





Abstract
Lipoid congenital adrenal hyperplasia (LCAH) is characterized by a disorder of steroidogenesis in both adrenal glands and gonads. 46,XX patients with classic LCAH usually have thelarche and menarche but show anovulatory menstruations and subsequent premature menopause. Only three patients with classic LCAH have been reported to successfully achieve delivery with the aid of assisted reproductive therapies for conception and progesterone replacement therapy during early pregnancy. In contrast, pubertal development and pregnancy outcomes in patients with nonclassic LCAH have not been fully elucidated.
We report four Japanese women who had a diagnosis of primary adrenal insufficiency during infancy or childhood and carried compound heterozygous STAR mutations (p.Gln258* and p.Arg188His, p.Gln258* and p.Met225Thr, and p.Gln258* and p.Arg272Cys). In all four patients, thelarche and menarche spontaneously occurred from 10 to 11 years of age and from 12 to 14 years of age, respectively. Subsequently, their menstruation cycles were regular at almost 1-month intervals. Patient 1 conceived naturally twice, and patient 2 conceived with the use of clomiphene citrate for ovulation induction. These two patients maintained the pregnancies without progesterone replacement therapy and successfully delivered children.
Patients with nonclassic LCAH maintain ovarian function, which enables normal pubertal development and a successful pregnancy outcome without progesterone replacement therapy.
Lipoid congenital adrenal hyperplasia (LCAH; OMIM 201710) is an autosomal recessive disorder caused by biallelic loss-of-function mutations in the STAR gene encoding steroidogenic acute regulatory protein (StAR) (1–3). As the first step of steroidogenesis, StAR facilitates cholesterol transfer from the outer to the inner mitochondrial membrane in steroidogenic cells of the adrenal glands and gonads (4). Patients with LCAH are divided into two groups (classic and nonclassic types) based on clinical severity. Patients with classic LCAH have severe impairment in adrenal and gonadal steroidogenesis due to functionally null STAR mutations (2). They are characterized by neonatal- or early infantile-onset primary adrenal insufficiency (PAI) and normal female external genitalia irrespective of their chromosomal sex. In contrast, patients with nonclassic LCAH usually show late infantile- or childhood-onset PAI with or without mineralocorticoid deficiency, because their adrenal glands preserve partial steroidogenic capacity due to the residual activity of StAR. 46,XY patients with nonclassic LCAH show normal or underdeveloped male external genitalia, because their testes preserve testosterone-producing capacity as well (5).
Patients with classic LCAH with a 46,XX karyotype present with spontaneous pubertal development and menarche. However, after menarche, the residual steroidogenic capacity is impaired by the accumulation of ovarian cholesterol esters, resulting in follicular atresia (2). Therefore, they have irregular anovulatory menstruations and eventually develop premature menopause (2, 3, 6). The histopathological analyses of their postpubertal ovaries showed increased macrophages in the stroma and progressive lipoid deposits in theca cells and macrophages (7, 8). Some patients with classic LCAH have large ovarian cysts, occasionally causing ovarian torsion as life-threatening acute abdomen (3, 7).
Based on these observations, achieving and maintaining pregnancy in patients with classic LCAH has been regarded as difficult. They cannot produce enough estrogens to induce the LH surge and subsequent ovulations. Even if they could conceive, they cannot synthesize enough progesterone to maintain pregnancy (9). In fact, only three pregnancy cases have ever been reported. In all the cases, conception was achieved with assisted reproductive technologies, and the pregnancies were maintained with progesterone replacement therapy (10–12).
In contrast, ovarian function in patients with nonclassic LCAH has not been fully elucidated, and there is no report of pregnancy and delivery. Nevertheless, the prognosis of ovarian function in patients with nonclassic LCAH could be more favorable than that in classic LCAH because of the residual steroidogenic capacity in the ovaries. In this report, we assessed the ovarian function of four Japanese women with nonclassic LCAH and describe successful pregnancies and deliveries without hormone replacement therapy in two patients.
