





Abstract
PAX8 is a transcription factor required for thyroid development, and its mutation causes congenital hypothyroidism (CH). More than 20 experimentally verified loss-of-function PAX8 mutations have been described, and all but one were located in the DNA-binding paired domain.
We report the identification and functional characterization of 3 novel truncating PAX8 mutations located outside the paired domain.
Three CH probands, diagnosed in the frame of newborn screening, had thyroid hypoplasia and were treated with levothyroxine. Next-generation sequencing-based mutation screening was performed. Functionality of the identified mutations were verified with Western blotting, intracellular localization assays, and transactivation assays with use of HeLa cells. Luciferase complementation assays were used to evaluate the effect of mutations on the interaction between PAX8 and its partner, NKX2-1.
Each proband had novel truncating PAX8 mutations that were I160Sfs*52, Q213Efs*27, and F342Rfs*85. Western blotting showed destabilization of the I160fs-PAX8 protein. Q213fs-PAX8 and F342fs-PAX8 showed normal protein expression levels and normal nuclear localization, but showed loss of transactivation of the luciferase reporter. By luciferase complementation assays, we showed that PAX8-NKX2-1 interaction was defective in Q213fs-PAX8. We also characterized the recombinant PAX8 proteins, and found that the protein sequence corresponding to exon 10 (363-400 aa residues) was essential for the PAX8-NKX2-1 interaction.
Clinical and molecular findings of 3 novel truncating PAX8 mutations located outside the paired domain were reported. Experiments using cultured cells and recombinant proteins showed that the C-terminal portion (ie, 363-400 aa) of PAX8 is required for the PAX8-NKX2-1 interaction.
Congenital hypothyroidism (CH) is the most common congenital endocrine disorder, affecting approximately 1 in 3000 to 4000 newborns (1). Thyroid hormone deficiency in early infancy leads to irreversible intellectual disability, but early diagnosis and treatment can prevent the disability. Clinically, CH patients can be classified into 2 major categories, including CH with decreased amount of thyroid tissues (thyroid dysgenesis; thyroid ectopia, aplasia, and hypoplasia) and CH with goiter (thyroid dyshormonogenesis). Thyroid dysgenesis accounts for approximately 80% of permanent and severe cases and thyroid dyshormonogenesis accounts for the remaining 10% to 20%. In relatively mild cases, the morphology of the thyroid gland is often normal and would be difficult to classify into either of the 2 categories.
Thyroid dysgenesis is generally considered to be a nongenetic disorder (2, 3). The best-characterized genetic form of thyroid dysgenesis is PAX8 mutations, which account for 1.4% to 3.3% of cases (4-6). CH due to heterozygous loss-of-function PAX8 mutations has been described in 30 unrelated families harboring a total of 22 experimentally verified mutations (4-24). Although the typical phenotype of CH due to PAX8 mutations is permanent CH with thyroid hypoplasia, hypothyroidism of mutation carriers may be subclinical or completely normal (ie, incomplete penetrance). All but one of those mutations have been located in the DNA binding domain called the paired domain. The exception is the p.Thr277* mutation, which was observed in a family showing incomplete penetrance (4).
PAX8 is a member of the PAX gene family, which is characterized by the presence of an evolutionarily conserved paired domain in the N terminus. In mice and humans, Pax8/PAX8 is expressed in the thyroid follicular cells (TFCs) from prenatal to adulthood (25). In early studies using the heterologous expression experiments, a synergistic interaction between rat Pax8 and another thyroid-specific transcription factor, rat Nkx2-1 (also known as thyroid transcription factor-1) was postulated (26, 27). Subsequently, the critical role of this interaction in TFC differentiation was elegantly demonstrated by generation of functional TFCs cells from mouse embryonic stem cells by forced expression of the 2 factors (28). In this differentiation experiments, the synergy between murine Pax8 and murine Nkx2-1 on messenger RNA expression of multiple thyroid-specific molecules such as thyrotropin (TSH) receptor, sodium iodine symporter, and thyroglobulin was observed. Although growing evidence indicate the importance of the Pax8–Nkx2-1 interaction, the molecular mechanism(s) of it remain largely unknown.
In the present study, we report the identification and functional characterization of 3 novel PAX8 mutations, all of which were truncating mutations located outside the paired domain. A series of functional analyses revealed a previously underrecognized role of the C-terminal portion of PAX8, which is essential for the PAX8–NKX2-1 interaction.
Materials and Methods
Clinical reports
A summary of clinical information is shown in Table 1. Three probands were born to nonconsanguineous Japanese parents and had no family history of thyroid disorders, including CH and autoimmune thyroid disorders. They had positive result in newborn screening (NBS) for CH, and were diagnosed as CH after the initial evaluation. They were promptly treated with levothyroxine (L-T4), and showed normal growth and intellectual development at their last clinical visits.
