





Abstract
Central precocious puberty (CPP) has been associated with loss-of-function mutations in 2 paternally expressed genes (MKRN3 and DLK1). Rare defects in the DLk1 were also associated with poor metabolic phenotype at adulthood.
Our aim was to investigate genetic and biochemical aspects of DLK1 in a Spanish cohort of children with CPP without MKRN3 mutations.
A large cohort of children with idiopathic CPP (Spanish PUBERE Registry) was studied. Genomic deoxyribonucleic acid was obtained from 444 individuals (168 index cases) with CPP and their close relatives. Automatic sequencing of MKRN3 and DLK1 genes were performed.
Five rare heterozygous mutations of MKRN3 were initially excluded in girls with familial CPP. A rare allelic deletion (c.401_404 + 8del) in the splice site junction of DLK1 was identified in a Spanish girl with sporadic CPP. Pubertal signs started at 5.7 years. Her metabolic profile was normal. Familial segregation analysis showed that the DLK1 deletion was de novo in the affected child. Serum DLK1 levels were undetectable (<0.4 ng/mL), indicating that the deletion led to complete lack of DLK1 production. Three others rare allelic variants of DLK1 were also identified (p.Asn134=; g.-222 C>A and g.-223 G>A) in 2 girls with CPP. However, both had normal DLK1 serum levels.
Loss-of-function mutations of DLK1 represent a rare cause of CPP, reinforcing a significant role of this factor in human pubertal timing.
Genomic imprinting has been demonstrated to play a relevant role in regulation of pubertal timing in humans. Several loss-of-function mutations of 2 maternally imprinted genes, MKRN3 and Delta-like 1 homolog (DLK1), were described in children with familial central precocious puberty (CPP) (1-4). Interestingly, a potential interaction between 2 imprinted regions where these CPP genes are located (chromosomes 15q11.2 and 14q32.2) was determined in vitro, suggesting a unique epigenetic regulation involving these loci (5).
Notably, loss-of-function mutations of MKRN3 are very well-established causes of familial PCC (first description in 2013; 800 studied patients) (1, 3). The frequency of these mutations is particularly high in Occidental countries (up to 46% in European studies) and to date, more than 30 different inactivating mutations have been described in girls and boys with familial CPP from different ethnic backgrounds (3). In contrast, DLK1 defects in children with CPP are recent and apparently uncommon, with scarce information on genotype–phenotype correlations.
DLK1, also known as preadipocyte factor 1 (Pref-1) and fetal antigen (FA1), is an epidermal growth factor-like (EGF-like) membrane-bound protein. It contains 6 tandem EGF-like repeats, a juxtamembrane region with a TACE (ADAM17)-mediated cleavage site, a transmembrane domain, and a short intracellular tail (6, 7). DLK1 is part of the Notch signaling pathway that controls many developmental processes. It is well known that DLK1 prevents the differentiation of preadipocytes into mature adipocytes (8-10) and Dlk1 knockout mice present obesity, growth retardation, and skeletal malformations (9). Conversely, mice expressing a Dlk1 transgene in adipose tissue are lean and show decreased fat pad weight (11). In humans, common allelic variants of the DLK1 gene have been associated with severe obesity (Trio families study) in children and early menarche (GWAS studies) in European women (12).