Case Reports
Patient 1
The clinical course of this patient was partially published in Japanese (13). A girl was born to nonconsanguineous healthy Japanese parents at full-term gestation, and her birth weight was 3320 g. She had skin pigmentation from the age of 4 years. At 6 years of age, she was referred to us for high fever, vomiting, diarrhea, and lethargy. Endocrine evaluations revealed a low serum cortisol level (4.1 µg/dL; reference range 5.6 to 21.3 µg/dL) and an extremely high plasma ACTH level (2110 pg/mL; reference range 7.7 to 22.1 pg/mL). Her serum aldosterone level and plasma renin activity were 255 (reference range 50 to 900) pg/mL and 2.1 (reference range 0.2 to 2.7) ng/mL/h, respectively. She received a diagnosis of ACTH resistance, and glucocorticoid replacement therapy was initiated. At the age of 10 years, fludrocortisone was added. She had normal growth and development and showed thelarche at the age of 10 years and 9 months. She had spontaneous menarche at the age of 13 years and 6 months, and her menstruations were regular at 40-day intervals. Her serum LH, FSH, estradiol, and progesterone levels are shown in an online repository (14). At the age of 24 years, her serum LH and estradiol levels were 120 and 796 mIU/mL, respectively, indicating that her estradiol secretion was sufficient to induce the LH surge. Her serum progesterone level during the luteal phase was 23.6 ng/mL at the age of 25 years, implying spontaneous ovulation. At the age of 27 years, she married and conceived naturally. She maintained a pregnancy without progesterone replacement therapy. We monitored her serum estradiol and progesterone levels during her pregnancy (Fig. 1) (16). Her serum progesterone level at 6 weeks and 5 days’ gestation was around the fifth percentile and then increased. She delivered a healthy girl weighing 2530 g at 37 weeks’ gestation by elective cesarean delivery. At the age of 30 years, she conceived naturally again and also maintained the second pregnancy without progesterone replacement therapy. Her serum estradiol and progesterone levels during her second pregnancy are also shown in Fig. 1 and an online repository (16). She delivered a healthy boy weighing 2768 g at 37 weeks’ gestation by elective cesarean delivery.
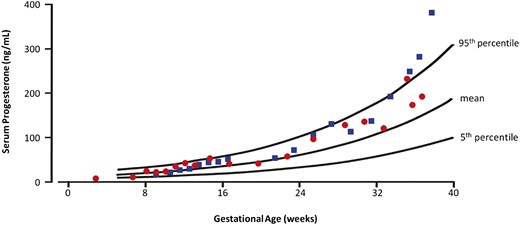
Serum progesterone levels in two pregnancies of patient 1 are shown. Red circles indicate serum progesterone levels in the first pregnancy, and blue squares indicate those in the second pregnancy. The 5th percentile, mean, and 95th percentile curves of serum progesterone levels in normal pregnancies are referred to in the publication of O’Leary et al. (15).
Patient 2
A girl was born to nonconsanguineous healthy Japanese parents at 39 weeks’ gestation without complications, and her birth weight was 3000 g (+0.41 SD). Her external genitalia were complete female type. She received a diagnosis of PAI in infancy, and glucocorticoid and mineralocorticoid replacement therapy was introduced. Unfortunately, an accurate age at onset and her laboratory data at diagnosis were unavailable. She showed thelarche at the age of 11 years. She had spontaneous menarche at the age of 14 years, and her menstrual cycle was regular at ~1-month intervals. However, she sometimes had irregular menstruations accompanied by hypermenorrhea and dysmenorrhea. In her early 20s, she received a diagnosis of endometriosis and underwent dilatation of the cervix and curettage of the endometrium. Postoperative ultrasonography showed that she had ovarian cysts, which were aspirated transvaginally. She married at the age of 28 years and then started ovulation induction with clomiphene citrate. The dosing regimen of the clomiphene therapy was 50 mg once daily for 5 days and was subsequently 100 mg once daily for 5 days. At the age of 30 years, she conceived after the fourth round of clomiphene therapy. She maintained her pregnancy without progesterone replacement therapy and delivered a healthy boy weighing 3035 g at 38 weeks’ gestation by elective cesarean delivery.
Patient 3
A girl was born to nonconsanguineous healthy Japanese parents at 39 weeks’ gestation without complications, and her birth weight and length were 3065 g (+0.61 SD) and 47 cm (−0.95 SD), respectively. Her external genitalia were complete female type. Her elder sister died with a high fever of unknown etiology at the age of 10 months. In early childhood, she was also repeatedly hospitalized because of lethargy with high fever. At the age of 4 years, she underwent an endocrine examination, which revealed a low serum cortisol level (7.3 μg/dL) along with a high plasma ACTH level (3200 pg/mL) and a normal serum 17α-hydroxyprogesterone level (0.1 ng/mL). Her plasma renin activity and serum aldosterone level were 3.2 ng/mL/h and <10 pg/mL, respectively. Based on these findings, she received a diagnosis of PAI, and glucocorticoid replacement therapy was started. Subsequently, fludrocortisone was added at the age of 6 years. At the age of 14 years, she had spontaneous menarche, and at the age of 21 years, her menstrual cycle was regular at ~1-month intervals. Her serum LH, FSH, and estradiol levels are shown in an online repository (14). Her serum LH and estradiol levels at the age of 19 years were 28.35 mIU/mL and 886 pg/mL, respectively, suggesting that her estradiol level would be sufficient to induce the LH surge. We did not assess serum progesterone levels during the luteal phase.