Clinical characteristics of the truncating PAX8 mutation carriers
| Family 1 I160Sfs*52 | Family 2 Q213Efs*27 | Family 3 F342Rfs*85 | |||
|---|---|---|---|---|---|
| Variable | Patient 1 | Mother of Patient 1 | Patient 2 | Patient 3 | Reference |
| Age, y, sex | 16, F | 45, F | 9, M | 24, F | |
| Blood-spot TSH at NBS, mU/L | 83 | NA | 98 | > 80 | |
| Information at diagnosis | |||||
| Age, y | 0.1 | 0.1 | 0.1 | ||
| Serum TSH, mU/L | 126 | NE | 399 | 176 | 1.7-9.1 |
| Serum free T4, ng/dL | 1.4 | NE | 0.28 | 0.81 | 0.9-2.3 |
| Serum Tg, ng/mL | 240 | NE | NA | > 320 | 3.8-56 |
| Thyroid ultrasonography | |||||
| Age at evaluation, y | 16 | 8 | 13 | ||
| Size, SDS | –2.3 | NE | –2.4 | –2.0 | |
| Information at last evaluation | |||||
| Age, y | 16 | 45 | 8 | 24 | |
| Height, cm (SDS) | 150 (–1.4) | 158 (0.0) | 130 (–0.1) | 159.7 (0.3) | |
| Weight, kg (SDS) | 44.3 (– .9) | NE | 28.9 (–0.0) | 60.5 (0.7) | |
| Serum TSH, mU/L | 9.9 | 1.5 | NE | 1.8 | 0.5-5.0 |
| Serum free T4, ng/dL | 1.30 | 1.33 | NE | 1.25 | 0.9-1.8 |
| Serum Tg, ng/mL | 11.1 | 13.9 | NE | NE | < 30 |
| L-T4 dose, µg/kg/d | 1.1 | Not treated | 1.5 | Not treated | |
| Family 1 I160Sfs*52 | Family 2 Q213Efs*27 | Family 3 F342Rfs*85 | |||
|---|---|---|---|---|---|
| Variable | Patient 1 | Mother of Patient 1 | Patient 2 | Patient 3 | Reference |
| Age, y, sex | 16, F | 45, F | 9, M | 24, F | |
| Blood-spot TSH at NBS, mU/L | 83 | NA | 98 | > 80 | |
| Information at diagnosis | |||||
| Age, y | 0.1 | 0.1 | 0.1 | ||
| Serum TSH, mU/L | 126 | NE | 399 | 176 | 1.7-9.1 |
| Serum free T4, ng/dL | 1.4 | NE | 0.28 | 0.81 | 0.9-2.3 |
| Serum Tg, ng/mL | 240 | NE | NA | > 320 | 3.8-56 |
| Thyroid ultrasonography | |||||
| Age at evaluation, y | 16 | 8 | 13 | ||
| Size, SDS | –2.3 | NE | –2.4 | –2.0 | |
| Information at last evaluation | |||||
| Age, y | 16 | 45 | 8 | 24 | |
| Height, cm (SDS) | 150 (–1.4) | 158 (0.0) | 130 (–0.1) | 159.7 (0.3) | |
| Weight, kg (SDS) | 44.3 (– .9) | NE | 28.9 (–0.0) | 60.5 (0.7) | |
| Serum TSH, mU/L | 9.9 | 1.5 | NE | 1.8 | 0.5-5.0 |
| Serum free T4, ng/dL | 1.30 | 1.33 | NE | 1.25 | 0.9-1.8 |
| Serum Tg, ng/mL | 11.1 | 13.9 | NE | NE | < 30 |
| L-T4 dose, µg/kg/d | 1.1 | Not treated | 1.5 | Not treated | |
Abbreviations: F, female; L-T4, levothyroxine; M, male; NA, not applicable; NBS, newborn screening; NE, not evaluated; SDS, SD score; T4, thyroxine; Tg, thyroglobulin; TSH, thyrotropin.
Clinical characteristics of the truncating PAX8 mutation carriers
| Family 1 I160Sfs*52 | Family 2 Q213Efs*27 | Family 3 F342Rfs*85 | |||
|---|---|---|---|---|---|
| Variable | Patient 1 | Mother of Patient 1 | Patient 2 | Patient 3 | Reference |
| Age, y, sex | 16, F | 45, F | 9, M | 24, F | |
| Blood-spot TSH at NBS, mU/L | 83 | NA | 98 | > 80 | |
| Information at diagnosis | |||||
| Age, y | 0.1 | 0.1 | 0.1 | ||
| Serum TSH, mU/L | 126 | NE | 399 | 176 | 1.7-9.1 |
| Serum free T4, ng/dL | 1.4 | NE | 0.28 | 0.81 | 0.9-2.3 |
| Serum Tg, ng/mL | 240 | NE | NA | > 320 | 3.8-56 |
| Thyroid ultrasonography | |||||
| Age at evaluation, y | 16 | 8 | 13 | ||
| Size, SDS | –2.3 | NE | –2.4 | –2.0 | |
| Information at last evaluation | |||||
| Age, y | 16 | 45 | 8 | 24 | |
| Height, cm (SDS) | 150 (–1.4) | 158 (0.0) | 130 (–0.1) | 159.7 (0.3) | |
| Weight, kg (SDS) | 44.3 (– .9) | NE | 28.9 (–0.0) | 60.5 (0.7) | |
| Serum TSH, mU/L | 9.9 | 1.5 | NE | 1.8 | 0.5-5.0 |
| Serum free T4, ng/dL | 1.30 | 1.33 | NE | 1.25 | 0.9-1.8 |
| Serum Tg, ng/mL | 11.1 | 13.9 | NE | NE | < 30 |
| L-T4 dose, µg/kg/d | 1.1 | Not treated | 1.5 | Not treated | |
| Family 1 I160Sfs*52 | Family 2 Q213Efs*27 | Family 3 F342Rfs*85 | |||
|---|---|---|---|---|---|
| Variable | Patient 1 | Mother of Patient 1 | Patient 2 | Patient 3 | Reference |
| Age, y, sex | 16, F | 45, F | 9, M | 24, F | |
| Blood-spot TSH at NBS, mU/L | 83 | NA | 98 | > 80 | |
| Information at diagnosis | |||||
| Age, y | 0.1 | 0.1 | 0.1 | ||
| Serum TSH, mU/L | 126 | NE | 399 | 176 | 1.7-9.1 |
| Serum free T4, ng/dL | 1.4 | NE | 0.28 | 0.81 | 0.9-2.3 |
| Serum Tg, ng/mL | 240 | NE | NA | > 320 | 3.8-56 |
| Thyroid ultrasonography | |||||
| Age at evaluation, y | 16 | 8 | 13 | ||
