Nodule <10 mm IL |
| 15 | F | Nonsense exon 10: c.1594CT (p.Arg532X) | NS | 41 | Lobectomy right middle lobe | N | 10 | 0 | Typical carcinoid | pT1cN0M0 | 10.7 | Nodules <10 mm IL and CL |
| 16 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del) | FS | 44 | Segmentectomy lower left lobe | Y (0/3) | 35 | 2 | Atypical carcinoid | pT2N0cM0 | 7.5 | No lung lesions |
| 17 | F | Nonsense exon 8: c.1192C > T (p.Gln398X) | NS | 46 | Wedge resection upper left lobe | N | 10 | 2 | Atypical carcinoid | pT1cN0M0 | 0.1 | ND |
| 18 | M | Frameshift exon 10: c.1430dupG (p.Glu478fs) | FS | 43 | Wedge resection lower left lobe | N | 5 | 1 | Typical carcinoid | pT1cN0M0 | 6.0 | No lung lesions |
| 19 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | NS | 43 | Wedge resection right middle lobe | Y (1/1) | 6 | ND | Typical carcinoid | pT1N1cM0 | 6.8 | Nodule ≥10 mm IL, nodule <10 mm CL |
| 20 | M | Frameshift exon 2: c.249_252del (p.Ile85fs) | FS | 38 | Lobectomy upper left lobe | Y (15/16) | 15 | 10 | Atypical carcinoidc | pT3N2cM0 | 0.7 | Liver metastasesk |
| 21 | M | Frameshift exon 2: c.249_252del (p.Ile85fs) | ND | 66 | Lobectomy upper left lobe | Y (1/6) | 15 | 6 | Atypical carcinoid | pT1N1cM0 | 5.8 | Nodules <10 mm IL and CL |
| 22 | M | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 56 | Lobectomy lower right lobe | Y (2/7) | 37 | 4 | Atypical carcinoid | pT4N2cM0 | 4.3 | Nodule <10 mm CL |
| 23 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 57 | Lobectomy right middle lobe | Y (2/?) | 19 | ND | Typical carcinoid | pT3N2cM0 | 2.9 | ND |
| 24 | F | Deletion whole gene: c.-110-?_1848+?del (p.?) | NS | 64 | Wedge resection lower left lobe | N | 9 | 2 | Atypical carcinoid | pT1cN0M0 | 0.8 | ND |
| 25 | F | Missense exon 10: c.1489C > T (p.Pro497Ser)b | NS | 27 | Partial resection upper left lobe | ND | ND | ND | Typical carcinoid | ND | 38.0 | Nodule <10 mm IL |
| 26 | F | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 54 | Lobectomy lower right lobe | Y (1/12) | 12 | 0 | Typical carcinoid | pT3N1cM0 | 6.0 | No lung lesions |
| 27 | F | Nonsense exon 2: c.270T > G (p.Tyr90*) | NS | 44 | Lobectomy upper left lobe | Y (4/4) | 14 | <1 | Typical carcinoid | pT3N2M0 | 2.2 | Nodules <10 mm IL and CL |
| 28 | F | Frameshift exon 10: c.1677_1684dup8 (p.Lys562fs)a | NS | 57 | CT-guided biopsy upper left lobe | ND | ND | ND | Typical carcinoid | cT1N0M0 | 5.0 | No lung lesions l |
| 29 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 43 | Wedge resection lower left lobe | ND | 10 | <2 | Typical carcinoid | pT1cN0M0 | 6.3 | Nodule <10 mm IL, nodule ≥10 mm CLm |
| Patient | Sex | Genetic Mutation | Smoking | Age at Diagnosis (yr) | Type of Surgery | Lymfadenectomy (Positive/Total Number of Lymph Nodes) | Tumor Size on PA (mm) | Mitotic Index (per 10 hpf) | WHO Classification | TNM Classification | Length of Follow-up (yr) | Follow-up Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | FS | 44 | Wedge resection lower left lobe | N | 7 | 1 | Typical carcinoid | pT1cN0M0 | 9.2 | Nodule ≥10 mm IL |
| 2 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 62 | Wedge resection lower left lobe | Y (0/ND) | 23 | 0 | Typical carcinoid | pT1N0cM0 | 13.2 | No lung lesions |
| 3 | F | Frameshift exon 10: c.1430dupG (p.Glu478fs) | NS | 42 | Wedge resection upper left lobe | N | 5 | ND | Typical carcinoid | pT1c(m)N0M0 | 14.0 | Nodule <10 mm IL, nodule ≥10 mm CLd |
| 4 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | NS | 42 | Wedge resection upper left lobe | N | 14 | 0 | Typical carcinoid | pT1N0M0 | 8.8 | Nodules <10 mm IL and CL |
| 5 | M | Deletion exon 1 to 3: c.-110-?_669+?del(p.?) | CS | 45 | Lobectomy upper right lobe | Y (0/11) | 18 | 1 | Typical carcinoid | pT1N0cM0 | 5.5 | Nodules ≥10 mm IL and CL; diede |
| 6 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 62 | Lobectomy upper left lobe | Y (0/11) | 30 | <1 | Typical carcinoid | pT1N0cM0 | 5.0 | No lung lesions; diedf |
| 7 | M | Frameshift exon 10: c.1561dup (p.Arg521fs) | CS | 38 | Wedge resection right middle lobe | N | 7 | <2 | Typical carcinoid | pT1cN0M0 | 5.6 | No lung lesions; I diedg |
