





Abstract
Enhanced levels of catecholestradiols, 2-hydroxyestradiol (2-OHE2) or 4-hydroxyestradiol (4-OHE2), are reported in endometriosis. During gestation, catecholestradiol activation of adrenergic receptors (AR) elevates estrogen receptor (ER)-independent proliferation of uterine arterial endothelial cells.
To investigate β-AR-mediated catecholestradiol effects on human endometrial stromal cell (HESC) and epithelial cell survival in endometriosis.
β-AR immunostaining of eutopic and ectopic endometria (n = 9). Assays for cell viability, 5-bromo-2′-deoxyuridine proliferation, apoptosis, quantitative PCR, and estrogenicity (alkaline phosphatase activity), as well as siRNA β-AR silencing and immunoblot analyses of cultured HESCs or Ishikawa cells treated with control or 2-OHE2 or 4-OHE2 ±β-AR antagonist or ±p38 MAPK inhibitor.
University research institution.
Women with or without endometriosis.
None.
β-AR expression in eutopic vs ectopic endometria and regulation of HESC survival by 2-OHE2 and 4-OHE2.
Eutopic and ectopic endometrial stromal and epithelial cells displayed β2-AR immunoreactivity with increased staining in the functionalis vs basalis layer (P < 0.05). Both 2-OHE2 and 4-OHE2 enhanced HESC and Ishikawa cell survival (P < 0.05), an effect abrogated by β-AR antagonist propranolol, but not ER antagonist ICI182,780. 2-OHE2 or 4-OHE2 failed to induce cell survival and estrogenic activity in ADRB2-silenced HESCs and in Ishikawa cells, respectively. Although 2-OHE2 inhibited apoptosis and BAX mRNA expression, 4-OHE2 induced proliferation and decreased apoptosis (P < 0.05). Both catecholestradiols elevated phospho-p38 MAPK levels (P < 0.05), which was blocked by propranolol, and p38 MAPK inhibitor reversed catecholestradiol-enhanced HESC survival.
Catecholestradiols increase endometrial cell survival by an ER-independent β-AR-mediated p38 MAPK activation, suggesting that agents blocking β-AR (e.g., propranolol) or inhibiting 2-OHE2- or 4-OHE2-generating enzymes (i.e., CYP1A1/B1) could treat endometriosis.
Endometriosis is a complex estrogen-dependent gynecologic disorder that contributes to infertility, chronic pelvic pain, dysmenorrhea, and dyspareunia. It affects about 15% of all reproductive-aged women and 20% to 50% of infertile women (1-4). It is defined by the presence of endometrial tissue, consisting of glands and/or stroma, outside the uterine cavity (5-7). Endometriosis is associated with increased local 17β-estradiol (E2) production resulting from enhanced aromatase expression by endometriotic stromal cells (8, 9), suggesting that constant, but local, autocrine, and paracrine actions of E2 lead to the development and maintenance of endometriotic lesions (3, 10, 11). Multiple studies support the concept that endometriotic cells exhibit enhanced cell survival that reflects an imbalance in cell proliferation and programmed cell death compared with normal endometrium (12-18). Cytochrome P450 enzymes metabolize E2 into bioactive metabolites called catecholestradiols, including 2- and 4-hydroxyestradiol (2-OHE2 and 4-OHE2, respectively), which exhibit structural similarities to catecholamines such as norepinephrine and epinephrine (19-21). The presence of a phenolic A ring “catechol” moiety shared between catecholamines and catecholestradiols mediates similar ligand properties and classifies them as adrenergic receptor “(AR)-mimetic” agents, a property not shared by E2 (22). 3D structural analyses demonstrate that this hydrophilic “catechol” conformation is essential for AR activation (23, 24). During gestation, 2-OHE2 and 4-OHE2 activation of mainly β2-ARs elevates uterine arterial endothelial cell proliferation by an estrogen receptor (ER)-independent pathway (22, 25).
Together with increased local E2 production, aberrant expression of E2 metabolizing enzymes (i.e., CYP1A1 and CYP1B1) is suggested to enhance local generation of 2-OHE2 and 4-OHE2 in women with endometriosis (26-28). Thus, the aim of our current study is to expand our understanding of the role(s) of catecholestradiols previously noted in enhancing uterine artery endothelial cell survival to assess their effects on human endometrial stromal cell (HESC) survival, which has been linked to the pathogenesis of endometriosis. We hypothesize that the ER-independent and β-AR-mediated effects of 2-OHE2 and 4-OHE2 contribute to HESC survival in endometriosis. Therefore, in this study, we evaluated endometriotic tissue expression of β2-ARs in vivo and determined whether 2-OHE2 and 4-OHE2 potentiate HESC viability via AR activation in vitro. Subsequently, mechanistic studies were performed to identify involvement of proliferation, apoptosis, and/or autophagy and relevant intracellular signaling pathways (i.e., MAPK) p38 and Erk1/2 as well as protein kinase B (or Akt) (29) in mediating catecholestradiol-β-AR-induced enhanced HESC viability.
Materials and Methods
Endometrial Tissue Collection
Consent forms and protocols were approved by the institutional review board of the University of South Florida. Endometrial biopsies were obtained from reproductive aged women (range, 32-42 years) undergoing hysterectomy or myomectomy for benign indications. A written informed consent describing the procedures and aims of the study was obtained from each patient before surgery in compliance with regulations concerning the use of human tissues. An endometrial sampling device was used to obtain biopsies from the fundal region of the uterine cavity, which were then transported to the laboratory in medium consisting of Hanks balanced salt solution (Gibco Laboratories, Gaithersburg, MD) and 1% antibiotic and antimycotic agents (Gemini Bio-Products, West Sacramento, CA).
For immunohistochemistry studies, serial paraffin sections were obtained from Yale Pathology Tissue Services under a protocol approved by the Yale Human Investigation Committee. Tissue specimens were derived from eutopic and ectopic endometrial samples (n = 9; 5 from proliferative and 4 from secretory phase) from women with endometriosis (range, 33-49 years) undergoing laparoscopy for infertility or pelvic pain.
Immunohistochemistry
As described previously (30), deparaffinization and rehydration of endometrial paraffin sections (5 µm) were followed by antigen retrieval by boiling in citrate buffer solution (10 mmol/L; pH 6.0) for 25 minutes and incubating with 0.25% trypsin-EDTA (Gibco) for 3 minutes. Endogenous peroxide activity was quenched in 3% H2O2 (in 50% methanol/50% distilled H2O) at room temperature (RT) for 20 minutes. After several washings in 0.1% Tris-buffered saline-tween 20 (TBS-T) and blocking with 10% normal horse serum (Vector Labs, Burlingame, CA) in a humidified chamber for 30 minutes at RT, sections were incubated with mouse monoclonal anti-human β2-AR IgG (1:200 dilution equal to 1 μg/mL, Santa Cruz Biotechnology, Santa Cruz, CA) or with nonspecific mouse IgG isotype-matched antibodies at the same concentrations in 5% normal horse serum overnight at 4°C to serve as negative controls. Myometrial vascular smooth muscle cells were used as a positive control and incubated with the same concentration of primary antibody used for endometrial tissues. After washing, the slides were incubated with biotinylated horse anti-mouse secondary IgG (1:400; Vector Labs) for 30 minutes at RT. The antigen-antibody complex was visualized by incubating with avidin-biotin-peroxidase complex for 30 minutes and diaminobenzidine (Vector Labs) as the chromogen for 4 minutes at RT. The slides were then counterstained with hematoxylin and mounted. β2-AR immunoreactivity was scored semiquantitatively by a histological score (HSCORE) value as described (31). Each slide was evaluated by 2 investigators blinded to the type and source of the tissues. The average HSCORE of both examiners was used.