In 2017, Dauber et al. (2). described a complex defect of DLK1 (∼14-kb deletion and 269-bp duplication) in 5 members of a multigenerational Brazilian family with nonsyndromic CPP by using linkage analysis and whole-genomic sequencing. This deletion included the 5′ untranslated region and the first exon of DLK1. Only family members who inherited the defect from their father were shown to have precocious puberty, consistent with the known pattern of imprinting of DLK1. This very rare genomic defect was the first description of severe isolated deficiency of DLK1 in humans. More recently, 3 novel frameshift mutations located at the extracellular domain of DLK1 (p.Gly199Alafs*11, p.Val271Cysfs*14, and p.Pro160Leufs*50) have been described in 5 women with familial CPP (4 Brazilian women and 1 English woman) (13). Metabolic abnormalities, such as overweight/obesity, early-onset glucose intolerance/type 2 diabetes mellitus, and hyperlipidemia, were more prevalent in the women with DLK1 mutations than in the idiopathic CPP group. Interestingly, 2 sisters who carried the p.Gly199Alafs*11 mutation also exhibited polycystic ovary syndrome and infertility (13). The high prevalence of metabolic alterations in adult women who experienced precocious menarche or CPP due to DLK1 defects suggests that DLK1 represents a new link between reproduction and metabolism.
A soluble form of DLK1 with a molecular weight of 50 kDa can be generated through TACE (ADAM17)-mediated cleavage of its extracellular domain. Serum DLK1 concentrations are undetectable in women with CPP caused by deleterious defects of DLK1, suggesting that this accessible biochemical measurement could be a potential screening assay for the diagnosis of familial CPP due to a rare deficiency of DLK1.
In the present study, comprehensive investigation of genetic and biochemical aspects of DLK1 was performed in a large cohort of Spanish children with CPP (Spanish PUBERE Registry) without MKRN3 mutations. This study represents a collaborative initiative between Brazilian and Spanish universities for continuous elucidation of the genetic basis of CPP in children. Novel DLK1 findings were demonstrated in the Spanish cohort that had been previously diagnosed with idiopathic CPP, reinforcing a significant role of this factor in human pubertal timing.
Patients and Methods
Spanish cohort
Spanish children with idiopathic CPP evaluated in 55 centers throughout the country were studied. This cohort constitutes the Spanish PUBERE Registry that emerged to address different issues related to CPP, such as epidemiological and clinical information (14). The PUBERE Registry is supported by the Spanish Society for Pediatric Endocrinology (Sociedad Española de Endocrinología Pediátrica).
Genomic deoxyribonucleic acid (DNA) was obtained from 444 individuals, including 168 cases with CPP (index cases) and their relatives. Idiopathic CPP was defined as girls diagnosed with progressive thelarche before the age of 8 years and boys with testicular volume greater than 4 mL (Prader orchidometer) before 9 years of age, with the diagnosis confirmed with luteinizing hormone releasing hormone (LHRH) testing; luteinizing hormone (LH) peak after LHRH stimulation (100 µg/m2) greater than 7 IU/L; bone age minus chronological age equals more than 1 year.
The CPP group was mainly composed of female patients (161 girls and 7 boys), with a mean chronological age at presentation of 6.87 years ± 0.84 in girls and 6.24 years ± 0.53 in boys; the mean bone age at diagnosis was 9.55 years ± 1.85 in girls and 9.35 years ± 0.65 in boys. Familial CPP cases were diagnosed around 20% of the Spanish cohort. No magnetic resonance imaging (MRI) lesions were identified in this group of CPP patients.
Sanger sequencing
DNA was collected from the index cases, their parents, and first- or second-degree family members when available. Genomic DNA was extracted from peripheral blood lymphocytes according to standard protocols (15). The MKRN3 sequencing was first performed and followed by DLK1 analysis in negative MKRN3 cases. The entire coding regions of MKRN3 (1 exon—GenBank accession number NM_005664) and DLK1 (5 exons—GenBank accession number NM_003836) were amplified by polymerase chain reaction (PCR) followed by purification and automatic sequencing of the products by using the Sanger sequencing method. DNA sequences obtained were compared to the human GenBank MKRN3 and DLK1 sequence using Sequencher sequence alignment software. Familial segregation analysis of potential pathogenic variants was performed by Sanger sequencing. PCR primers and conditions are available upon request.
DLK1 serum measurements
Serum DLK1 levels were measured in the affected family members and pubertal controls by using a soluble DLK1 enzyme-linked immunosorbent assay from Immuno-Biological Laboratories, Inc. (IBL-America, Minneapolis, MN). The intra- and interassay variations were 5.6% and 8.1%, respectively. The sensitivity limit was 0.34 ng/mL.