| 8 | F | Missense exon 4: c.683TC (p.Leu228Pro) | NS | 54 | Lobectomy upper right lobe | Y (1/>6) | 15 | 5 | Atypical carcinoid | pT1N1cM0 | 23.3 | Nodule <10 mm CL |
| 9 | M | Nonsense exon 6: c.819TG (p.Y273X) or c.819TA(p.Tyr273X) | NS | 41 | Wedge resection in upper and lower left lobe | N | ND | <5 | Atypical carcinoid | pT1cN0pM1 | 12.8 | Nodule <10 mm CL |
| 10 | F | Frameshift exon 3: c.653_660del (p.Ala218fs) | FS | 24 | Segmentectomy lower left lobe | N | 13 | ND | Typical carcinoid | pT1cN0M0 | 36.2 | Nodules ≥10 mm CLj |
| 11 | F | Nonsense exon 8: c.1074CG (p.Tyr358X) | NS | 23 | Bilobectomy of middle and lower right lobes | Y (0/6) | 25 | 2 | Atypical carcinoid | pT1N0cM0 | 11.3 | No lung lesions |
| 12 | M | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | ND | 37 | Lobectomy right middle lobe | ND | ND | ND | Typical carcinoid | ND | 36.2 | Nodule <10 mm IL, nodule ≥10 mm CL; diedh |
| 13 | F | Splice mutation intron 4: c.799-9GA(p.?) | NS | 38 | Lobectomy upper left lobe | Y (1/3) | 20 | ND | Typical carcinoid | pT1N1cM0 | 8.2 | Nodules <10 mm IL and CL |
| 14 | F | Nonsense exon 2: c.377GA (p.Trp126X) | FS | 54 | Lobectomy upper right lobe | Y (0/2) | 12 | <2 | Typical carcinoid | pT1N0cM0 | 7.3 | Nodule <10 mm IL |
| 15 | F | Nonsense exon 10: c.1594CT (p.Arg532X) | NS | 41 | Lobectomy right middle lobe | N | 10 | 0 | Typical carcinoid | pT1cN0M0 | 10.7 | Nodules <10 mm IL and CL |
| 16 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del) | FS | 44 | Segmentectomy lower left lobe | Y (0/3) | 35 | 2 | Atypical carcinoid | pT2N0cM0 | 7.5 | No lung lesions |
| 17 | F | Nonsense exon 8: c.1192C > T (p.Gln398X) | NS | 46 | Wedge resection upper left lobe | N | 10 | 2 | Atypical carcinoid | pT1cN0M0 | 0.1 | ND |
| 18 | M | Frameshift exon 10: c.1430dupG (p.Glu478fs) | FS | 43 | Wedge resection lower left lobe | N | 5 | 1 | Typical carcinoid | pT1cN0M0 | 6.0 | No lung lesions |
| 19 | F | Frameshift exon 2: c.249_252del (p.Ile85fs) | NS | 43 | Wedge resection right middle lobe | Y (1/1) | 6 | ND | Typical carcinoid | pT1N1cM0 | 6.8 | Nodule ≥10 mm IL, nodule <10 mm CL |
| 20 | M | Frameshift exon 2: c.249_252del (p.Ile85fs) | FS | 38 | Lobectomy upper left lobe | Y (15/16) | 15 | 10 | Atypical carcinoidc | pT3N2cM0 | 0.7 | Liver metastasesk |
| 21 | M | Frameshift exon 2: c.249_252del (p.Ile85fs) | ND | 66 | Lobectomy upper left lobe | Y (1/6) | 15 | 6 | Atypical carcinoid | pT1N1cM0 | 5.8 | Nodules <10 mm IL and CL |
| 22 | M | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 56 | Lobectomy lower right lobe | Y (2/7) | 37 | 4 | Atypical carcinoid | pT4N2cM0 | 4.3 | Nodule <10 mm CL |
| 23 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 57 | Lobectomy right middle lobe | Y (2/?) | 19 | ND | Typical carcinoid | pT3N2cM0 | 2.9 | ND |
| 24 | F | Deletion whole gene: c.-110-?_1848+?del (p.?) | NS | 64 | Wedge resection lower left lobe | N | 9 | 2 | Atypical carcinoid | pT1cN0M0 | 0.8 | ND |
| 25 | F | Missense exon 10: c.1489C > T (p.Pro497Ser)b | NS | 27 | Partial resection upper left lobe | ND | ND | ND | Typical carcinoid | ND | 38.0 | Nodule <10 mm IL |
| 26 | F | Frameshift exon 10: c.1561dup (p.Arg521fs) | FS | 54 | Lobectomy lower right lobe | Y (1/12) | 12 | 0 | Typical carcinoid | pT3N1cM0 | 6.0 | No lung lesions |
| 27 | F | Nonsense exon 2: c.270T > G (p.Tyr90*) | NS | 44 | Lobectomy upper left lobe | Y (4/4) | 14 | <1 | Typical carcinoid | pT3N2M0 | 2.2 | Nodules <10 mm IL and CL |
| 28 | F | Frameshift exon 10: c.1677_1684dup8 (p.Lys562fs)a | NS | 57 | CT-guided biopsy upper left lobe | ND | ND | ND | Typical carcinoid | cT1N0M0 | 5.0 | No lung lesions l |
| 29 | F | In-frame deletion exon 2: c.358_360del (p.Lys120del)a | NS | 43 | Wedge resection lower left lobe | ND | 10 | <2 | Typical carcinoid | pT1cN0M0 | 6.3 | Nodule <10 mm IL, nodule ≥10 mm CLm |
Patients 1–16 have already been reported in our earlier study on neuroendocrine tumors of thymus and lung (7).