Isolation and Culture of Primary Human Endometrial Stromal Cells and Culture of Ishikawa Cells
HESCs were isolated and maintained in a monolayer culture, as described previously (32). Briefly, minced human endometrial tissues were digested by collagenase B and deoxyribonuclease for 1 hour at 37°C with agitation, then passed through a 70-μm wire sieve (BD Biosciences, Franklin Lakes, NJ) to remove epithelial cells. HESCs were plated in plastic flasks (75 cm2) and maintained in culture medium consisting of phenol red-free DMEM ( Gibco) and 5% charcoal stripped bovine calf serum (Gemini) plus 1% antibiotic and antimycotic agents (Gemini) at 37°C in 5% CO2. Culture medium was changed every 72 hours until confluence. Cells were then passaged and seeded on culture dishes and grown for subsequent experiments. After the first passage, HESC cultures contain 0% to 7% epithelial cells, no endothelial cells, and 0.2% macrophages as characterized previously (33). Ishikawa cells (endometrial adenocarcinoma cell line) were thawed from frozen stocks and grown to 80% confluence in T-75 plastic flasks containing the same DMEM culture medium and passaged to 96-well plates for the experiments at 37°C in 5% CO2.
Experimental Treatments
Dosages of agents were based on previous concentration- and time-dependent studies (10, 22). HESCs at 70% to 80% confluence were serum starved for 24 hours before treatment. HESCs were treated with vehicle (control) or 10–8 and 10-10 M E2 (Sigma-Aldrich, St. Louis, MO) or 2-OHE2 or 4-OHE2 (Steraloids, Inc, Newport, RI) ± non-subtype-specific antagonists (2 × 10-5 M, 1-hour pretreatment) of α-ARs (phentolamine; Santa Cruz) or β-ARs (propranolol; Sigma-Aldrich) for 15 and 30 minutes to be used in Western blot analysis or 48 hours to be used in 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) cell viability, 5-bromo-2′-deoxyuridine (BrdU) proliferation, or ELISA-based apoptosis assays. Parallel experiments included HESC cultures treated with control or E2 or 2-OHE2 or 4-OHE2 (10–8 M) ± 10–5 M SB203580 (30-minute pretreatment; p38 MAPK inhibitor; MedChemExpress, Monmouth Junction, NJ) for 48 hours for XTT cell viability and for 6 or 96 hours (treatment media were replenished after 48 hours) for quantitative-PCR (qPCR) analyses (Table 1).
List of PCR probes with corresponding TaqMan assay identification
| Primer/probe name | TaqMan assay identification |
|---|---|
| ADRB2 | Hs00240532_s1 |
| PCNA | Hs00696862_m1 |
| MKI67 | Hs01032433_m1 |
| BAX | Hs00180269_m1 |
| BAD | Hs00188930_m1 |
| BCL2 | Hs00608023_m1 |
| IL-8 | Hs00174103_m1 |
| CCL2 | Hs00234140_m1 |
| CDH2 | Hs00983056_m1 |
| SNAI2 | Hs00161904_m1 |
| VEGFA | Hs00900055_m1 |
| ACTB | Hs99999903_m1 |
| Primer/probe name | TaqMan assay identification |
|---|---|
| ADRB2 | Hs00240532_s1 |
| PCNA | Hs00696862_m1 |
| MKI67 | Hs01032433_m1 |
| BAX | Hs00180269_m1 |
| BAD | Hs00188930_m1 |
| BCL2 | Hs00608023_m1 |
| IL-8 | Hs00174103_m1 |
| CCL2 | Hs00234140_m1 |
| CDH2 | Hs00983056_m1 |
| SNAI2 | Hs00161904_m1 |
| VEGFA | Hs00900055_m1 |
| ACTB | Hs99999903_m1 |
List of PCR probes with corresponding TaqMan assay identification
| Primer/probe name | TaqMan assay identification |
|---|---|
| ADRB2 | Hs00240532_s1 |
| PCNA | Hs00696862_m1 |
| MKI67 | Hs01032433_m1 |
| BAX | Hs00180269_m1 |
| BAD | Hs00188930_m1 |
| BCL2 | Hs00608023_m1 |
| IL-8 | Hs00174103_m1 |
| CCL2 | Hs00234140_m1 |
| CDH2 | Hs00983056_m1 |
| SNAI2 | Hs00161904_m1 |
| VEGFA | Hs00900055_m1 |
| ACTB | Hs99999903_m1 |
| Primer/probe name | TaqMan assay identification |
|---|---|
| ADRB2 | Hs00240532_s1 |
| PCNA | Hs00696862_m1 |
| MKI67 | Hs01032433_m1 |
| BAX | Hs00180269_m1 |
| BAD | Hs00188930_m1 |
| BCL2 | Hs00608023_m1 |
| IL-8 | Hs00174103_m1 |
| CCL2 | Hs00234140_m1 |
| CDH2 | Hs00983056_m1 |
| SNAI2 | Hs00161904_m1 |
| VEGFA | Hs00900055_m1 |
| ACTB | Hs99999903_m1 |
Cell survival assays
XTT, a yellow tetrazolium salt, is cleaved to a soluble orange formazan by metabolically active cells to detect viable cells. Serum starved HESCs at passages 1 through 5 or Ishikawa cells were cultured in 96-well plates (5 × 103 cells/well). After 24 or 48 hours of treatment, HESCs or Ishikawa cells (n = 4 with at least duplicate) were incubated with 50 μL XTT solution/well (Cell Signaling Tech, Topsfield, MA) for 4 hours at 37°C. The absorbance was read at 450 nm.
For apoptosis detection, cytoplasmic histone-associated-DNA-fragments (mono- and oligonucleosomes) were quantitated using Cell Death Detection ELISA Plus Kit (Sigma-Aldrich). Colorimetric absorbance was measured at 405 nm after treatments of HESCs (n = 3 with quadruplicate) for 48 hours. A cell proliferation assay kit (Cell Signaling Tech) assessed BrdU incorporation levels into newly synthesized DNA. Following treatments of HESCs (n = 3 in quadruplicate), BrdU solution was added and incubated at 37°C for 48 hours. Colorimetric absorbance was then measured at 450 nm.