| Size, SDS | –2.3 | NE | –2.4 | –2.0 | |
| Information at last evaluation | |||||
| Age, y | 16 | 45 | 8 | 24 | |
| Height, cm (SDS) | 150 (–1.4) | 158 (0.0) | 130 (–0.1) | 159.7 (0.3) | |
| Weight, kg (SDS) | 44.3 (– .9) | NE | 28.9 (–0.0) | 60.5 (0.7) | |
| Serum TSH, mU/L | 9.9 | 1.5 | NE | 1.8 | 0.5-5.0 |
| Serum free T4, ng/dL | 1.30 | 1.33 | NE | 1.25 | 0.9-1.8 |
| Serum Tg, ng/mL | 11.1 | 13.9 | NE | NE | < 30 |
| L-T4 dose, µg/kg/d | 1.1 | Not treated | 1.5 | Not treated | |
Abbreviations: F, female; L-T4, levothyroxine; M, male; NA, not applicable; NBS, newborn screening; NE, not evaluated; SDS, SD score; T4, thyroxine; Tg, thyroglobulin; TSH, thyrotropin.
Patient 1 (a 16-year-old girl; PAX8 c.478 + 4_7del) was born at 36 weeks of gestation. She was noticed to have lymphangioma of her neck during the fetal period and was admitted to a neonatal intensive care unit soon after birth. She received sclerotherapy for the lymphangioma at age 19 days. Because of the positive result in NBS, she was referred to us at age 17 days. Poor activity, poor weight gain, and umbilical hernia were noted. She had a high serum TSH level with a normal serum free thyroxine (T4) level (Table 1). At age 6 years, her thyroid function was reassessed with discontinuation of therapy, revealing a high serum TSH level (17.5 mU/L; reference, 0.5-5.0 mU/L) with a normal serum free T4 level (1.2 ng/dL; reference, 0.9-1.8 ng/dL). Thyroid ultrasonography showed thyroid hypoplasia. Thyroidal 123I uptake was normal (15.7% at 24 hours; reference, 8%-40%). During the follow-up period, slightly high serum creatinine levels (0.61 mg/dL; age-specific reference, 0.35-0.58 mg/dL) were noted when she was age 11 years. She also had a high serum cystatin C level (0.98 mg/L; age-specific reference, 0.61-0.95 mg/L), and was diagnosed as stage 2 chronic kidney disease. Ultrasonography of the kidneys were unremarkable. At the last clinical visit, she was doing well with 1.1 µg/kg/day of L-T4. Reevaluation of her thyroid function with discontinuation of the therapy showed subclinical hypothyroidism (see Table 1). The mother of Patient 1 was born before the implementation of NBS for CH in Japan. Adult height was normal, and she had no symptoms suggesting hypothyroidism. After the identification of the PAX8 mutation in her, the first thyroid function test was performed, revealing normal thyroid function (see Table 1).
Patient 2 (an 8-year-old boy, PAX8 p.Q213fs) was born at term after an uneventful pregnancy. He had a high blood-spot TSH level in NBS, and came to us at age 14 days. He had an umbilical hernia, jaundice, and open posterior fontanel. Overt hypothyroidism was confirmed by a blood test (see Table 1). Thyroid ultrasonography at age 8 years (conducted under the therapy) showed a normoechoic hypoplastic thyroid gland. He had a history of stage 1 neuroblastoma, which was diagnosed at age 4 months, and received surgical resection 1 week after the diagnosis. Computed tomography scan of the abdomen showed no congenital abnormalities of the kidneys and urinary tract. At the last visit, he was doing well with L-T4 therapy (1.5 µg/kg/day). The father of patient 2 also had the identical frameshift PAX8 mutation. Thyroid function test of the father has not been conducted.
Patient 3 (a 24-year-old woman; PAX8 p.F342fs) was born at term after an uneventful term pregnancy. She had a high blood-spot TSH level at NBS. She came to us at age 9 days with no CH-related symptoms. Blood examination showed a high serum TSH level and a low serum free T4 level (see Table 1). At age 4 years, we reassessed her thyroid status with transient discontinuation of therapy. She had subclinical hypothyroidism (TSH 6.6 mU/L, free T4 1.3 ng/dL) with a slightly high serum thyroglobulin level (55 ng/mL; reference < 30 ng/mL). Thyroid ultrasonography showed a hypoplastic gland with low echogenicity. She had normal 123I uptake (25.8% at 24 hours). She had been treated with L-T4 to age 24 years, when her thyroid function was reevaluated with discontinuation of therapy. Her serum TSH levels remained within the normal range (TSH 1.8-2.8 mU/L; N = 3). Treatment has since been discontinued.