Microsatellite analysis
The biological paternity was confirmed by the AmpFlSTR® Identifiler® PCR Amplification Kit, a short tandem repeat multiplex assay that amplifies 15 tetranucleotide repeat loci and the Amelogenin gender-determining marker using manufacture instruction (Life Technologies, Carlsbad, CA).
In silico analysis
In silico analysis of splice regions was performed by NNSplice (16), Human Splice Finder (17), FSplice (Softberry, Mount Kisco, NY) and NetGene2 (18). The promoter region of DLK1 was studied using RNAfold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). The 5′ untranslated region messenger ribonucleic acid (mRNA) of DLK1 was submitted with default parameters to predict the potential secondary structure for the wildtype and the DLK1 g.-223 G>A and g.-222 C>A mutant mRNA. Potential minimum free energy (MFE) structures, centroid structures, and positional entropies were obtained (19). The American College Medical Genetics and Genomics classification and the pathogenicity sites prediction was performed by Varsome (20).
Results
MKRN3 analysis
Loss-of-function mutations of MKRN3 are frequent genetic causes of familial CPP with paternal inheritance and they were first excluded. Five rare variants were identified, including 4 missense mutations and 1 frameshift mutation, in girls with CPP. Two variants have previously been associated with CPP phenotype (Table 1) (21).
Rare allelic variants of MKRN3 identified in patients with CPP from a Spanish cohort
| Variant | Phenotype | |||||||
|---|---|---|---|---|---|---|---|---|
| cDNA | Protein | Allelic frequencya | Familial segregation | Previous description in CPP | ACMG classification | Sex | Chronological age of puberty onset | Bone age advancement |
| c.1249T>C (rs745560329) | p.Phe417Leu | MAF < 0.01 | Father | (21) | Pathogenic | Female | 6.8 | 1.1 |
| c.724C>T (rs1277371835) | p.Arg242Trp | MAF < 0.001 | NA | NA | Pathogenic | Female | 6.8 | 2.6 |
| c.475_476insC (rs763195944) | p.Ala162Glyfs | MAF < 0.01 | Father | (1) | Pathogenic | Female | 6.5 | 2.1 |
| c.1252T>C (rs1470111765) | p.Tyr418His | MAF < 0.001 | Father | NA | Likely pathogenic | Female | 6.4 | 0.5 |
| c.890T>C (rs147605349) | p.Met297Arg | MAF < 0.01 | Father | NA | Likely pathogenic | Female | 7.3 | 1.7 |
| Variant | Phenotype | |||||||
|---|---|---|---|---|---|---|---|---|
| cDNA | Protein | Allelic frequencya | Familial segregation | Previous description in CPP | ACMG classification | Sex | Chronological age of puberty onset | Bone age advancement |
| c.1249T>C (rs745560329) | p.Phe417Leu | MAF < 0.01 | Father | (21) | Pathogenic | Female | 6.8 | 1.1 |
| c.724C>T (rs1277371835) | p.Arg242Trp | MAF < 0.001 | NA | NA | Pathogenic | Female | 6.8 | 2.6 |
| c.475_476insC (rs763195944) | p.Ala162Glyfs | MAF < 0.01 | Father | (1) | Pathogenic | Female | 6.5 | 2.1 |
| c.1252T>C (rs1470111765) | p.Tyr418His | MAF < 0.001 | Father | NA | Likely pathogenic | Female | 6.4 | 0.5 |
| c.890T>C (rs147605349) | p.Met297Arg | MAF < 0.01 | Father | NA | Likely pathogenic | Female | 7.3 | 1.7 |
Abbreviations: ACMG, American College of Medical Genetics; CPP, central precocious puberty; MAF minor allele frequency; NA, not available
aHighest minor allelic frequency observes including 1000 genome Phase 3, NHLBI Exome Sequencing Project and gnomAD.