Abbreviations: CL, contralateral lung; CS, current smoker; CT, computed tomography; F, female; FS, former smoker; hpf, high-power field; IL, ipsilateral lung; LCNEC, large cell neuroendocrine carcinoma; (m), multiple tumors; M, male; N, no; ND, not determined; NS, never smoked; PA, pathology; RTx, radiotherapy; TNM, TNM Classification of Malignant Tumors; WHO, World Health Organization; Y, yes; yr, year.
aBased on genetic analysis of family members.
bVariant of uncertain significance.
cHigh-grade tumor difficult to classify as either atypical carcinoid or LCNEC. Based on histological, immunohistochemical, and molecular findings, it was concluded that this tumor was best classified as a high-grade atypical carcinoid (see the Results section).
dThe contralateral nodule was removed by a lobectomy of the middle right lobe. Histopathological examination revealed a typical carcinoid (diameter 34 mm) with a mitotic index <1 and ipsilateral positive hilar lymph nodes (TNM classification: pT2N1Mx).
eCause of death: adenocarcinoma of unknown origin.
fCause of death: prostate carcinoma.
gCause of death: metastatic thymic NET.
hCause of death: complicated surgery (not MEN1-related).
iPatient received additional radiotherapy (unknown dose).
jThe contralateral nodules were removed by a lobectomy of the middle right lobe and segment resection of the right upper lobe. Histopathological examination showed a typical carcinoid (largest lesion: diameter 14 mm) with a mitotic index <1 (TNM classification: pT1N0M1). Follow-up imaging afterwards revealed a nodule <10 mm in the right lung.
kPatient received additional radiotherapy (60 Gy). After 12 months, new liver metastases were found, which were histopathologically proven, showing an atypical carcinoid with a Ki67 of 16%.
lPatient received radiotherapy on the lesion in the upper left lobe (55 Gy) and on another—not biopsied—lesion in the lower left lobe (60 Gy).
mThe contralateral nodule was removed by a wedge resection of the lower right lobe. Histopathological examination revealed a typical carcinoid (diameter 10 mm) with a mitotic index <1. Two lymph nodes were removed without tumor localization (TNM classification: pT1N0Mx)
A total of 50 patients were diagnosed with 1 (lesion suspected of) lung NET, 43 patients were diagnosed with 2, 8 patients were diagnosed with 3, and 1 patient was diagnosed with 4 (lesions suspected of) lung NETs, respectively. The baseline tumor size at diagnosis—defined as the largest nodule diameter at the 1st abnormal CT scan—was <10 mm in 125 lesions and ≥10 mm in 27 lesions. The tumor size was not described in 12 lung NET lesions. A total of 75 lesions were identified in the left lung, compared with 89 lesions located in the right lung.
Growth analysis
Nineteen patients were excluded from the growth analysis due to the lack of sequential data. Additionally, 5 lung lesions were excluded because of a baseline tumor size ≥20 mm. Three tumors ≥20 mm were surgically removed. Pathology reports confirmed a lung NET in all cases. The 2 remaining tumors were not removed due to synchronic metastatic disease (n = 1) and apparent shrinkage in a partial cystic tumor, withholding immediate surgery (n = 1). Recurrence after surgery has occurred in 1 patient. Two patients with a baseline tumor of ≥20 mm had a concurrent smaller (<20 mm) lesion suspect of lung NET that was included in the analysis. Therefore, a total of 114 lesions suspect of lung NET in 80 patients were included in the tumor growth analysis. The median baseline tumor diameter was 5 mm (IQR: 3.0–6.3 mm, range 1–17mm).
The increase of tumor diameter was 6.0% per year, equivalent to a doubling time of 11.8 years. The individual tumor growth is illustrated in Fig. 2. Genotype, gender, smoking status, the age at diagnosis of lung NET, and baseline tumor size did not significantly affect tumor growth (Table 2). Operated lung NETs were associated with a significantly higher growth rate than other lesions (P < 0.0005).