All absorbance results were normalized by subtracting the blank measurement and relative colorimetric activity was calculated as a fold change of the control.
Alkaline Phosphatase Assay
To bioassay estrogenic potency of 2-OHE2 and 4-OHE2, we used a well-established technique using induction of alkaline phosphatase activity by E2 in Ishikawa cells (34). This technique is based on a colorimetric assay of alkaline phosphatase enzyme activity in 96-well microtiter plates. We confirmed that Ishikawa cells express ER-α and ER-β by qPCR analysis. Thereafter, alkaline phosphatase activity was assayed exactly as described (34, 35).
Western blot
Total protein from HESC cultures (n = 3) treated with experimental conditions for 15 and 30 minutes was extracted using a 1:1 dilution with 2× Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA) and 5% β-mercaptoethanol. Samples (13 μL) were loaded on 4% to 15% Tris-HCl Ready Gels (Bio-Rad), electrophoretically separated, and electroblotted onto nitrocellulose membrane (Bio-Rad). The membranes were incubated with 10% nonfat dry milk in TBS-T for 1 hour to reduce nonspecific binding. Subsequently, the membranes were incubated overnight at 4°C with primary antibodies against β-actin, LC3B, or total or phosphorylated forms of p38 or Erk1/2 MAPK and Akt (all antibodies from Cell Signaling Tech, Table 2) in TBS-T containing 5% BSA. The membranes were washed several times with TBS-T and incubated with peroxidase labeled anti-rabbit secondary antibody (Vector Labs) diluted at 1:7500 and anti-biotin horseradish peroxidase-linked antibody (Cell Signaling Tech) diluted at 1:1000 in 2.5% nonfat dry milk in TBS-T. The proteins were visualized by light emission on film (Denville Scientific, Inc, Holliston, MA) using chemiluminescence substrate (ThermoScientific, Waltham, MA). Immunoblot bands were quantified using Image Studio Lite. β-actin bands from stripped/reprobed membranes were used as loading control.
List of antibodies with corresponding dilution, molecular weight, and company catalog number
| Antibody | Dilution | Molecular weight (kDa) | Catalog no. (company) |
|---|---|---|---|
| β2-AR | 1:200 | 56-85 | sc-81577 (Santa Cruz) |
| Phospho-p38 MAPK | 1:1000 | 43 | 4631 (Cell Signaling Tech) |
| Total p38 MAPK | 1:1000 | 40 | 9212 (Cell Signaling Tech) |
| Phospho-Erk1/2 | 1:1000 | 42, 44 | 9106 (Cell Signaling Tech) |
| Total Erk1/2 | 1:1000 | 42, 44 | 4348 (Cell Signaling Tech) |
| Phospho-Akt | 1:2000 | 60 | 4060 (Cell Signaling Tech) |
| Total Akt | 1:1000 | 60 | 8596 (Cell Signaling Tech) |
| LC3B | 1:1000 | 14 (LC3-I), 16 (LC3-II) | 3868 (Cell Signaling Tech) |
| β-actin | 1:1000 | 45 | 8457 (Cell Signaling Tech) |
| Antibody | Dilution | Molecular weight (kDa) | Catalog no. (company) |
|---|---|---|---|
| β2-AR | 1:200 | 56-85 | sc-81577 (Santa Cruz) |
| Phospho-p38 MAPK | 1:1000 | 43 | 4631 (Cell Signaling Tech) |
| Total p38 MAPK | 1:1000 | 40 | 9212 (Cell Signaling Tech) |
| Phospho-Erk1/2 | 1:1000 | 42, 44 | 9106 (Cell Signaling Tech) |
| Total Erk1/2 | 1:1000 | 42, 44 | 4348 (Cell Signaling Tech) |
| Phospho-Akt | 1:2000 | 60 | 4060 (Cell Signaling Tech) |
| Total Akt | 1:1000 | 60 | 8596 (Cell Signaling Tech) |
| LC3B | 1:1000 | 14 (LC3-I), 16 (LC3-II) | 3868 (Cell Signaling Tech) |
| β-actin | 1:1000 | 45 | 8457 (Cell Signaling Tech) |
List of antibodies with corresponding dilution, molecular weight, and company catalog number
| Antibody | Dilution | Molecular weight (kDa) | Catalog no. (company) |
|---|---|---|---|
| β2-AR | 1:200 | 56-85 | sc-81577 (Santa Cruz) |
| Phospho-p38 MAPK | 1:1000 | 43 | 4631 (Cell Signaling Tech) |
| Total p38 MAPK | 1:1000 | 40 | 9212 (Cell Signaling Tech) |
| Phospho-Erk1/2 | 1:1000 | 42, 44 | 9106 (Cell Signaling Tech) |
| Total Erk1/2 | 1:1000 | 42, 44 | 4348 (Cell Signaling Tech) |
| Phospho-Akt | 1:2000 | 60 | 4060 (Cell Signaling Tech) |
| Total Akt | 1:1000 | 60 | 8596 (Cell Signaling Tech) |
| LC3B | 1:1000 | 14 (LC3-I), 16 (LC3-II) | 3868 (Cell Signaling Tech) |
| β-actin | 1:1000 | 45 | 8457 (Cell Signaling Tech) |
| Antibody | Dilution | Molecular weight (kDa) | Catalog no. (company) |
|---|---|---|---|
| β2-AR | 1:200 | 56-85 | sc-81577 (Santa Cruz) |
| Phospho-p38 MAPK | 1:1000 | 43 | 4631 (Cell Signaling Tech) |
| Total p38 MAPK | 1:1000 | 40 | 9212 (Cell Signaling Tech) |
| Phospho-Erk1/2 | 1:1000 | 42, 44 | 9106 (Cell Signaling Tech) |
| Total Erk1/2 | 1:1000 | 42, 44 | 4348 (Cell Signaling Tech) |
| Phospho-Akt | 1:2000 | 60 | 4060 (Cell Signaling Tech) |
| Total Akt | 1:1000 | 60 | 8596 (Cell Signaling Tech) |
| LC3B | 1:1000 | 14 (LC3-I), 16 (LC3-II) | 3868 (Cell Signaling Tech) |
| β-actin | 1:1000 | 45 | 8457 (Cell Signaling Tech) |
RNA Extraction and Analysis by qPCR
Total RNA was isolated from cultured HESCs using RNeasy Mini kit (Qiagen, Germantown, MD) and quantified by OD260/280 with Nano-Drop ND-1000 UV-vis Spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE). Reverse transcription and qPCR were performed as described previously (36). The expression of ADRB2, PCNA, MKI67, BAX, BAD, BCL2, IL-8, CCL2, CDH2, SNAI2, VEGFA, and ACTB were each determined by qPCR using TaqMan Gene Expression Assays (all probes from Taqman by Applied Biosystems, Foster City, CA, Table 1) according to the manufacturer’s protocol. All samples were analyzed in duplicate and the average was used. Expression of the target mRNAs was normalized to ACTB levels and the 2−ΔΔCt (cycle threshold) method was used to calculate relative expression levels. Results are reported as fold change of gene expression levels.