Rare allelic variants of MKRN3 identified in patients with CPP from a Spanish cohort
| Variant | Phenotype | |||||||
|---|---|---|---|---|---|---|---|---|
| cDNA | Protein | Allelic frequencya | Familial segregation | Previous description in CPP | ACMG classification | Sex | Chronological age of puberty onset | Bone age advancement |
| c.1249T>C (rs745560329) | p.Phe417Leu | MAF < 0.01 | Father | (21) | Pathogenic | Female | 6.8 | 1.1 |
| c.724C>T (rs1277371835) | p.Arg242Trp | MAF < 0.001 | NA | NA | Pathogenic | Female | 6.8 | 2.6 |
| c.475_476insC (rs763195944) | p.Ala162Glyfs | MAF < 0.01 | Father | (1) | Pathogenic | Female | 6.5 | 2.1 |
| c.1252T>C (rs1470111765) | p.Tyr418His | MAF < 0.001 | Father | NA | Likely pathogenic | Female | 6.4 | 0.5 |
| c.890T>C (rs147605349) | p.Met297Arg | MAF < 0.01 | Father | NA | Likely pathogenic | Female | 7.3 | 1.7 |
| Variant | Phenotype | |||||||
|---|---|---|---|---|---|---|---|---|
| cDNA | Protein | Allelic frequencya | Familial segregation | Previous description in CPP | ACMG classification | Sex | Chronological age of puberty onset | Bone age advancement |
| c.1249T>C (rs745560329) | p.Phe417Leu | MAF < 0.01 | Father | (21) | Pathogenic | Female | 6.8 | 1.1 |
| c.724C>T (rs1277371835) | p.Arg242Trp | MAF < 0.001 | NA | NA | Pathogenic | Female | 6.8 | 2.6 |
| c.475_476insC (rs763195944) | p.Ala162Glyfs | MAF < 0.01 | Father | (1) | Pathogenic | Female | 6.5 | 2.1 |
| c.1252T>C (rs1470111765) | p.Tyr418His | MAF < 0.001 | Father | NA | Likely pathogenic | Female | 6.4 | 0.5 |
| c.890T>C (rs147605349) | p.Met297Arg | MAF < 0.01 | Father | NA | Likely pathogenic | Female | 7.3 | 1.7 |
Abbreviations: ACMG, American College of Medical Genetics; CPP, central precocious puberty; MAF minor allele frequency; NA, not available
aHighest minor allelic frequency observes including 1000 genome Phase 3, NHLBI Exome Sequencing Project and gnomAD.
DLK1 analysis
A heterozygous deletion (c.401_404 + 8del) affecting exon 4 and intron 4-5 of DLK1 was identified in a girl with sporadic CPP (Patient 1). The deletion comprised the splice site junction. Both of her parents had a wildtype sequence of exon 4 of DLK1 (Fig. 1A). The biological paternity was confirmed using microsatellite analysis in this case.
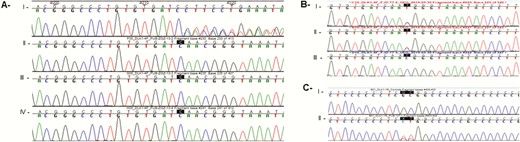
Sequencing data of DLK1 gene showing 3 rare allelic variants. (A) DLK1 deletion c.401_404 + 8del identified in patient 1 (index case): I, index; II, father; III, mother; IV, sister; all of them were wildtype. (B) Patient 2 with the synonymous DLK1 c.402C>T p.Asn134= (rs150016759): I, index case; II, father; III, mother wildtype. (C) Patient 3 with DLK1 promoter region alteration g.-222 C>A and g.-223 G>A. I, control; II, index case.