Potential determinants of tumor growth
| Tumor Growtha | |
|---|---|
| Statistical significance and regression coefficienta | |
| Overall tumor growth (β, 95% CI) | 1.060 (1.038–1.083) |
| Effect modifiers (P-value for interaction) | |
| Gender | P = 0.437 |
| Male, n = 34 (β, 95% CI) | 1.071 (1.036–1.108) |
| Female, n = 46 (β, 95% CI) | 1.053 (0.975–1.138) |
| Age at lung NET diagnosis | P = 0.356 |
| Reference value for age = 0 | 1.096 (1.019–1.178) |
| Change per year (β, 95% CI) | 0.999 (0.997–1.001) |
| Smoking statusb | P = 0.199 |
| Never smoked, n = 39 (β, 95% CI) | 1.065 (0.985–1.152) |
| Former or current smoker, n = 19 (β, 95% CI) | 1.036 (0.999–1.074) |
| Genotype | P = 0.120 |
| Nonsense/frameshift exon 2,9,10 mutations n = 28 (β, 95% CI) | 1.036 (0.999–1.074) |
| Other mutations,c n = 50 (β, 95% CI) | 1.074 (0.990–1.164) |
| Genotype | P = 0.408 |
| JunD interacting domain mutations,d n = 25 (β, 95% CI) | 1.071 (1.033–1.109) |
| Other mutations,d n = 45 (β, 95% CI) | 1.050 (0.968–1.140) |
| Genotype | P = 0.106 |
| CHES1 interacting domain mutations,e n = 20 (β, 95% CI) | 1.031 (0.996–1.066) |
| Other mutations,e n = 50 (β, 95% CI) | 1.068 (0.988–1.156) |
| Genotype | P = 0.447 |
| Missense mutations,f n = 15 (β, 95% CI) | 1.054 (1.026–1.082) |
| Nonsense/frameshift mutations,f n = 40 (β, 95% CI) | 1.076 (0.992–1.167) |
| Baseline tumor size | P = 0.147 |
| Diameter < median, n = 55 (β, 95% CI) | 1.057 (1.033–1.081) |
| Diameter ≥ median, n = 59 (β, 95% CI) | 1.071 (1.028–1.116) |
| Tumor Growtha | |
|---|---|
| Statistical significance and regression coefficienta | |
| Overall tumor growth (β, 95% CI) | 1.060 (1.038–1.083) |
| Effect modifiers (P-value for interaction) | |
| Gender | P = 0.437 |
| Male, n = 34 (β, 95% CI) | 1.071 (1.036–1.108) |
| Female, n = 46 (β, 95% CI) | 1.053 (0.975–1.138) |
| Age at lung NET diagnosis | P = 0.356 |
| Reference value for age = 0 | 1.096 (1.019–1.178) |
| Change per year (β, 95% CI) | 0.999 (0.997–1.001) |
| Smoking statusb | P = 0.199 |
| Never smoked, n = 39 (β, 95% CI) | 1.065 (0.985–1.152) |
| Former or current smoker, n = 19 (β, 95% CI) | 1.036 (0.999–1.074) |
| Genotype | P = 0.120 |
| Nonsense/frameshift exon 2,9,10 mutations n = 28 (β, 95% CI) | 1.036 (0.999–1.074) |
| Other mutations,c n = 50 (β, 95% CI) | 1.074 (0.990–1.164) |
| Genotype | P = 0.408 |
| JunD interacting domain mutations,d n = 25 (β, 95% CI) | 1.071 (1.033–1.109) |
| Other mutations,d n = 45 (β, 95% CI) | 1.050 (0.968–1.140) |
| Genotype | P = 0.106 |
| CHES1 interacting domain mutations,e n = 20 (β, 95% CI) | 1.031 (0.996–1.066) |
| Other mutations,e n = 50 (β, 95% CI) | 1.068 (0.988–1.156) |
| Genotype | P = 0.447 |
| Missense mutations,f n = 15 (β, 95% CI) | 1.054 (1.026–1.082) |
| Nonsense/frameshift mutations,f n = 40 (β, 95% CI) | 1.076 (0.992–1.167) |
| Baseline tumor size | P = 0.147 |
| Diameter < median, n = 55 (β, 95% CI) | 1.057 (1.033–1.081) |
| Diameter ≥ median, n = 59 (β, 95% CI) | 1.071 (1.028–1.116) |
β stands for the regression coefficient from the linear mixed models analysis, denoting growth as change in tumor size (factor) per year. Statistical significance is shown in bold.
Abbreviation: CHES1, checkpoint kinase 1; CI, confidence interval; NET, neuroendocrine tumor.
aTumor growth was assessed using multilevel linear mixed models analysis, accounting for clustering of observations within lung tumors within patients. Logarithmic-transformed lung NET diameter was used as a dependent variable and follow-up time was used as main fixed effect. Potential determinants of tumor growth were treated as additional fixed (interacting) covariates.
bData on smoking status were available in 58/80 patients included in the growth analysis (72.5%).
cAll other mutations included. Patients without genetic analysis or with a CDKN1B mutation were treated as missings (n = 2).
dOnly patients with pathogenic germline nonsense, frameshift, missense mutations, and in-frame deletions included. JunD interacting domain: codons 1–40, 139–242, and 323–428.
eOnly patients with pathogenic germline nonsense, frameshift, missense mutations, and in-frame deletions included. CHES1 interacting domain: codons 428–610.
fOnly patients with pathogenic germline nonsense, frameshift, and missense mutations included.