Mutation detection
From January 2006 to December 2019, a total of 920 genomic DNA samples derived from Japanese patients with primary CH were collected at the Department of Molecular Endocrinology, National Research Institute for Child Health and Development (Tokyo, Japan) and the Department of Pediatrics, Keio University School of Medicine (Tokyo, Japan). In the present study, CH was defined as an individual with one or more confirmed high serum TSH measurements (compared to the age-specific normal range) in infancy. Transient as well as permanent CH patients were included. Patients with exogenous factors affecting thyroid functions, including maternal Graves disease with or without antithyroid drug usage during pregnancy and excessive maternal or neonatal iodine exposure were excluded. Some mutation-carrying patients have been reported on elsewhere (14-16, 29-33), whereas the 3 mutation-carrying patients focused on in this study (I160Sfs*52, Q213Efs*27 and F342Rfs*85) have not been published. Eleven known genes associated with primary CH (DUOX2, DUOXA2, FOXE1, IYD, NKX2-1, PAX8, SLC5A5, SLC26A4, TG, TPO, and TSHR) were analyzed with the next-generation sequencing technique as previously described (33). The presence of variants detected by next-generation sequencing was confirmed by polymerase chain reaction (PCR)-based Sanger sequencing. We also performed parental genotyping by PCR-based Sanger sequencing. Three mutations were searched in publicly available variant databases, including 4.7K JPN (https://jmorp.megabank.tohoku.ac.jp/202001/variants), the 1000 Genomes Project database (http://www.1000genomes.org/), and the genome aggregation database (gnomAD) (https://gnomad.broadinstitute.org/).
Plasmids, cell culture, and expression experiments
A luciferase reporter encoding the promoter sequence of TG (TG-luc) and expression vectors for PAX8 (NM_003466) and NKX2-1 (NM_004387) have been described previously (14). Nucleotide-level PAX8 mutations (Q40P, Q201fs, F342fs) were introduced with a standard site-directed mutagenesis technique. C-terminal exon deletions (ΔEx10-12 [R364*] and ΔEx11-12 [S401*]) were created by inserting a termination codon. For evaluation of the intronic PAX8 mutation (c.478 + 4_7del), the 3.0-kb genomic sequence corresponding to PAX8 exons 4 to 7 (hg19; chr2:113 999 127-114 002 201) was PCR-amplified from a control DNA sample, and was inserted into the previously mentioned vector using the Gibson assembly technique (NEBuilder HiFi DNA Assembly Master Mix; New England Biolabs). The c.478 + 4_7del mutation was introduced via site-directed mutagenesis. For immunodetection assays (eg, Western blot and immunofluorescence), we constructed doxycycline-inducible expression vectors encoding N-terminal 3xFLAG-tagged PAX8 by inserting the PAX8 sequence into a previously described inducible stable vector (34). Culture and transfection of HeLa cells were performed as previously described (35). Inducible stable cells were established as previously described (34).
Cells transfected with each intron-containing PAX8 vector (wild-type [WT] or c.478 + 4_7del]) were harvested at 48 hours after transfection, and subject to total RNA isolation with AllPrep DNA/RNA/Protein Mini Kit (Qiagen). The RNA and was then converted to complementary DNA, and PCR-based sequencing was performed with primers 5’- CAA CCA TTC AAC CTC CCT ATG GAC A -3’ and 5’- GTC CAG TTC TAG CTC CAC CCC TTC C -3’.
For Western blot analysis, inducible stable cells were treated with or without 1 μg/mL doxycycline for 24 hours. For the protein stability assays, doxycycline-treated cells were incubated with 10 mM MG132 (Sigma-Aldrich) or vehicle (dimethyl sulfoxide) for an additional 12 hours. Whole-cell lysate was obtained with 1% Triton X-100 in Tris-buffered saline (TBS) supplemented with a protease inhibitor cocktail. The lysates were separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and Western blotting was performed with anti-FLAG M2 antibody (Sigma-Aldrich). For visualization of subcellular localization of each PAX8 protein (WT, Q201fs, and F342fs), inducible stable cells grown on a glass-bottom dish were fixed with cold methanol, and blocking was performed with 5% bovine serum albumin in TBS with 0.4% Tween 20 at room temperature for 1 hour. Immunofluorescence was performed with anti-FLAG M2 antibody and Alexa Fluor 555-conjugated goat antimouse immunoglobulin G antibody (Thermo Fisher Scientific). Nuclei were stained with Hoechst 33342. Cells were observed under an Olympus FV1000-D confocal microscope.
For luciferase reporter assays, cells grown in 96-well plates with 70% to 80% confluence were transiently transfected with 90 ng of TG-luc, and 10 ng of each PAX8 expression vector (WT or mutant) with or without 1 ng of the NKX2-1 expression vector. Forty-eight hours after transfection, we measured luciferase activities using ONE-Glo Luciferase Assay System (Promega) and a FlexStation 3 microplate reader (Molecular Devices). Experiments were conducted in quadruplicate, and were repeated at least 3 times. The activity data were expressed as mean ± SEM.
Luciferase complementation assay
We constructed an Escherichia coli expression vector encoding the fusion protein of PAX8, the Small BiT (SmBiT) sequence (Promega), and the FLAG tag (PAX8-SmBiT-FLAG) using the Gibson assembly technique. Similarly, a chimeric construct with FLAG-tagged NKX2-1 and the Large BiT (LgBiT) sequence (Promega) was created (NKX2-1-LgBiT-FLAG). As negative controls, vectors encoding monomeric enhanced green fluorescent protein (mEGFP) were created (mEGFP-SmBiT-FLAG and mEGFP-LgBiT-FLAG). For the decoy experiments, maltose-binding protein (MBP) sequence was fused to partial PAX8 sequence corresponding to exons 10 to 12 (363-449 aa residues) or exon 10 (363-400 aa) with C-terminal FLAG tag (MBP-PAX8Ex10-12-FLAG and MBP-PAX8Ex10-FLAG). MBP-FLAG was used as a negative control.