A rare heterozygous synonymous allelic variant was identified in a girl (Patient 2) with sporadic CPP (Fig. 1B). The variant c.402C>T p.Asn134= (rs150016759) has a minor allele frequency less than 0.01 in the normal population and 0.0008825 in European population (according to gnomAD data). In silico analysis showed that this synonymous variant was located in a potential donor 5′ splice region, characterized by a conserved sequence GU at the 5′ end of the intron 4-5. Although the alteration does not modify the amino acid sequence, this nucleotide change could disrupt RNA splicing resulting in the loss of exons or the inclusion of introns, leading to an altered protein-coding sequence. Segregation analysis showed that it was inherited from her father and was absent in her mother.
Two consecutive rare allelic variants (g.-222 C>A and g.-223 G>A) in the DLK1 promoter region were identified in an adopted girl (Patient 3) with CPP (Fig. 1C). According to the Encyclopedia of DNA Elements (ENCODE consortium), both variants affected regions that are DNA sequences for potential binding of 4 transcription factors—Retinoic Acid Receptor Alpha (RARA), E2F Transcription Factor 1 (E2F1), Eomesodermin (EOMES), and Retinoic Acid Receptor Gamma (RARG). Therefore, both variants could impair gene transcription, increasing or reducing its expression. The allele frequencies of the g.-222 C>A (rs1442192598) and g.-223 G>A (rs1186489952) variants were 0.00002449 and 0.00002222, respectively. These variants were not identified in European or Latin American populations. As this patient was adopted, her biological parents were not available for genetic analysis.
In silico analysis was performed using the software RNA Fold, which predicts the secondary structures of single-stranded RNA. The software calculates the energy necessary to form the secondary structure of the RNA based on MFE algorithms. The MFE of a RNA molecule can be affected by 3 properties of nucleotides in the sequence: their number, composition, and arrangement. The lower the thermodynamic energy of the structure, the more stable it generally is. The secondary structure is directly associated to the RNA function. The results suggested a potential change in the MFE necessary for the formation of the second RNA structure and centroid secondary structure.
Clinical features of girls with rare DLK1 variants
Patient 1.
The girl with a heterozygous deletion (c.401_404 + 8del) affecting exon 4 and intron 4-5 of DLK1 showed progressive CPP. Pubertal signs appeared at 5.7 years of age. The first clinical visit took place at 6.3 years of age and the physical examination showed thelarche III, pubarche III, height 125.5 cm (1.6 SDS [standard deviation score]), weight 30.7 kg (2.1 SDS), body mass index (BMI) 19.5 kg/m2 (1.5 SDS), bone age 10.5 years with target height of 146.6 cm. Personal antecedents were uneventful. Biochemical analysis confirmed early activation of the central gonadotrophic axis. Basal and peak LH levels after LHRH stimulation were 1.7 mIU/mL and 32.77 mIU/mL, respectively, and basal and peak follicle-stimulating hormone (FSH) after LHRH stimulation were 6.32 mIU/mL and 19.89 mIU/mL, respectively. MRI of the hypothalamic–pituitary region was normal and pelvic ultrasound demonstrated increased ovarian volume with follicles consistent with CPP. The patient was treated with gonadotropin-releasing hormone (GnRH) analogues during 3.8 years (6.3 to 10.1 years) with no side effects. Menarche occurred at 11.2 years and at 15.9 years her height was 153.5 cm (–1.4 SDS), weight 41.6 kg, BMI 17.6 kg/m2 (–1.2 SDS), and she has regular menses without hirsutism, obesity, or hyperandrogenism.
Patient 2.