siRNA Transfection
HESCs (n = 3) were seeded either in 6-well plates (2 × 105 cell/well) for silencing of ADRB2 expression to confirm transcriptional suppression by qPCR or in 96-well plates (1 × 104 cell/well) for XTT assay to be performed after ADRB2-siRNA transfection. HESCs were transfected with either ADRB2-siRNA (Santa Cruz; SC-39866) to knockdown ADRB2 mRNA expression or nonspecific scramble (control) siRNA (Invitrogen, Carlsbad, CA) by using lipofectamine RNAiMax transfection reagent (Invitrogen) based on the manufacturer’s instruction. Briefly, ADRB2-siRNA was diluted in Opti-MEM (Gibco, Grand Island, NY) at different concentrations (10 and 20 nM), then mixed with diluted transfection reagent. After 15 minutes, complexes were added into cells and incubated for 2 hours before adding growth media. Seventy-two hours after transfection, cells were washed with ice-cold PBS and store at –80°C. Transfection efficiency was performed by qPCR using Taq-Man gene expression assay for ADRB2 with ACTB as internal control. For XTT assay, after 24 hours of control-siRNA or ADRB2-siRNA (20 nM) transfection, cells were treated with 10–8 M of 2-OHE2 or 4-OHE2 for 48 hours.
Statistical Analysis
Statistical analyses were performed with Sigmaplot version 11.0 (Systat Software, Inc, San Jose, CA). Normality testing was performed using the Kolmogorov-Smirnov test. Parametric and nonparametric data were compared using 1-way ANOVA or Kruskal-Wallis ANOVA on ranks followed by post hoc Tukey test for all pairwise comparisons, as deemed appropriate. Statistical significance was defined a priori as P < 0.05 and data are presented as means ± standard error of the mean.
Results
In Situ Regulation of β2-AR in Eutopic and Ectopic Endometrial Samples
Endometrial stromal and epithelial cells in women with endometriosis displayed moderate to weak β2-AR immunoreactivity (Fig. 1A). β2-AR immunostaining intensities were similar between paired eutopic and ectopic HESCs and did not show cycle-dependent expression as detected by HSCORE analysis (Fig. 1A). Human endometrial epithelial cells displayed stronger β2-AR immunostaining in eutopic compared with ectopic tissue during the secretory phase with similar immunostaining evident in the proliferative phase (Fig. 1A and B). Comparison of the basalis with the functionalis layers of eutopic endometrium revealed stronger β2-AR staining in both stromal and epithelial cells in the functionalis layer (Fig. 1A and C; P < 0.05). Specificity of immunostaining was confirmed by replacing the primary antibody with an isotype-matched normal IgG incubation of endometrial samples, which revealed no detectable immunoreactivity (Fig. 1D). Incubation of myometrial tissues with mouse monoclonal anti-human β2-AR IgG (1:200 dilution) resulted in moderate-to-strong staining of vascular smooth muscle cells as well as myometrial smooth muscle cells (Fig. 1E).
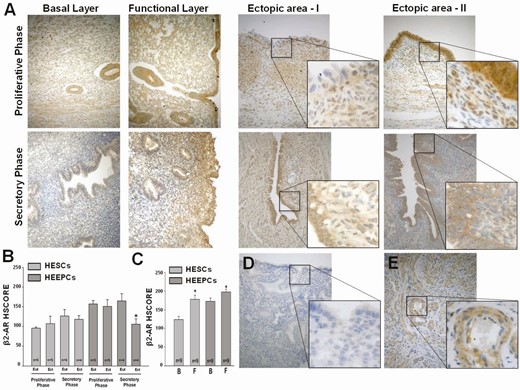
Expression of β2-AR in eutopic and ectopic endometrial tissues. (A) Representative β2-AR immunostaining in endometrial sections from patients with endometriosis during the proliferative and secretory phases in the basal and functional layers of eutopic endometrium as well as their paired ectopic endometrial tissues. Bars in graphs represent means ± standard error of the mean for β2-AR HSCOREs in human endometrial stromal and epithelial cells in eutopic vs ectopic tissues according to (B) menstrual cycle phase or according to (C) location within basalis vs functionalis layers of eutopic endometrium. (D) Negative control of eutopic endometrium displays no immunoreactivity, whereas (E) positive control of myometrium shows moderate-to-strong immunostaining of smooth muscle cells within the tunica media layer of arterioles as well as myometrial smooth muscle cells. *P < 0.05 vs eutopic endometria in secretory phase or vs corresponding basalis layer. AR, adrenergic receptor; B, basalis layer of endometrium; Ect, ectopic; Eut, eutopic; F, functionalis layer of endometrium; HEEPC, human endometrial epithelial cell; HESC, human endometrial stromal cell; HSCORE, histological score. Original magnification 20× with insets 40× (A, D, E).
In Vitro Regulation of HESC Survival by Catecholestradiols
In culture, HESCs treated with control or E2 or 2-OHE2 or 4-OHE2 (10–8 M) for 6 hours (n = 4) showed comparable ADRB2 mRNA levels (P > 0.05; Fig. 2), consistent with our in situ observations of β2-AR protein expression in eutopic and ectopic stromal cells.
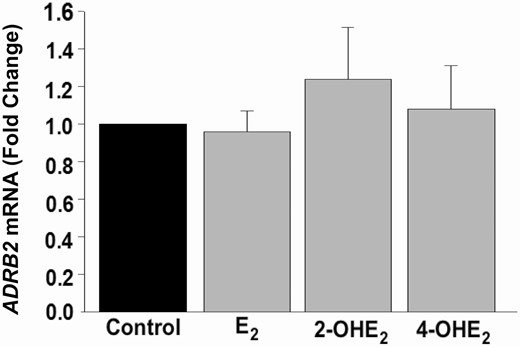
In vivo Gene Expression of ADRB2 in HESCs. qPCR analysis of ADRB2 mRNA expression in control or 10–8 M E2 or 2-OHE2- or 4-OHE2-treated HESC (n = 4) cultures for 6 hours. Bars represent means ± standard error of the mean as fold changes vs control. 2-OHE2, 2-hydroxyestradiol; 4-OHE2, 4-hydroxyestradiol; E2, 17β-estradiol; HESC, human endometrial stromal cell; qPCR, quantitative PCR.
Previously, Jobe et al (22) showed catecholestradiol responses via ARs, specifically β2-AR, regulate uterine artery endothelial cell proliferation during gestation. Based on these observations, XTT assays (n = 6 with at least triplicate) were first performed following concentration-dependent incubations with physiologic steroid levels. 2-OHE2 and 4-OHE2 at 10-10 M and 10-8 M significantly increased HESC survival compared with control (P < 0.01; Fig. 3A). Interestingly, 4-OHE2 was more potent than E2 at 10-10 M (P < 0.05; Fig. 3A).