Each plasmid was transformed into E coli BL21(DE3) (Nippon Gene). After growth in Terrific Broth at 37°C to OD600 = 0.4, protein expression was induced by 0.4 mM of isopropyl-β-d-thiogalactopyranoside at 37°C for 4 hours. Harvested cells (approximately 1 g) were lysed with 1 mg/mL lysozyme in TBS (10 mL) at 4°C for 30 minutes, and then solubilized by addition of Triton X-100 (final concentration, 1%) and incubation at 4°C for 20 minutes. The recombinant proteins were purified using the ANTI-FLAG M2 Affinity Agarose Gel (Sigma-Aldrich), leaving 300 µL of resin. The column was washed with 3 column volumes (CVs) of TBS with medium salt (0.3 M NaCl), 3 CVs of TBS with high salt (1 M NaCl), and 3 CVs of TBS with medium salt. The bead-bound protein was finally eluted by competitive elution with 600 µL of TBS containing 100 μg/mL 3xFLAG peptide. For the luciferase complementation assays, each protein sample was adjusted to final concentration at 30 nM in a total volume of 20 µL. Ultimately, the solution included 0.3 M NaCl, 20 mM Tris-HCl (pH 8.0), 30 nM LgBiT-tagged protein, 30 nM SmBiT-tagged protein, 1.0% dextran, and 0.3% Nano-Glo Luciferase Assay Reagent (Promega). After 3 minutes’ incubation at room temperature, the luminescence of each protein solution was measured on a FlexStation 3 plate reader. Experiments were conducted in triplicate, and were repeated 3 times. The activity data were expressed as mean ± SEM.
Literature review of PAX8 mutation carriers
Data of serum TSH levels of experimentally verified PAX8 mutation carriers were collected from 19 papers that were published by January 2020 (4-7, 9-11, 13-20, 22, 36). A total of 66 mutation carriers were identified, and serum TSH level data obtained under no L-T4 therapy were collected. Among the 63 mutation carriers with missense or truncating mutations, 38 had TSH data in the infantile period (ie, age ≤ 1 year) alone, 19 had data in the postinfantile period (ie, age > 1 year) alone, and 6 had data in both periods. We used the Mann-Whitney U test to compare the serum TSH levels between missense mutation carriers and truncating mutation carriers.
Ethics
This study was approved by the ethics committees of the National Center for Child Health and Development and the Keio University School of Medicine. Written informed consent for molecular studies and publication of anonymized medical information was obtained from the study participants and/or their parents.
Results
Mutation detection
We conducted next-generation sequencing-based systematic mutation screening in a large cohort of Japanese CH patients (N = 920) and found 18 patients (belonging to 14 families) with PAX8 mutations, including 9 with a missense mutation, 5 with in-frame indel mutations, 3 with frameshift indel mutations (p.D46Sfs*24, p.Q213Efs*27, and p.F342Rfs*85), and 1 with a splice site mutation (c.478 + 4_7del). Clinical and molecular characteristics of 7 patients (5 families: p.L16P, p.F20S, p.D46Sfs*24, p.K80_A83dup, and p.R133Q) have already been published (14-16). In the present study, we focused on one novel splice site mutation (patient 1 c.478 + 4_7del) and 2 novel frameshift mutations (patient 2 c.637_638del, p.Q213Efs*27; and patient 3 c.1018_1022dup, p.F342Rfs*85) that have not yet been reported. The 3 mutations were found in 3 CH probands as a heterozygous mutation (Fig. 1A). None of them were registered in publicly available variant databases. The 3 mutations were located outside the paired domain (see Fig. 1A). The pedigrees of the mutation carriers are shown in Fig. 1B. Parental genotyping revealed that the mutations were transmitted from the parent (patient 1 and patient 2), or occurred de novo (patient 3). The mother of patient 1 had a normal serum TSH level (1.5 mU/L). The father of patient 2 has not been assessed for the thyroid function.
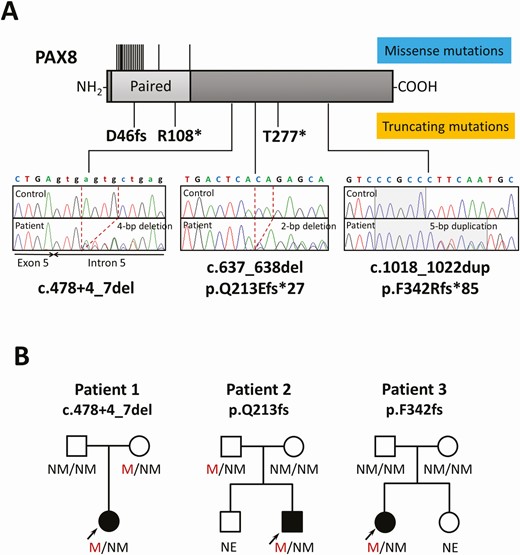
Identification of 3 novel truncating PAX8 mutations. A, A schematic diagram of the PAX8 protein and electropherograms of each mutation-carrying patient are shown. The deleted nucleotides are shown by dashed lines and the duplicated nucleotides are shown by the shaded area. The location of previously described mutations is indicated by the black lines (upper, missense mutations; lower, truncating mutations) B, Pedigrees of the 3 mutation-carrying patients. Black symbols indicate individuals affected with congenital hypothyroidism. M, mutated; NE, not evaluated; NM, not mutated.