The heterozygous synonymous allelic variant (c.402C>T p.Asn134=) was identified in a girl with sporadic CPP and in her father. Pubertal signs appeared at 7 years of age. The first clinical visit took place at 7.25 years of age and the physical examination showed thelarche II, pubarche II, height 131.2 cm (2.2 SDS), weight 31 kg (1.8 SDS), BMI 18 kg/m2 (0.65 SDS), and bone age 8 years and 10 months. Personal antecedents were uneventful. Biochemical analysis confirmed early activation of the central gonadotrophic axis. Basal and peak LH levels after LHRH stimulation were <0.07 mIU/mL and 7.7 mIU/mL, respectively, and basal and peak FSH after LHRH stimulation were 2.1 mIU/mL and 19 mIU/mL, respectively. MRI of the hypothalamic–pituitary region was normal and pelvic ultrasound demonstrated increased ovarian volume. The patient was treated with GnRH analogues during 2.8 years (7.4-10 years) with no side effects. Menarche occurred at 11 years and at 13 years her height was 163.5 cm (1.1 SDS), growth velocity <2 cm during the last year), BMI 22 kg/m2 (0.9 SDS) and she has regular menses without hirsutism, obesity, or hyperandrogenism.
Patient 3.
Two consecutive rare allelic variants (g.-222 C>A and g.-223 G>A) in the DLK1 promoter region were identified in an adopted girl with CPP. Hence, genetic and biochemical information from her parents was not available. Pubertal signs appeared at 6 years and 6 months of age. The first clinical visit took place at 7.1 years of age and the physical examination showed thelarche II, pubarche II, height 126.6 cm (1.19 SDS), BMI 16.6 kg/m2 (0.06 SDS), bone age 9 years and 6 months. Personal antecedents were uneventful. Biochemical analysis confirmed early activation of the central gonadotrophic axis. Basal and peak LH levels after LHRH stimulation were <0.37 mIU/mL and 7.86 mIU/mL, respectively, and basal and peak FSH after LHRH stimulation were 2.45 mIU/mL and 6.95 mIU/mL, respectively. Pelvic ultrasound demonstrated increased ovarian volume. The patient was treated with GnRH analogues during 3 years (7.6-10.6 years) with no side effects. Menarche occurred at 11 years and 11 months, and at 12 years and 7 months her height was 158.3 cm (0.7 SDS), BMI 27.42 kg/m2 (2.46 SDS), and she has regular menses with obesity and physical signs of hyperandrogenism.
Circulating DLK1 concentrations
To investigate the effect of the novel variants on DLK1 production, serum DLK1 levels were measured. DLK-1 serum levels were normalized in 62 patients from a group of 250 healthy girls and boys throughout development. The control group included prepubertal (Tanner I; n = 31) and pubertal patients at the start of puberty (Tanner II; n = 31). We have shown in the pubertal group only Tanner II (Fig. 2).
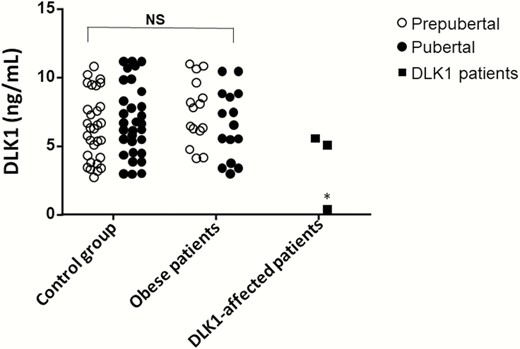
DLK1 serum levels in controls, obese patients, 1 patient with CPP due to DLK1 deletion and 2 patients with CPP and variants in DLK1. *P < .001 with control group.
Patient 1 who carried the c.401_404 + 8del deletion had undetectable DLK1 levels (<0.4 ng/mL), supporting that the deletion led to a complete lack of DLK1 production in this individual. The serum DLK1 levels of her father, mother, and sister were within normal levels (ranged 6.36-8.98 ng/mL). Patient 2 with c.402C>T p.Asn134= (rs150016759) had normal levels of DLK1 (6.11 ng/mL), as did her father (5.05 ng/mL). Patient 3 with 2 consecutive rare allelic variants (g.-222 C>A and g.-223 G>A) in the DLK1 promoter region also had normal levels of DLK1 (5.55 ng/mL). Because the patient was adopted, DLK1 levels in the biological parents were not available.