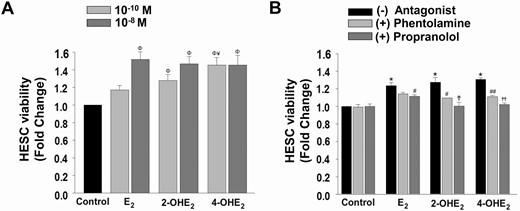
Regulation of HESC viability by E2or 2-OHE2or 4-OHE2. (A) XTT cell viability index in HESC monolayers (n = 6) treated with control (vehicle) or E2 or 2-OHE2 or 4-OHE2 at 10-10 M and 10-8 M. (B) Parallel experiments (n = 4) evaluating inhibitory effects of phentolamine (α-AR antagonist; 2 × 10-5 M 1-hour pretreatment) or propranolol (β-AR antagonist; 2 × 10-5 M 1-hour pretreatment) on 10-8 M 2-OHE2 or 4-OHE2-induced cell viability. Bars represent means ± standard error of the mean as fold changes vs control. *P < 0.05 or ΦP < 0.01 vs control; ¥P < 0.05 vs E2. Partial inhibition (#P < 0.05, ##P < 0.001), defined as statistically significant attenuation of the catecholestradiol-induced survival response with addition of an AR antagonist. Complete inhibition (ϮP < 0.05, ϮϮP < 0.001), defined as statistically significant abrogation of the catecholestradiol-induced survival response with addition of an AR antagonist to the survival levels of control treatment. 2-OHE2, 2-hydroxyestradiol; 4-OHE2, 4-hydroxyestradiol; AR, adrenergic receptor; E2, 17β-estradiol; HESC, human endometrial stromal cell; XTT: 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide.
To further investigate whether the HESC survival effects of 2-OHE2 or 4-OHE2 are β-AR-mediated, similar experiments (n = 4 with duplicate) were performed following pretreatment with a non-subtype-specific AR antagonist, phentolamine (α-blocker; 2 × 10-5 M) or propranolol (β-blocker; 2 × 10-5 M). These analyses revealed that propranolol completely abrogated 2-OHE2- and 4-OHE2-enhanced HESC viability to the level of control (P < 0.05 and P < 0.001, respectively; Fig. 3B) and partially inhibited E2-enhanced HESC survival (P < 0.05). Additionally, phentolamine partially inhibited the catecholestradiol-induced increase in HESC survival (P < 0.05) but did not affect the E2-induced increase in HESC survival. Therefore, subsequent experiments focused on β-AR mediated 2-OHE2- and 4-OHE2 effects on HESC survival.
β-AR-mediated, ER-independent, Catecholestradiol-induced Survival of Ishikawa Cells
Presence of β2-AR immunostaining in epithelial cells in eutopic and ectopic endometrial tissues was further assessed in vitro using Ishikawa cell cultures. Ishikawa cells were treated for 24 or 48 hours with control or E2 or 2-OHE2 or 4-OHE2 (10–8 M) following with or without 1 hour of pretreatment with propranolol (β-blocker; 2 × 10-5 M) or ICI182,780 (nonspecific ER antagonist; 10–6 M). XTT analysis (n = 3) detected, similar to HESCs, enhanced cell survival in 2-OHE2- and 4-OHE2-treated Ishikawa cells when compared with control (Fig. 4A, P < 0.05). Pretreatment with β-AR antagonist completely abrogated catecholestradiol-induced cell survival (P < 0.05). To assess whether the possible effects of catecholestradiols were mediated via ERs, the cells were pretreated with nonspecific ER-antagonist ICI182,780. XTT analysis confirmed the inhibitory actions of ICI182,780 on E2 (P < 0.05), but not on 2-OHE2- and 4-OHE2-treated Ishikawa cells (Fig. 4A).
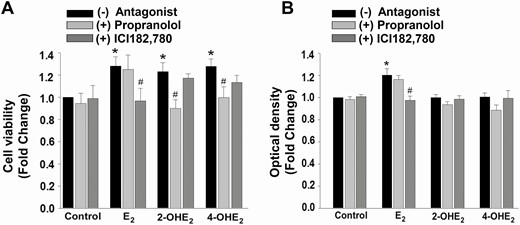
Cell survival effect of catecholestradiols in Ishikawa cells is not associated with estrogen receptors. Effect of propranolol (β-adrenergic receptor antagonist; 2 × 10-5 M 1-hour pretreatment) or ICI182,780 (nonspecific antagonist of estrogen receptors; 10-6 M 1-hour pretreatment) on catecholestradiol-induced Ishikawa cell survival treated with control (vehicle) or 10-8 M E2 or 2-OHE2 or 4-OHE2 as measured by XTT assay (A, n = 3) and estrogenicity via induction of alkaline phosphatase activity (B, n = 9). Bars represent means ± standard error of the mean as fold changes vs control. *P < 0.05 vs control; #P < 0.05 treatment with steroid plus antagonist vs treatment with corresponding steroid-only. 2-OHE2, 2-hydroxyestradiol; 4-OHE2, 4-hydroxyestradiol; E2, 17β-estradiol; ER, estrogen receptor; HESC, human endometrial stromal cell; XTT, 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide.
Furthermore, estrogenic alkaline phosphatase activity revealed that E2, but neither 2-OHE2- nor 4-OHE2-treated Ishikawa cells stimulated alkaline phosphatase activity (Fig. 4B, P < 0.05). The E2-induced alkaline phosphatase activity was abrogated by ICI182,780 pretreatment (P < 0.05), but not by propranolol pretreatment (Fig. 4B).
Regulation of Proliferation and Apoptosis Contributes to Catecholestradiol-induced HESC Survival
To elucidate whether the induction of HESC survival in response to catecholamine treatment reflects proliferation and/or apoptosis, BrdU proliferation and ELISA-based cell death detection assays were performed in parallel experiments. Following 48 hours of experimental treatment, an increased proliferation index in response to E2 and 4-OHE2 (P < 0.05 vs control; Fig. 5A) and decreased apoptosis levels in HESCs treated with 2-OHE2 and 4-OHE2 (P < 0.01 vs control; Fig. 5B) were detected. These results indicate that elevated HESC survival in response to 2-OHE2 treatment is associated solely with reduced apoptosis, whereas induction of HESC survival by 4-OHE2 reflects contributions of both proliferative and anti-apoptotic mechanisms.