Potential determinants of tumor growth
| Tumor Growtha | |
|---|---|
| Statistical significance and regression coefficienta | |
| Overall tumor growth (β, 95% CI) | 1.060 (1.038–1.083) |
| Effect modifiers (P-value for interaction) | |
| Gender | P = 0.437 |
| Male, n = 34 (β, 95% CI) | 1.071 (1.036–1.108) |
| Female, n = 46 (β, 95% CI) | 1.053 (0.975–1.138) |
| Age at lung NET diagnosis | P = 0.356 |
| Reference value for age = 0 | 1.096 (1.019–1.178) |
| Change per year (β, 95% CI) | 0.999 (0.997–1.001) |
| Smoking statusb | P = 0.199 |
| Never smoked, n = 39 (β, 95% CI) | 1.065 (0.985–1.152) |
| Former or current smoker, n = 19 (β, 95% CI) | 1.036 (0.999–1.074) |
| Genotype | P = 0.120 |
| Nonsense/frameshift exon 2,9,10 mutations n = 28 (β, 95% CI) | 1.036 (0.999–1.074) |
| Other mutations,c n = 50 (β, 95% CI) | 1.074 (0.990–1.164) |
| Genotype | P = 0.408 |
| JunD interacting domain mutations,d n = 25 (β, 95% CI) | 1.071 (1.033–1.109) |
| Other mutations,d n = 45 (β, 95% CI) | 1.050 (0.968–1.140) |
| Genotype | P = 0.106 |
| CHES1 interacting domain mutations,e n = 20 (β, 95% CI) | 1.031 (0.996–1.066) |
| Other mutations,e n = 50 (β, 95% CI) | 1.068 (0.988–1.156) |
| Genotype | P = 0.447 |
| Missense mutations,f n = 15 (β, 95% CI) | 1.054 (1.026–1.082) |
| Nonsense/frameshift mutations,f n = 40 (β, 95% CI) | 1.076 (0.992–1.167) |
| Baseline tumor size | P = 0.147 |
| Diameter < median, n = 55 (β, 95% CI) | 1.057 (1.033–1.081) |
| Diameter ≥ median, n = 59 (β, 95% CI) | 1.071 (1.028–1.116) |
| Tumor Growtha | |
|---|---|
| Statistical significance and regression coefficienta | |
| Overall tumor growth (β, 95% CI) | 1.060 (1.038–1.083) |
| Effect modifiers (P-value for interaction) | |
| Gender | P = 0.437 |
| Male, n = 34 (β, 95% CI) | 1.071 (1.036–1.108) |
| Female, n = 46 (β, 95% CI) | 1.053 (0.975–1.138) |
| Age at lung NET diagnosis | P = 0.356 |
| Reference value for age = 0 | 1.096 (1.019–1.178) |
| Change per year (β, 95% CI) | 0.999 (0.997–1.001) |
| Smoking statusb | P = 0.199 |
| Never smoked, n = 39 (β, 95% CI) | 1.065 (0.985–1.152) |
| Former or current smoker, n = 19 (β, 95% CI) | 1.036 (0.999–1.074) |
| Genotype | P = 0.120 |
| Nonsense/frameshift exon 2,9,10 mutations n = 28 (β, 95% CI) | 1.036 (0.999–1.074) |
| Other mutations,c n = 50 (β, 95% CI) | 1.074 (0.990–1.164) |
| Genotype | P = 0.408 |
| JunD interacting domain mutations,d n = 25 (β, 95% CI) | 1.071 (1.033–1.109) |
| Other mutations,d n = 45 (β, 95% CI) | 1.050 (0.968–1.140) |
| Genotype | P = 0.106 |
| CHES1 interacting domain mutations,e n = 20 (β, 95% CI) | 1.031 (0.996–1.066) |
| Other mutations,e n = 50 (β, 95% CI) | 1.068 (0.988–1.156) |
| Genotype | P = 0.447 |
| Missense mutations,f n = 15 (β, 95% CI) | 1.054 (1.026–1.082) |
| Nonsense/frameshift mutations,f n = 40 (β, 95% CI) | 1.076 (0.992–1.167) |
| Baseline tumor size | P = 0.147 |
| Diameter < median, n = 55 (β, 95% CI) | 1.057 (1.033–1.081) |
| Diameter ≥ median, n = 59 (β, 95% CI) | 1.071 (1.028–1.116) |
β stands for the regression coefficient from the linear mixed models analysis, denoting growth as change in tumor size (factor) per year. Statistical significance is shown in bold.
Abbreviation: CHES1, checkpoint kinase 1; CI, confidence interval; NET, neuroendocrine tumor.
aTumor growth was assessed using multilevel linear mixed models analysis, accounting for clustering of observations within lung tumors within patients. Logarithmic-transformed lung NET diameter was used as a dependent variable and follow-up time was used as main fixed effect. Potential determinants of tumor growth were treated as additional fixed (interacting) covariates.
bData on smoking status were available in 58/80 patients included in the growth analysis (72.5%).
cAll other mutations included. Patients without genetic analysis or with a CDKN1B mutation were treated as missings (n = 2).
dOnly patients with pathogenic germline nonsense, frameshift, missense mutations, and in-frame deletions included. JunD interacting domain: codons 1–40, 139–242, and 323–428.
eOnly patients with pathogenic germline nonsense, frameshift, missense mutations, and in-frame deletions included. CHES1 interacting domain: codons 428–610.
fOnly patients with pathogenic germline nonsense, frameshift, and missense mutations included.
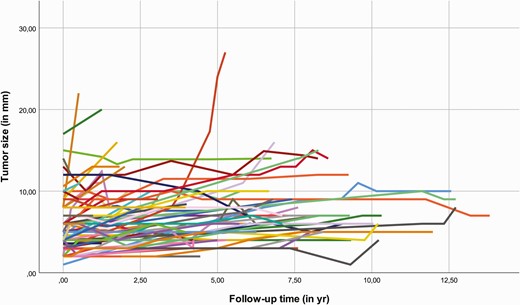
Individual growth of lesions suspect of lung NET. Abbreviations: NET, neuroendocrine tumor; mm, millimeter; yr, years.