Discussion
Loss-of-function mutations of DLK1 represent the most recently recognized genetic cause of familial CPP with paternal inheritance. Here, novel findings of DLK1, including a pathogenic deletion leading to a DLK1 deficiency, were identified in Spanish girls with progressive and nonsyndromic CPP. Interestingly, none of them had history of premature sexual development on their paternal side, as we had expected for a condition associated with maternal imprinting gene defects. Indeed, patient 1 was a de novo heterozygous deletion (c.401_404 + 8del) affecting exon 4 and intron 4-5 of DLK1 gene. Her biological parents had no similar DLK1 abnormality, indicating that she is a true sporadic case of CPP associated with a DLK1 defect. The functional impact of this deletion was clearly demonstrated by undetectable serum DLK1 levels.
DLK1 exists in both transmembrane and shed forms. Soluble DLK1 arises by proteolytic extracellular domain clipping through the ADAM 17/TACE enzyme. There are 2 processing sites in the extracellular domain, 1 located near the fourth EGF repeat and the other in the region proximal to the transmembrane domain, resulting in the larger (50 kDa) and smaller (25 kDa) soluble forms (7, 22). Among the 6 pathogenic variants of DLK1 described in patients with CPP so far, 4 of them (the new deletion described here and 3 other frameshift pathogenic allelic variant—p.Gly199Alafs*11, p.Val271Cysfs*14, and p.Pro160Leufs*50) are located in the extracellular domain (Fig. 3) (13). More specifically, only 2 consecutive exons (4 and 5) that encode the extracellular (EFG-4 to 6) and transmembrane domain were affected in these distinct families with CPP, suggesting that this particular region could be a hot spot for pathogenic allelic variants. These particular DLK1 defects were associated with undetectable DLK1 serum levels. Notably, Villanueva et al. (23) reported that Dlk1 expression in the mouse hypothalamus occurred mainly as a cleaved protein and is expressed in dendrites of neurons in multiple nuclei (arcuate, paraventricular, supraoptic, suprachiasmatic, dorsomedial, and lateral hypothalamic), suggesting a relevant neuroendocrine regulation for this soluble DLK1 in postnatal development.
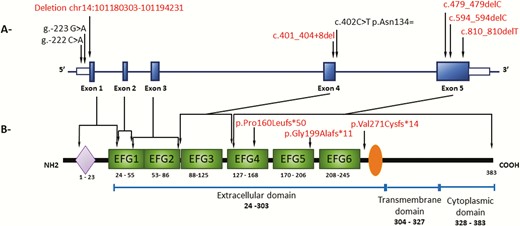
Schematic representation of the human DLK1 gene (A) and human DLK1 protein (B) respectively. (A) Human DLK1 gene (transcript ENST00000341267.9). Blue boxes indicate the coding sequence of the 5 exons of the gene in human. Open boxes indicate the 5′ and 3′ untranslated region of the gene, respectively. The localization of the allelic variant identified in patient with familial CPP are indicated by arrows. In red are the pathogenic allelic variants already described (2, 13) and the 1 described in this work. (B) Human DLK1 protein structure (P80370). The purple lozenge indicates the signal peptide; green boxes the 6 EGF-like repeats. Orange ellipse indicates the extracellular TACE (ADAM17) proteolytic cleavage domain. The numbers represent the amino acid positions of the indicated domains.
The long-term evaluation of the first described women with familial precocious menarche (<9 years) or CPP due to DLK1 variants showed multiple metabolic alterations, such as overweight/obesity, type 2 diabetes mellitus, and hyperlipemia, in agreement with the known physiological metabolic functions of DLK1 (an inhibitor of adipogenesis and fat accumulation). Patient 1 who harbored a pathogenic DLK1 defect, started pubertal development at ~5 years with no additional medical problems and she had a satisfactory response to the classical GnRH analog treatment, suggesting a typical CPP at initial diagnosis. No weight excess, metabolic alterations, or syndromic features were noticed in this affected girl with the DLK1 pathogenic defect. However, she is a teenage girl (15 years) and close clinical follow-up is recommended.