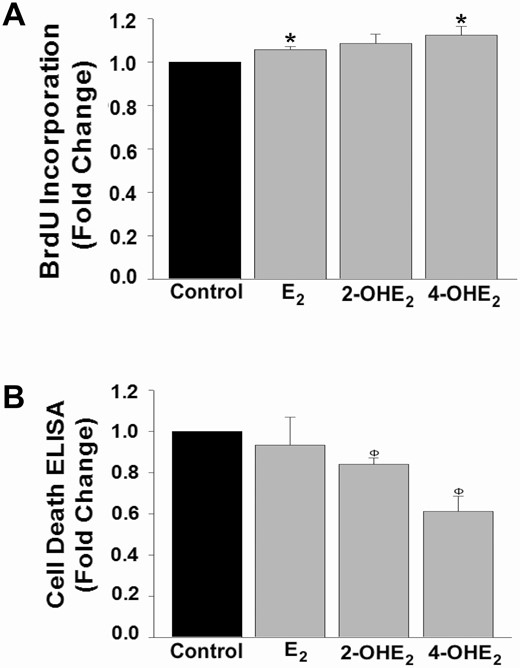
Catecholestradiol-induced HESC survival is via proliferative and anti-apoptotic mechanisms. (A) BrdU incorporation levels or (B) cell death detection ELISA in HESC cultures (n = 3) treated 48 hours with control or 10-8 M E2 or 2-OHE2 or 4-OHE2. Bars represent means ± standard error of the mean as fold changes vs control. *P < 0.05 or ΦP < 0.01 vs control. 2-OHE2, 2-hydroxyestradiol; 4-OHE2, 4-hydroxyestradiol; BrdU, 5-bromo-2′-deoxyuridine; E2, 17β-estradiol; HESC, human endometrial stromal cell.
We also measured transcription levels of several markers of proliferation (PCNA, MKI67), apoptosis (BAX, BAD), or anti-apoptosis (BCL2). Based on our qPCR analysis of cultured HESCs incubated with control or 2-OHE2 or 4-OHE2 (10–8 M) for either 6 hours (n = 4) or 96 hours (n = 5), we observed only statistically significant decreased BAX expression in response to 2-OHE2 compared with control at 96 hours (P < 0.01; Fig. 6). No other significant alterations in mRNA regulation including PCNA, MKI67, BAD, or BCL2 (data not shown) were detected at either time point.
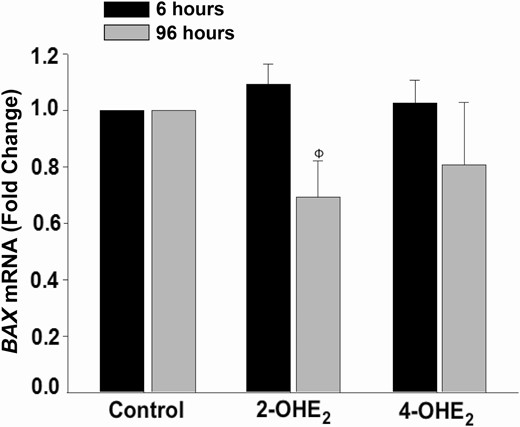
2-OHE2treatment decreases BAX mRNA expression in HESC cultures.BAX mRNA expression by qPCR under treatment with control or 2-OHE2 or 4-OHE2 (10–8 M) for 6 hours (n = 4) or 96 hours (n = 5). Bars represent means ± standard error of the mean as fold changes vs control. ΦP < 0.01 vs control at 96 hours. 2-OHE2, 2-hydroxyestradiol; 4-OHE2, 4-hydroxyestradiol; HESC: human endometrial stromal cell; q PCR, quantitative PCR.
ADRB2 Silencing Decreases 2-OHE2- or 4-OHE2- induced Survival Rates in HESCs
To understand whether survival effects of 2-OHE2 and 4-OHE2 are specifically mediated by β2-AR, we first performed 10- and 20-nM ADRB2-siRNA transfections to silence ADRB2 expression in HESCs, with the expectation of obtaining ≥ 50% suppression on ADRB2 mRNA. After 72 hours of siRNA transfection, compared with scramble-siRNA, qPCR analysis confirmed that ADRB2-siRNA transcriptional suppression efficiency was 44% and 65% at 10- and 20-nM concentrations, respectively (P < 0.01; Fig. 7A), which led us to use 20 nM ADRB2-siRNA concentration for subsequent treatments.
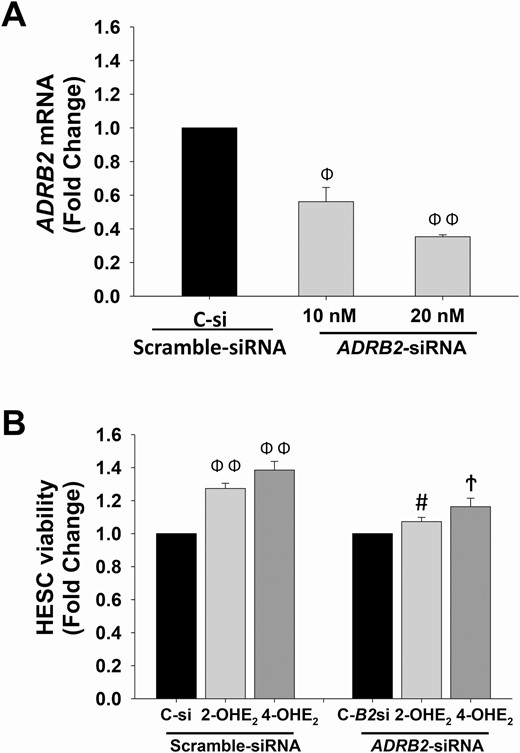
ADRB2-siRNA transfection blocks 2-OHE2- or 4-OHE2-induced HESC survival. (A) ADRB2 mRNA expression levels in HESCs (n = 3) treated with either scramble-siRNA (control) or ADRB2-siRNA at 10 nM and 20 nM for 72 hours. (B) XTT cell viability index in HESCs (n = 3) pretreated with scramble-siRNA or ADRB2-siRNA (20 nM) for 24 hours, and followed by 10-8 M of 2-OHE2 or 4-OHE2 treatment for 48 hours. Bars represent means ± standard error of the mean. ΦP < 0.01 or ΦΦP ≤ 0.001 vs scramble-siRNA (C-si) treated control, #P < 0.05 vs C-si treated 2-OHE2, ϮP < 0.01 vs C-si treated 4-OHE2. 2-OHE2, 2-hydroxyestradiol; 4-OHE2, 4-hydroxyestradiol; C-B2si, ADRB2-siRNA; HESC, human endometrial stromal cells; XTT, 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide.
Next, we performed an XTT assay to evaluate cell survival rates in 2-OHE2- or 4-OHE2-treated and ADRB2 silenced HESCs. Compared with scramble-siRNA-treated control, 2-OHE2 (P < 0.001) and 4-OHE2 (P < 0.01) treatments significantly increased HESC survival, whereas in ADRB2 silenced groups, 2-OHE2 and 4-OHE2 treatments failed to produce a significant effect on HESC survival. Moreover, there was a significant decrease in 2-OHE2 (P < 0.05) and 4-OHE2 (P < 0.01) treatments in scramble-siRNA versus ADRB2-siRNA-treated HESCs (Fig. 7B).