Survival analysis
Twelve patients diagnosed with 1 or multiple lesions suspected of lung NET died during follow-up (11.8%); their cause of death was not related to the lung NET. The overall 15-year survival rate after diagnosis of lung NET was 78.0% (95% confidence interval [CI]: 64.6–94.2%, see Figure 3; the overall 10-year survival rate was 87.8% [95% CI: 80.1–96.3%]). The survival of operated patients was not significantly different from nonoperated patients (P = 0.18). Moreover, gender, smoking status, genotype, baseline tumor size, tumor classification, and lymph node involvement did not significantly influence survival (data not shown).
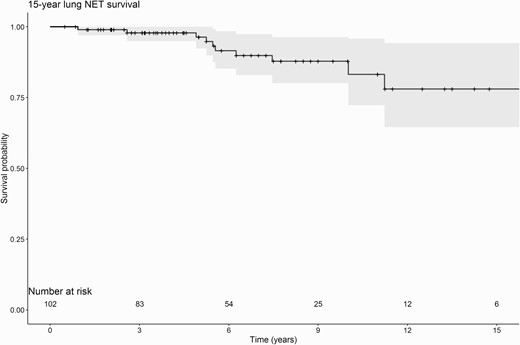
Fifteen-year lung NET survival rate. Abbreviation: NET, neuroendocrine tumor.
Description of the case with an exceptional tumor course
A 31-year-old male MEN1 patient (patient 20) was initially diagnosed with lung NET based on thoracic imaging, which showed 3 small intrapulmonary nodules (5 mm) that were suspicious for NETs. For the first 6 years of follow-up, the nodules showed a gradual growth over the years up to a tumor diameter of 11 mm (corresponding with a doubling time of 5.1 years). However, 1 nodule located in the left upper lobe started to expand rapidly from 11 to 16 mm within 12 months, with new irregular tumor margins. Functional imaging (Gallium-68 DOTATATE) showed no somatostatin receptor uptake by the tumor, but a number of mediastinal lymph nodes (station 2L, 5 and 6) showed pathological uptake. Lobectomy with lymph node dissection followed soon after. Histological examination revealed a high-grade neuroendocrine neoplasm, which was difficult to classify as either atypical carcinoid or LCNEC. There was extensive vasoinvasive growth, an intralobular satellite lesion, and tumor-positive mediastinal and hilar lymph nodes. The tumor was resected with free margins. An endobronchial ultrasound performed postoperatively showed 6 tumor-positive lymph nodes in mediastinal stations 2L and 4L. Therefore, the patient received adjuvant radiotherapy (60 Gy in 30 sessions). Follow-up CT thorax and liver showed no local recurrence for 9 months postsurgery. After 12 months, new extensive liver metastases were found, which were histopathologically confirmed, showing an atypical carcinoid with a Ki67 of 15%.
To further elucidate whether this high-grade neuroendocrine neoplasm should be classified as either atypical carcinoid or LCNEC, extensive analyses were performed on the resected lung NET tissue. Histological analysis showed a tumor with a nested growth pattern composed of rather monotonous cells with round to oval nuclei and clumped chromatin (see Supplemental Material, Fig. S1A) (21). Mitotic figures were frequently seen (>10 per high-power fields), and the Ki67 labelling index was 75%. By immunohistochemistry, the tumor was strongly positive for chromogranin A, synaptophysin, and transcription termination factor 1 (TTF1), and it was negative for somatostatin receptor type 2a (SSTR2a). P53 immunohistochemistry revealed a wild-type expression pattern. There was no loss or ATRX or DAXX. Menin immunohistochemistry (see Supplemental Material, Fig. S1B) showed loss of expression in the tumor cells (21).
At initial assessment, NGS did not reveal any mutations. In addition, Reverse transcription polymerase chain reaction (RT-PCR) did not show cMET exon 14 skipping. FISH did not reveal a translocation of the RET gene or alternative lengthening of telomeres. Single nucleotide peptide array was performed to further investigate the somatic second hit inactivation of MEN1 and to confirm immunohistochemical menin loss, but this did not reveal loss of the MEN1 locus. Finally, whole genome sequencing was performed, which indeed revealed a somatic inactivating c.333dupT (p.Val112fs) MEN1 mutation of unknown clinical relevance, suggesting that this mutation—additional to the known germline frameshift c.249_252del (p.Ile85fs) mutation of the patient—was responsible for the loss of a functional MEN1 gene in the tumor. Based on histological, immunohistochemical, and molecular findings, in particular the somatic second hit inactivation of the MEN1 gene, and lack of mutations associated with LCNEC, it was concluded that this tumor was best classified as a high-grade atypical carcinoid related to the MEN1 syndrome.
Interestingly, WGS also showed a likely pathogenic c.3127A > G (p.Met1043Val) mutation in the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) gene, associated with the PI3K-AKT-mTOR pathway. In retrospect, this mutation was also found in the NGS output with a allele frequency <1%. Further analysis of the WGS data showed that the c.333dupT (p.Val112fs) MEN1 mutation was unlikely to have a subclonal origin, whereas the c.3127A > G (p.Met1043Val) PIK3CA mutation was probably subclonal (see Supplementary Material, Fig. S2) (21). The variation in allele frequencies of the PIK3CA mutation between different tumor samples supports this conclusion. Although it was not possible to indisputably determine the order of events, it seems plausible to assume that the PIK3CA mutation occurred after the somatic MEN1 mutation, leading to accelerated tumor growth.