Serum DLK1 levels were measured in healthy Spanish boys and girls throughout development to establish its normal values. Furthermore, DLK1 serum levels were studied in prepubertal and pubertal children with severe obesity. No differences were found between controls and obese children (Fig. 2). Patient 2 harbored a rare polymorphism c.402C>T p.Asn134= (rs150016759) that was inherited from her father. Besides being a rare variant and being suggested as a potential loss of splice site by in silico analysis, the DLK1 serum levels in this patient and in her parents were at normal range. Similar data were noticed in Patient 3, who carried 2 potential regulatory variants into the 5′ untranslated region of DLK1 gene.
Human DLK1 is encoded by a paternally expressed gene located on the long arm of chromosome 14 (14q32.2), within a locus associated with Temple syndrome, an imprinting disorder mainly characterized during childhood by pre- and/or postnatal growth failure, hypotonia, and small hands and feet (24). Notably, 80% to 90% of patients with Temple syndrome have been described with early puberty or CPP (24), which seems to be much more common than the frequency of early puberty in imprinting disorders affecting other imprinted loci. Moreover, patients with Temple syndrome may develop overweight/obesity during childhood, and hyperlipidemia and type 2 diabetes as young adults. Interestingly, Abi Habib et al. (25) identified barely detectable levels of serum DLK1 in patients with Temple syndrome with epimutations or paternal deletions at chromosome 14q32.2.
The prevalence of DLK1 pathogenic variants appears to be significantly lower than MKRN3 defects causing familial CPP. Among the genetic analysis of a total of 168 Spanish girls with idiopathic CPP (including familial and sporadic cases), only 1 girl presented a pathogenic DLK1 defect. Interestingly, this Spanish cohort was screened for MKRN3 mutations and 3.5% had potentially pathogenic variants in MKRN3.
Here, we have described the first de novo DLK1 mutation associated with a true case of sporadic CPP in a girl with progressive and nonsyndromic premature sexual development phenotype. In conclusion, DLK1 defects associated with familial and sporadic CPP are very rare and can be associated with potential metabolic consequences at adulthood. Furthermore, our results indicate that the genomic deletion in DLK1 led to the lack of serum DLK1, while the other 2 variants found in DLK1 did not. In order to establish a genotype–phenotype correlation of these 2 rare variants in patients with CPP, it remains to be determined whether their protein product is biologically active.
Abbreviations
- ACMG
American College of Medical Genetics
- BMI
body mass index
- CPP
central precocious puberty
- DNA
deoxyribonucleic acid
- EGF
epidermal growth factor
- FSH
follicle-stimulating hormone
- GnRH
gonadotropin-releasing hormone
- LH
luteinizing hormone
- LHRH
luteinizing hormone releasing hormone
- MFE
minimum free energy
- MRI
magnetic resonance imaging
- mRNA
messenger ribonucleic acid
- PCR
polymerase chain reaction
Acknowledgments
Financial Support: This work was supported by grant 2008.1.1677.5.4 (to A.C.L) from the Universidade de São Paulo (USP); grant 13/03236-5 (to A.C.L), and 2017/23892-5 (to L.R.M) from Fundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp); grant 403525/2016-0 (to A.C.L) from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq); Fondo de Investigación del Instituto de Salud Carlos III (fondos FEDER); grant PI019/0166 (to J.A) and CIBER de fisiopatología y nutrición (CIBEROBN) (to J.A.), Madrid, Spain.
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability
All data generated or analyzed during this study are included in this published article or in the data repositories listed in References.
References
Author notes
Drs. Montenegro, Labarta, Latronico, and Argente contributed equally to this article.

{kind=link}
{kind=link}
{kind=link}