2-OHE2 or 4-OHE2 Preferentially Activates p38 MAPK Intracellular Signaling in a β-AR-dependent Manner
To identify signaling pathways that mediate catecholestradiol-induced HESC survival, we measured activation (phosphorylation) and total levels of relevant signaling pathways. Our prior studies suggest that catecholestradiols stimulate proliferation of pregnant uterine artery endothelial cells (P-UAECs) via β2-AR as a result of increased phosphorylation of Erk1/2, p38, and JNK MAPKs (22, 29). Therefore, we evaluated cultured HESCs (n = 3) incubated with control medium or medium containing 10–8 M E2 or 2-OHE2 or 4-OHE2 for 15 and 30 minutes ± β-AR antagonist propranolol (2 × 10-5 M, 1-hour pretreatment) and observed that phosphorylation levels of p38 MAPK were increased by either 2-OHE2 or 4-OHE2, but not by E2, at 15 and 30 minutes posttreatment (Fig. 8A and B). Furthermore, β-AR blockade mitigated the catecholestradiol-induced p38 MAPK phosphorylation (Fig. 8A and B). However, neither phosphorylation nor total levels of Erk1/2 MAPK or Akt signaling cascades displayed a difference among treatment groups (data not shown).
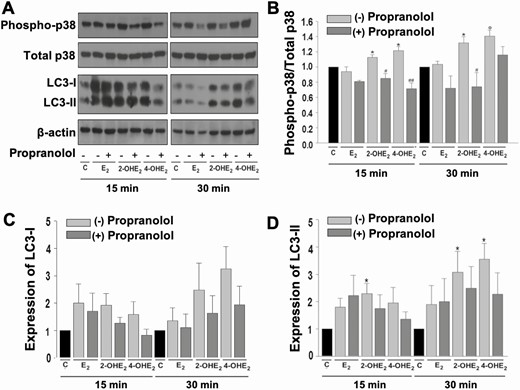
Catecholestradiols activate p38 MAPK signaling in HESCs in a β-AR-dependent manner. (A) Representative immunoblot analysis of phosphorylation (active, aka phospho-) and total p38 MAPK, LC3-I, LC3-II, and β-actin levels in confluent HESCs treated with control or 10-8 M E2 or 2-OHE2 or 4-OHE2 ± 2 × 10-5 M propranolol (1-hour pretreatment) for 15 and 30 minutes. Graphs represent differential expression of (B) phospho-p38 MAPK vs total p38 MAPK, (C) LC3-I/β-actin, and (D) LC3-II/β-actin. Bars represent means ± standard error of the mean (n = 3) as fold changes vs control. *P < 0.05 and ΦP < 0.01 vs control; #P < 0.05 or ##P < 0.01 vs corresponding steroid treatment alone. 2-OHE2, 2-hydroxyestradiol; 4-OHE2, 4-hydroxyestradiol; AR, adrenergic receptor; C, control; E2, 17β-estradiol; HESC, human endometrial stromal cell.
An association between p38 MAPK signaling and activation of LC3-II-mediated cytoprotective autophagy has been demonstrated in previous studies (37, 38). Therefore, we next assessed LC3-I and LC3-II protein expression via Western blot. LC3-I expression did not change significantly in any treatment condition (Fig. 8A and C); however, treatment with 2-OHE2 at 15 and 30 minutes or 4-OHE2 at 30 minutes significantly increased LC3-II expression and pretreatment with the β-AR antagonist propranolol abolished this significant increase (Fig. 8A and D).
p38 MAPK Mediates 2-OHE2- or 4-OHE2-induced HESC Survival
To confirm that catecholestradiol-induced cell survival is functionally mediated by p38 MAPK signaling, HESC cultures were treated with control or E2 or 2-OHE2 or 4-OHE2 (10–8 M) in the presence and absence of the p38 MAPK specific inhibitor, SB203580 (10–5 M, 30-minute pretreatment). Treatment with E2 resulted in increased cell survival responses compared with control (P < 0.05), and this E2 response was not significantly suppressed by SB203580. XTT analysis at 48 hours posttreatment confirmed complete abrogation of both 2-OHE2- and 4-OHE2-enhanced cell survival response by SB203580 (P < 0.05 and P < 0.001, respectively; Fig. 9).
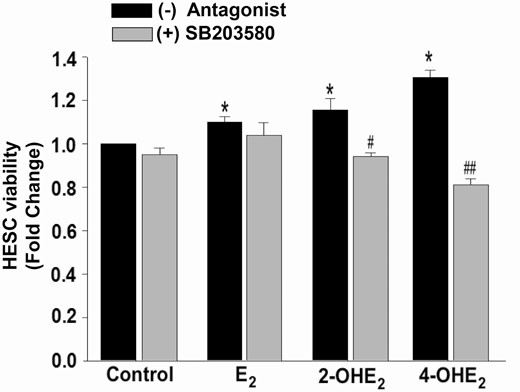
Inhibition of p38 MAPK signaling reverses 2-OHE2- or 4-OHE2-induced HESC survival. XTT assay demonstrating viability of HESCs treated with control or 10-8 M E2 or 2-OHE2 or 4-OHE2 ± p38 MAPK inhibitor (10-5 M SB203580 for 30 minutes pretreatment) for 48 hours. SB203580 inhibitor completely abrogates 2-OHE2 and 4-OHE2-induced HESC survival. Bars represent means ± standard error of the mean (n = 4 in triplicate) as fold changes vs control. *P < 0.05 vs control; #P < 0.05 or ##P < 0.01 vs corresponding steroid alone treatment. 2-OHE2, 2-hydroxyestradiol; 4-OHE2, 4-hydroxyestradiol; E2, 17β-estradiol; HESC, human endometrial stromal cell; XTT: 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide.
Endometriotic loci have an excess inflammatory milieu (39, 40) with increased p38 MAPK activation contributing to induction of inflammation, angiogenesis, and cell differentiation (10, 41), which led us to perform parallel studies conducted in the presence or absence of p38 MAPK inhibition to assess mRNA expression of markers of inflammation (IL-8, CCL2), mesenchymal-epithelial transition (CDH2, SNAI2), or angiogenesis (VEGFA). Cultured HESCs were treated with vehicle (control) or E2 or 2-OHE2 or 4-OHE2 (10–8 M) ± SB203580 (10–5 M, 30-minute pretreatment) for 6 hours; however, no differential expression of IL-8, CCL2, CDH2, SNAI2, or VEGFA was detected (data not shown).
Discussion
Catecholestradiols, 2-OHE2 and 4-OHE2, are biologically active E2 metabolites that bind ARs with high affinity (42-44). Our group has previously demonstrated that 2-OHE2 and 4-OHE2, unlike E2, induced proliferation of P-UAECs is via an ER-independent, β2-AR-mediated regulatory pathway (22). In addition to increased E2 production by aromatase in endometriotic cells, aberrant expression of E2-metabolizing enzymes, including CYP1A1 and CYP1B1, enhances local generation of catecholestradiols in ectopic endometrium (26-28). However, no studies have investigated the direct effect of 2-OHE2 and 4-OHE2 on HESC cultures. The results presented here demonstrate that catecholestradiols activate ARs to enhance human endometrial stromal and epithelial cells, as demonstrated with the Ishikawa cell line, viability. These results support the hypothesis that catecholestradiols contribute to the establishment and/or progression of endometriosis.