Discussion
In the present analyses with a longer follow-up compared with most previous studies, the indolent behavior of MEN1-related lung NETs is confirmed. Approximately 1/5 MEN1 patients (22.9%) were diagnosed with lesions highly suspect of lung NET(s). The high overall 15-year survival rate and the absence of lung NET-related mortality in the present study emphasizes the relatively benign characteristics of MEN1-related lung NETs. Overall, tumor growth was even lower than previously reported (6.0% per year in the current study vs 17.0% per year in our previous study). The lung lesions seemed to remain stable over longer periods of time, and growth even slowed down in some lesions, explaining the differences in outcomes of the present study when compared with our earlier results in partly the same patient cohort (7). Further investigations of the case with a remarkable sudden growth and aggressive tumor biology revealed a somatic PIK3CA driver mutation, which probably led to subclonal expansion and could explain the sudden deviant course of disease.
Comparison with literature
The prevalence of histopathologically proven lung NETs in our cohort (6.5%) is comparable to earlier findings in our (4.9%) and other cohorts (4.7–6.6%) (6, 8–10). The higher prevalence of lesions radiologically suspect of lung NET in this study (22.9%) compared with the results from our previous study (13.3%) can be explained by the larger proportion of patients under regular thoracic surveillance. Similar frequencies of lung nodules found on CT scans have been described in German and Tasman cohorts (29.3% and 26.0%, respectively) (9, 26). The extremely low prevalence of lung NETs in the subgroup of patients without MEN1 or CDKN1B mutation (2.1%) illustrates the differences in the phenotype and clinical course between mutation-positive and mutation-negative patients, as described previously (27).
Growth analysis showed an overall indolent course (tumor doubling time ± 12 years). Most lesions suspected of lung NET did not demonstrate significant progression, and some lesions even decreased at long-term follow-up. Through this mechanism, the longer follow-up time in this study could explain the lower overall growth rate compared with our earlier findings in 2014. Unfortunately, molecular mechanisms regulating the growth of lung NETs have not yet been revealed. Furthermore, operated lung NETs seemed to grow significantly faster than nonoperated lung NETs in this study. Obviously, these results should be interpreted with caution because a larger tumor size and growth rate often are an indication for surgery. This indication bias could explain the different growth rates between these 2 groups rather than a difference in the type of pathology. Moreover, the fact that the mitotic index was low in most of the fast-growing and/or larger lesions necessitating surgery underlines the benign course of MEN1-related lung NETs in general. In contrast to our previous results, we were unable to confirm gender-related differences in tumor growth in the current study. Further research in other cohorts is needed to determine the true role of gender in the growth of lung NETs.
Other studies on the growth rate of pulmonary nodules in MEN1 patients showed conflicting results. In a study of 75 MEN1 patients by Bartsch et al, pulmonary nodules showed (slight) progression in only 4 MEN1 patients (18% of patients with pulmonary nodules). None grew larger than 10 mm (median follow-up 67 months) (9). In contrast, results from the Tasman cohort including 50 MEN1 patients suggested a much more aggressive course of pulmonary nodules by demonstrating tumor progression in 54% of patients with lung nodules. However, in this study, tumors were identified using fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG PET/CT)-scans, and tumor growth was mainly seen in FDG-avid lesions. Moreover, pulmonary metastases from other malignancies were not excluded. The more aggressive growth could therefore be a reflection of the use of different selection criteria (26). Moreover, as the Tasman MEN1 population all share a common found mutation (NM_130799.2:c.446-3 C > G heterozygous), the differences in genetic background could also have contributed to the dissimilar course of disease between the 2 cohorts.
The excellent prognosis of lung NETs found in our study is comparable to findings in other cohorts (6, 8–10). In the largest cohort of histopathologically proven lung NETs to date (n = 51), overall survival was also not significantly decreased in patients with a lung NET. However, mainly poorly differentiated and aggressive lung tumors were the cause of death in 7 patients. The presence of atypical carcinoid and lymph node involvement tended to be associated with a higher mortality in the GTE cohort, while operated patients lived significantly longer. Furthermore, synchronous metastases were associated with shorter survival (10). We could not reproduce these associations in our cohort, which might be explained by differences in cohort setting (population-based or not), cohort size, lung NET definition, and/or selection criteria for surgery.
Extensive molecular analysis of the only high-grade neuroendocrine tumor in this cohort revealed that the somatic mutation of PIK3CA may have caused an aggressive course of the lung NET in patient 20. PIK3CA encodes the catalytic subunit of phosphatidyl 3-kinase (PI3K), an intracellular central mediator of cell survival signals. PIK3CA mutations are associated with numerous cancer types and is most frequently found in endometrial (24–46%), breast (20–32%), and bladder cancer (20–27%) (28). PIK3CA mutations are also described in squamous lung cancers (5–10%), in which they possibly lead to resistance to antiepidermal growth factor receptor therapy (29). To our knowledge, the frequency and impact of PIK3CA mutations in lung NETs has not been described to date.