Functional characterization of the 3 PAX8 mutations
The mutation detected in patient 1 (c. 478 + 4_7del) was located near the boundary of exon 5 and intron 5, but not in the essential splice site. We constructed an expression vector in which the genomic sequence corresponding to PAX8 exons 4 to 7 (including introns) was inserted, and tested the effect of the c. 478 + 4_7del mutation on splicing of the PAX8 messenger RNA (Fig. 2A). Reverse transcriptase–PCR using RNA prepared from transiently transfected HeLa cells showed a larger PCR product in mutant-expressing cells than in WT-expressing cells (see Fig. 2A). The amplified fragment had a partial intron 5 sequence inserted between exons 5 and 6, predicting the production of frameshifted protein (p.I160Sfs*52). Collectively, we report 3 truncating PAX8 mutations in the present study (Fig. 2B).
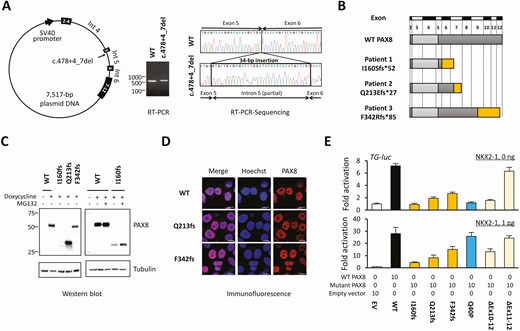
Characterization of the 3 truncating PAX8 mutations. A, A schematic diagram of the PAX8-expressing plasmid that contains the full genomic sequence corresponding exons 4 to 7 (left panel). Reverse transcriptase–polymerase chain reaction (RT-PCR) showed a slightly larger PCR product in mutant-expressing cells (middle panel). Sequencing of the RT-PCR product showed an insertion of a 34-bp sequence between exons 5 and 6 (right panel). B, Schematic diagrams of wild-type (WT)-PAX8 and the 3 truncating mutants identified in this study. C, Images of Western blotting are shown. The protein expression level of I160fs-PAX8 was low (left), which was partially recovered by a proteasome inhibitor MG132 (right). D, Subcellular localization analysis showed comparable localization of WT-PAX8 and 2 mutants. The scale bars indicate 10 μm. E, The upper panel shows the transactivation assays with no coexpression of NKX2-1. I160fs-PAX8 was nonfunctional, whereas Q213fs-PAX8 and F342fs-PAX8 had residual activities. The lower panel shows transactivation assays with coexpression of NKX2-1. Recovery of transactivation capacity (“rescue”) was observed in Q40P-PAX8. This “rescue” was not observed in Q213fs-PAX8, F342fs-PAX8, or ΔEx10-12-PAX8. The data are representative of 3 independent experiments with similar results. Bars indicate SEM.
Western blotting showed that the protein expression level of I160fs-PAX8 was detectable but very low, whereas those of Q213fs-PAX8 and F342fs-PAX8 were not reduced as compared with WT-PAX8 (Fig. 2C). As for I160fs-PAX8, treating cells with the proteasome inhibitor MG132 partially recovered the protein expression level (see Fig. 2C), indicating that the mutant protein was degraded via the proteasome-dependent pathway. Visualization of subcellular localization showed that Q213fs-PAX8 and F342fs-PAX8 were localized in the nucleus normally (Fig. 2D). Transactivation capacities of the mutant proteins were assessed, with or without coexpression of NKX2-1. For better characterization of the truncating PAX8 mutations, a previously reported p.Q40P mutation (with DNA-binding defect) (7) and C-terminal deletion mutants (ΔEx10-12-PAX8 and ΔEx11-12-PAX8) were also analyzed. In the NKX2-1-absent condition, I160fs-PAX8 showed negligible transactivation on TG-luc (Fig. 2E). Q213fs-PAX8 and F342fs-PAX8 showed partial transactivation (14.8 ± 3.7% and 27.0 ± 3.0% activity relative to WT-PAX8, respectively). As for the C-terminal deletion mutants, the transactivation capacity of ΔEx10-12-PAX8 was very low, whereas that of ΔEx11-12-PAX8 was normal (see Fig. 2E). NKX2-1 has been known to “rescue” the transcriptional defects of missense PAX8 mutations (10). Actually, we confirmed the “rescue” in Q40P-PAX8 (see Fig. 2E, sky-blue bars). Contrastingly, defective transactivation of Q213fs-PAX8 and F342fs-PAX8 as well as ΔEx10-12-PAX8 were not rescued by NKX2-1 (see Fig. 2E, yellow and beige bars), suggesting that the C-terminal portion of PAX8, corresponding to exons 10 to 12, is required for the PAX8–NKX2-1 interaction.