Sampson’s theory of retrograde menstruation represents the most widely accepted explanation contributing to the pathogenesis of endometriosis (45), which requires implantation and growth of endometrial tissues at extrauterine sites. In situ results in the current study demonstrate that endometriotic epithelial and stromal cells of both eutopic and ectopic tissues display an overall comparable presence of β2-AR, which is also maintained in in vitro conditions as confirmed by our qPCR analysis of HESC cultures. Furthermore, compared with the basalis layer, the functionalis layer of the endometrium displays enhanced β2-AR immunoreactivity, which directly supports the potential contribution of ARs to retrograde-menstruation-related implantation and propagation of endometriotic cells at ectopic loci. Weak to moderate β2-AR immunoreactivity was also detected in spiral artery vascular smooth muscle cells within the endometrium, which requires further investigation into whether spiral artery β2-ARs have spatial and/or temporal changes in expression throughout the menstrual cycle.
Previous studies reported detection of the expression of several other AR isoforms in human reproductive organs, including α2C-AR, which is upregulated by decidualization stimuli in stromal cell cultures obtained from either normal endometria or endometriotic cysts and modulates trophoblast syncytialization and migration in the placenta (46, 47). Therefore, enhanced β2-AR immunoreactivity in the functionalis layer may not only be involved in the pathogenesis of endometriosis but may also affect embryo implantation and trophoblast invasion in patients with endometriosis. We observed nuclear and cytoplasmic β2-AR immunostaining. Several studies previously noted that both α-AR and β-AR can be found in the nucleus (48-50). Future studies are warranted to determine nuclear versus membrane function responses catecholestradiols via β2-AR to identify their role especially in endometriosis.
Endometriotic cells exhibit enhanced cell survival compared with normal endometrium as reflected by both increased proliferation and reduced apoptosis (12-18) suggesting that approaches to disrupt mechanisms that promote cell survival are likely to prove effective in managing endometriosis. Our current in vitro results revealed that physiological levels of 2-OHE2 and 4-OHE2 increase HESC and Ishikawa cell viability via a nonestrogenic pathway as identified by low estrogenicity (alkaline phosphatase activity) and persistence of stimulation of cell survival despite ER antagonism by ICI182,780. Complete and partial inhibition of catecholestradiol-stimulated HESC viability by the AR blockers propranolol and phentolamine, respectively, indicates that these catecholestradiol effects are primarily β-AR and partially α-AR mediated. Silencing of ADRB2 confirmed catecholestradiol contribution to HESC viability via the β2-AR. Moreover, marginal down-regulation of E2-induced HESC survival by a β-AR blocker potentially reflects endogenous metabolism of E2 to catecholestradiols in HESC cultures.
Further analysis into the cell survival mechanisms demonstrated that 2-OHE2-mediated cell survival is primarily mediated by an anti-apoptotic mechanism confirmed by decreased expression of BAX, a pro-apoptosis protein, in HESCs after 96 hours of treatment with 2-OHE2. On the other hand, 4-OHE2-mediated cell survival comprises both proliferative and anti-apoptotic mechanisms, which is likely explained by the combined modest alterations in proliferative and anti-apoptotic markers at the transcriptional level because 4-OHE2 weakly induces MKI67 and inhibits BAX at levels that did not achieve statistical significance.
Consistent with our results, studies in rodents observed endometriotic angiogenesis and proliferation via a β2-AR agonist as well as decreased implant volume and recurrence risk after propranolol administration (51-53). Propranolol is a US Food and Drug Administration-approved medication that in pregnancy is used to treat hyperthyroidism, migraines, and, less commonly, hypertension (54, 55). Furthermore, propranolol does not impair fertility and is not linked to enhanced risk of major congenital anomalies in pregnancy (56-58), thus providing a safe and potentially effective option for endometriosis treatment, especially in patients desiring fertility. Further investigation into the efficacy of this treatment in clinical trials is warranted.
The p38 MAPK cascade regulates a variety of biological processes, including inflammation, autophagy, as well as cell growth and differentiation (41). In P-UAECs, p38 MAPK signaling is required for catecholestradiol-induced proliferation (29). The current study expanded our investigation to HESCs by demonstrating that catecholestradiol-β-AR interactions induce HESC survival via activation of p38 MAPK signaling, which likely contributes to endometriosis development (10, 32). Our laboratory previously demonstrated increased p38 MAPK activity in eutopic and ectopic endometria with increased phospho-p38 MAPK immunostaining in epithelial and stromal cells in the functionalis compared to basalis layer (10), which parallels the β2-AR immunostaining pattern in our present study. This premise is further supported by our present findings that 2-OHE2 and 4-OHE2 rapidly activate p38 MAPK, as well as LC3-II, signaling pathways in a β-AR-dependent manner because the β-AR antagonist, propranolol, reversed these catecholestradiol-induced activations. These observations indicate that catecholestradiols regulate HESC survival via β-AR-mediated p38 MAPK signaling and activation of LC3-II-mediated cytoprotective autophagy. Therefore, enhanced cytoprotective autophagic responses likely contribute to the catecholestradiol-induced proliferative and anti-apoptotic mechanisms (37, 59) and, additionally, may serve as an explanation for our modest alterations in proliferative and anti-apoptotic markers at the RNA level. Moreover, XTT cell survival analyses with p38 MAPK inhibition further reaffirm that catecholestradiol-induced HESC survival is mediated by activation of p38 MAPK signaling. Last, the failure of catecholestradiol to regulate transcription levels of IL8, CCL2, CDH2, SNAI2, and VEGFA in HESCs indicate a cell survival-specific role rather than promotion of inflammation, mesenchymal-epithelial transition, and/or angiogenesis.
In conclusion, these findings indicate that 2-OHE2 or 4-OHE2 induce endometrial stromal and epithelial cell survival in a β-AR-dependent and ER-independent manner by activating the p38 MAPK signaling cascade. These observations suggest novel treatment options to prevent endometriotic cell implantation and/or progression of endometriosis through inhibition of β-ARs (i.e., propranolol) and/or of E2-metabolizing enzymes that generate 2-OHE2 or 4-OHE2 alone or in combination with current endometriosis therapeutics (e.g., progestins, aromatase inhibitors).
Abbreviations
- 2-OHE2
2-hydroxyestradiol
- 4-OHE2
4-hydroxyestradiol
- AR
adrenergic receptor
- BrdU
5-bromo-2′-deoxyuridine
- E2
17β-estradiol
- ER
estrogen receptor
- HESC
human endometrial stromal cell
- HSCORE
histological score
- P-UAEC
pregnant uterine artery endothelial cell
- qPCR
quantitative PCR
- RT
room temperature
- TBS-T
Tris-buffered saline-tween 20
- XTT
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide.
Acknowledgments
The authors thank Maya Okuka for her excellent technical assistance and the participants that contributed to the study.
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}