Strengths
Because patients were selected from the national MEN1 database, including >90% of the Dutch MEN1 population, it is safe to assume that our study results are generalizable to the entire MEN1 population—at least in the Netherlands. Furthermore, standardized longitudinal data collection reduced the risk of information bias. Thirdly, the additional follow-up time and larger cohort size enabled us to study the natural course of MEN1-related lung NETs more accurately compared with our earlier study and previous studies in other MEN1 cohorts. Moreover, the reliability of the results has been further increased by the larger proportion of patients undergoing regular thoracic imaging (58.2% in our previous report vs 78.9% in our current cohort).
Limitations
However, some limitations must be kept in mind when interpreting these results. First of all, the retrospective design of this study could have affected growth analyses. These analyses were dependent on data from imaging studies performed during routine patient care. Although imaging protocols and radiology reports for lung NETs have not been standardized for this clinical study, all participating UMCs have a team dedicated to NETs and have employed dedicated thoracic radiologists. Most patients had all their follow-up scans in the same center, thereby reducing variation. Moreover, to avoid an overestimation of accuracy of the outcomes, we took the aspect of longitudinal observations clustered within patients into account in the mixed models analysis.
This study included cases radiologically suspect of lung NET without pathological confirmation. This may have introduced a risk of overestimating the prevalence of lung NET by including lesions that were not truly lung NET, because the interpretation of abnormalities on imaging studies is partly subjective. Combining the interpretation of a senior radiologist, the high number of follow-up scans (including functional imaging studies) and any biopsy results largely mitigated these risks.
One might argue that the lesions found on the CT scans are diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH). However, about half of the patients with DIPNECH complain of cough and dyspnea, often combined with signs of inflammation, bronchial obstruction, and mosaic attenuation on radiological imaging (30, 31). These entities were not seen in our patient cohort. Furthermore, to our knowledge, the combination of DIPNECH and MEN1 is limited to only 1 patient in the literature to date (30). Based on these considerations, we are confident that it is very unlikely that a diagnosis of DIPNECH has been missed.
Tumor growth was expressed as the change in the largest diameter of the lesions. It is important to realize that such lesions are in fact 3-dimensional objects, with an estimated volume of: * π * (radius)3 in the case of spherical-shaped lesions. This means that doubling of the largest diameter of a spherical lesion is associated with a proportional 8-fold increase in the volume of the lesion. The increasing availability of volumetric analysis in radiology allows for better estimation of the true tumor volume change over time—and thereby biological behavior—of lung nodules in the future.
Despite the increasing use of nuclear imaging in MEN1 patients, its exact role in the surveillance and follow-up is yet to be determined (32–35). Although lung NETs are sporadically mentioned in some studies on nuclear imaging in MEN1 patients, none have focused on its diagnostic value in lung lesions in MEN1 patients specifically. Unfortunately, the setting and retrospective nature of our study prevented us from investigating these matters.
Clinical implications
Results from this study confirm the benign nature of MEN1-related lung NETs, reflected by low tumor growth, excellent survival, and the lack of lung NET-related mortality. At long-term follow-up, tumor growth remained limited over time. From this perspective, these findings suggest justification of less frequent thoracic screening than currently advised (every 1 to 2 years) (11). This seems to be especially true for patients with clinically diagnosed MEN1 without a pathogenic MEN1 mutation, given the very low prevalence of lung NETs in this group. The results in the subgroup of clinically diagnosed MEN1 patients are in accordance with the recent evidence that clinically diagnosed MEN1 patients rarely develop a 3rd MEN1-related manifestation (27, 36). However, as illustrated by 1 high-grade neuroendocrine tumor (atypical carcinoid), periodic screening remains essential to detect unanticipated accelerated tumor growth in time. Unfortunately, there are still no accurate clinical predictors for growth. A lower thoracic screening frequency appears to be safe at the group level, but might result in failure of timely recognition of aggressively behaving tumors in some individual cases. Nevertheless, the number needed to screen for timely identification of individual aggressive cases is high. Therefore, a personalized screening program should be discussed with individual patients, balancing between the absolute risk individual patients are willing to take and the intensity of screening and exposure to ionizing radiation.
Additionally, although uncommon in MEN1 patients, thymus NET generally show a very aggressive course of disease and must be considered when discussing thoracic imaging in MEN1 patients (7). In our cohort, a pathologically proven thymus NET was found in 14 MEN1 patients (3.1%). Thoracic imaging led to the diagnosis in all but one, illustrating the possible additional yield of thoracic surveillance. This must be kept in mind when reviewing the frequency of thoracic imaging with MEN1 patients.
Surgical resection is considered the 1st treatment of choice in MEN1-related lung NETs (11). Tumor size and location have been suggested to be important factors when timing surgery (20). The low growth rate and lack of beneficial effect of surgery on prognosis in this study support a watch-and-wait policy for small lung NETs. However, in case of accelerated tumor growth during follow-up, surgery should be performed without delay.
Conclusion
Overall, MEN1-related lung NETs are slow-growing and have an excellent prognosis. However, unanticipated accelerated tumor growth does occur sporadically. Because no accurate risk factors for tumor growth can be described, periodic screening programs should be based on well-informed decision-making with the individual patient, balancing between the low absolute risk of an aggressive tumor in individuals and the potential harms of frequent thoracic imaging.
Acknowledgments
Financial Support: This research did not receive any specific grant or fellowship.
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability
Restrictions apply to some or all the availability of data generated or analyzed during this study to preserve patient confidentiality or because they were used under license. The corresponding author will on request detail the restrictions and any conditions under which access to some data may be provided.
References

{kind=link}
{kind=link}
{kind=link}