Luciferase complementation assay for analysis of PAX8–NKX2-1 interaction
To test the hypothesis that PAX8 interacts with NKX2-1 via its C-terminal portion, we first tried coimmunoprecipitation experiments using HeLa cells expressing PAX8 and NKX2-1 but failed (data not shown). We then moved on to a luciferase complementarity assay that was expected to detect a relatively weak interaction. Luciferase complementation assay is a method for the quantification of protein-protein interactions, in which interaction between the bait and the prey brings split luciferase fragments in close proximity to form a functional protein whose activity can be measured (37). We used the E coli expression system to produce recombinant proteins that were PAX8 or NKX2-1 fused to the split luciferase (SmBiT and LgBiT) (38) (Fig. 3A and 3B). Mixing 2 protein solutions (PAX8-SmBiT and NKX2-1-LgBiT) along with the luciferase substrate produced a 60-fold higher luminescence compared with mixing of a negative control pair (mEGFP-SmBiT and mEGFP-LgBiT) (Fig. 3C). A similar luminescence was obtained with a mixture of Q40P-PAX8-SmBiT and NKX2-1-LgBiT. Contrastingly, Q213fs-PAX8-SmBiT and ΔEx10-12-PAX8-SmBiT produced significantly low luminescence with NKX2-1-LgBiT (21.3 ± 1.8% and 19.2 ± 0.7% luminescence relative to WT-PAX8-SmBiT), suggesting the impairment of the PAX8–NKX2-1 interaction. For further verification of the PAX8–NKX2-1 interaction via the C-terminal portion, we created the “decoy” recombinant proteins (MBP-PAX8Ex10-12 and MBP-PAX8Ex10) (see Fig. 3B) to inhibit the luciferase complementation of PAX8-SmBiT and NKX2-1-LgBiT. As we expected, significant inhibition was observed by a 10-fold excess of the “decoy” (Fig. 3D), indicating that PAX8 interacts with NKX2-1 via the 34-amino acid protein sequence encoded by exon 10.
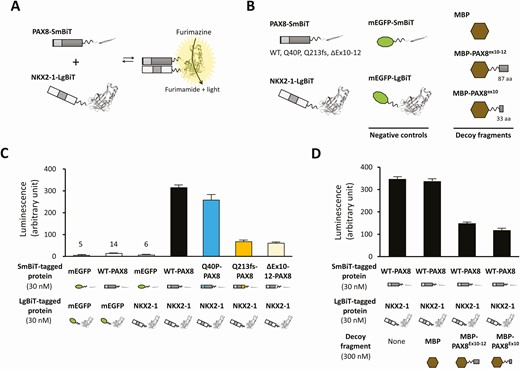
Luciferase complementation assay-based evaluation of the PAX8-NKX2-1 interaction. A, Schematic diagrams showing the luciferase complementation assay of PAX8-SmBiT and NKX2-1-LgBiT. In this split luciferase system, LgBit and SmBiT form functional luciferase if PAX8 and NKX2-1 interact. B, Schematic diagrams of recombinant proteins are shown. C, The luciferase complementation assays showed that the pair of wild-type (WT)-PAX8-SmBiT and NKX2-1-LgBiT showed 60-fold higher activity than the negative control pair. Q213fs-PAX8-SmBiT and ΔEx10-12-PAX8-SmBiT showed low luciferase activity when mixed with NKX2-1-LgBiT. D, The result of the “decoy” experiment is shown. Significant inhibition of the luciferase activity derived from the pair of PAX8-SmBiT and NKX2-1-LgBiT is shown by adding 10-fold excess concentration of the “decoy.”
Clinical phenotype of congenital hypothyroidism patients with truncating PAX8 mutations
Three out of the 4 truncating PAX8 mutation carriers had normal or near-normal serum TSH levels (1.5, 1.8, and 9.9 mU/L) (see Table 1). The observation prompted us to systematically investigate mutation type-dependent serum TSH levels using the literature data (Supplementary Table S1) (39). Missense mutation carriers and truncating ones both tended to have high serum TSH levels in the infantile period (Fig. 4). In the postinfantile period, serum TSH levels of missense mutation carriers showed a wide distribution, but the levels mostly exceeded 10 mU/L. The levels among truncating mutation carriers were significantly lower than missense mutation carriers (P < .001) in this period. Within-normal-range TSH levels were seen only among truncating mutation carriers (see Fig. 4).
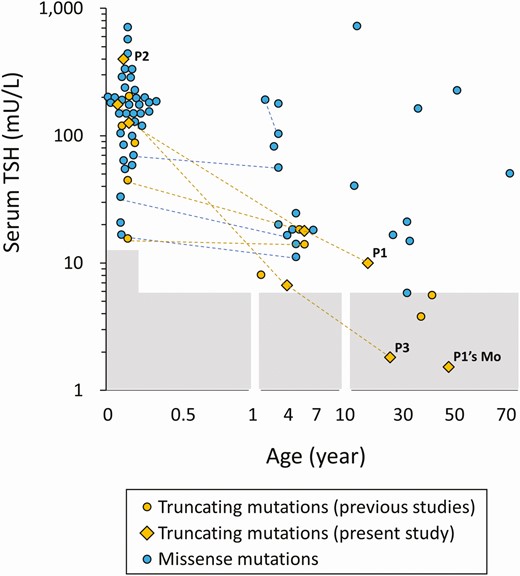
Distribution of serum thyrotropin (TSH) levels of PAX8 mutation carriers. The serum TSH levels, which were measured with no levothyroxine therapy, were plotted by age according to the types of mutations (blue symbols, missense mutations; yellow symbols, truncating mutations). The shaded areas indicate the reference range.
Discussion
We identified 3 novel truncating PAX8 mutations outside the paired domain in CH patients. One of the mutations was the first intronic PAX8 mutation. Twenty-one out of the 22 PAX8 previously reported mutations have been located in the paired domain. The amino acid sequences of the paired domain are highly conserved among PAX family genes, suggesting they are under strong structural constraints. Contrasting to the paired domain, protein sequences of the other regions of PAX family transcription factors are diverse. Given the diverse roles of PAX family transcription factors in embryogenesis and organogenesis (40), these C-terminal portions are likely to have unique functions for each molecule. As exemplified in this study, binding to transcription partner(s) via the C-terminal portion would be an example of such unique roles.
The 3 probands definitely had high serum TSH levels in the newborn period, and they commonly had marginal thyroid hypoplasia. These features were consistent with previously described phenotypes of the mutation carriers. However, we noticed that the serum TSH levels of the truncating mutation carriers tended to be low as compared with missense mutation carriers in the postinfantile period. At present, the reason why truncated mutation carriers show relatively mild phenotype in later life is unclear. The dominant-negative effect of missense PAX8 mutations, suggested in one previous study (10), could be an explanation, but we could not observe such an effect in our experiments using HeLa cells (data not shown).
In addition to the TFCs, PAX8 is expressed in renal epithelial cells along with another PAX family transcription factor, PAX2 (25). Hence, possible roles of PAX8 in renal development have been discussed (41). To date, 5 PAX8 mutation-carrying CH patients with renal complications have been described, including 2 with unilateral kidney agenesis (9, 19), 2 with unilateral ureterocele (17), and 1 with an incomplete horseshoe kidney (17). In this study, we incidentally found patient 1 to have stage 2 chronic kidney disease during the follow-up of CH. Prevalence of early-stage chronic kidney disease in the pediatric population is unknown because it is often asymptomatic and therefore underreported (42). Accumulation of data on renal function of PAX8 mutation carriers will be necessary to draw reliable conclusions about the possible relationship between the genetic defect and early-stage chronic kidney disease.
Kozmik et al compared multiple alternative transcript variants of PAX8 and found that the C-terminal sequence corresponding to exons 10 to 12 (363-449 aa residues) was essential for the transactivation of the paired domain reporter (43). Poleev and colleagues used the GAL4 fusion protein system to compare the transactivating capacity of PAX8-derived fragments, and further localized the “activation domain” to the sequence corresponding to exons 10 to 11 (363-425 aa) (44). Two groups independently reported that the C-terminal region including the “activation domain” is required for synergistic transactivation of the rat Tpo promoter (26) and the rat Tg promoter (27) with the rat Nkx2-1 protein. In this study, we provide 2 lines of evidence for the importance of the sequence corresponding to exon 10 (363-400 aa), including (i) the difference in transactivation capacity between ΔEx10-12-PAX8 and ΔEx11-12-PAX8, and (ii) the ability of MBP-PAX8Ex10 to interfere with luciferase complementation of PAX8-SmBiT and NKX2-1-LgBiT. Our data indicate that the exon 10 sequence is involved not only in the PAX8–NKX2-1 interaction but also in NKX2-1–independent transactivation of the TG promoter.
Among the 3 truncating PAX8 mutations identified in this study, the I160fs mutation was inactivated by protein destabilization. The remaining 2 (Q213fs-PAX8 and F342fs-PAX8) showed stable protein expression, but had limited transactivating capacity on TG-luc. This loss of function is probably explained by the loss of the exon 10 sequence. The characteristic loss of rescue by NKX2-1 coexpression is also likely explained by the loss of the exon 10 sequence, because the PAX8–NKX2-1 interaction requires the sequence.
Assessing the interaction of 2 (or more) transcription factors has been an important topic in molecular genetics research. The most widely used approach is the coimmunoprecipitation assay, but it has at least 2 limitations. One is the requirement of a relatively strong affinity between the analyzed proteins. There are many biological interactions that are weak, transient, and/or reversible. Another limitation is the lack of quantifiability. The effects of amino acid alterations on protein-protein interactions may be modest, and it is therefore difficult to detect such effects reliably using coimmunoprecipitation experiments. In this study, we introduced the luciferase complementation assay to quantitatively evaluate the PAX8–NKX2-1 interaction. This assay allowed us to not only show the direct interaction between the 2 factors, but also to measure the effect of sequence modifications. We emphasize the versatility of our approach, which can be easily extended to analyses of other transcription factors.
In conclusion, we report clinical and molecular findings of 3 novel truncating PAX8 mutations that were located outside the paired domain. In the literature and in this report, CH patients with a truncating PAX8 mutation had normal or slightly elevated serum TSH levels after infancy, raising the possibility that these patients might have a milder phenotype than those with a missense PAX8 mutation. Functional characterization of the truncating mutations led us to identify the functionally important sequence in the C-terminal portion (363-400 aa), which is likely to be involved both in the PAX8–NKX2-1 interaction and NKX2-1–independent transactivation of target genes.
Abbreviations
- CH
congenital hypothyroidism
- CV
column volume
- LgBiT
Large BiT
- L-T4
levothyroxine
- MBP
maltose-binding protein
- mEGFP
monomeric enhanced green fluorescent protein
- NBS
newborn screening
- PCR
polymerase chain reaction
- SmBiT
Small BiT
- T4
thyroxine
- TBS
Tris-buffered saline
- TFC
thyroid follicular cell
- TSH
thyrotropin
- WT
wild-type
Acknowledgments
Financial Support: This work was supported in part by Japan Society for the Promotion of Science KAKENHI (19K22607).
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability
The data sets generated and analyzed during the present study are not publicly available but are available from the corresponding author on reasonable request.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}