Patient 4
A girl was born to nonconsanguineous Japanese healthy parents at 39 weeks’ gestation without complications, and her birth weight was 3450 g (+1.74 SD). Her external genitalia were complete female type. Her younger brother, who had normal external genitalia, died at the age of 2 years of acute encephalopathy accompanied by hypoglycemia and hyponatremia. At the age of 6 years, she had a febrile seizure and subsequent impaired consciousness. Her laboratory findings showed hyponatremia (117 mEq/L), hyperkalemia (5.2 mEq/L), hypoglycemia (46 mg/dL), a low serum cortisol level (5.1 µg/dL), and a high plasma ACTH level (>1300 pg/mL). Her adrenal glands were not detected on abdominal MRI. She received a diagnosis of adrenal hypoplasia, and glucocorticoid and mineralocorticoid replacement therapy was started. She showed thelarche at the age of 10 years. She had menarche at the age of 12 years, and her menstruations were regular at 28-day intervals at the age of 19 years. Her serum LH, FSH, estradiol, and progesterone levels are shown in an online repository (14).
Genetic analysis of STAR
We obtained written informed consent from the patients or their parents for molecular studies, which were approved by the Ethics Committee of Keio University School of Medicine. We extracted genomic DNA from their peripheral lymphocytes and analyzed STAR with PCR-based Sanger sequencing. The primer sequences and PCR conditions are available upon request. We identified compound heterozygous mutations (p.Gln258* and p.Met225Thr) in patient 2. Nakae et al. (17) reported that STAR-Met225Thr retained 43% of wild-type STAR activity in vitro. We identified compound heterozygous mutations in the other patients (p.Gln258* and p.Arg272Cys in patients 1 and 3 and p.Gln258* and p.Arg188His in patient 4) and predicted that STAR-Arg188His would lose hydrogen bonds of Arg188 with Thr167 and Glu169, based on three-dimensional modeling (18). We performed structural modeling with the same method and predicted that STAR-Arg272Cys would lose hydrogen bonds of Arg272 with Asp106 and Val125 (19).
Discussion
We report four Japanese 46,XX patients with PAI who carried STAR mutations. Based on the review of previously reported cases, 46,XX patients with late infantile- or childhood-onset PAI or 46,XY patients with PAI and normal or underdeveloped male external genitalia are classified as having nonclassic LCAH (5). Patients 1, 3, and 4 received a diagnosis of PAI at the ages of 4 to 6 years, consistent with this classification. The age at onset of PAI was uncertain for patient 2. Previously, Nakae et al. (17) reported that a 46,XY patient carrying the same compound heterozygous mutations as patient 2 (p.Gln258* and p.Met225Thr) developed PAI at the age of 10 months and had clitoromegaly accompanied by a moderate testosterone-producing capacity in response to human chorionic gonadotropin. The combined residual activity of STAR-Met225Thr (43% of wild-type protein) and STAR-Gln258* (null) would be equivalent with that of homozygous STAR-Val187Met (21.6%) or STAR-Arg188Cys (13.6%), which was identified in patients with nonclassic LCAH (5). These data indicate that all four of our patients probably had nonclassic LCAH.
Women with classic LCAH usually undergo spontaneous pubertal development and show regular menses initially. However, their menses are anovulatory and become irregular within several years after menarche, and their serum LH levels are elevated after the onset of puberty (7, 20). In contrast, a 32-year-old woman with nonclassic LCAH, who carried compound heterozygous STAR mutations (p.Thr44Hisfs*3 and p.Gly221Ser), had regular menses and normal serum gonadotropin levels comparable to those seen in the early follicular phase (21). Similarly, our four female patients showed sufficient estrogen- and progesterone-producing capacity, and their LH levels were not elevated, despite the presence of PAI. The reason for this difference in age of onset between PAI and ovarian failure is still unknown but could result from the difference in the contribution of the StAR-dependent steroidogenic pathway to total steroidogenic capacity or in the secondary decline of the StAR-dependent steroidogenic pathway due to accumulated cholesterol ester. Because gonadotropin stimulation is a potential deleterious factor of ovarian failure, we speculate that the suppression of LH elevation might delay or alleviate the further compromise of ovarian function.
To our knowledge, patient 1 is the first reported case of successful natural conception in a woman with LCAH. Female patients with classic LCAH usually have anovulatory menstruations caused by impaired estradiol production that is insufficient for inducing the LH surge (20). Three previously reported patients with classic LCAH needed clomiphene therapy or in vitro fertilization to achieve conception (10–12). In our report, patient 2 also received clomiphene citrate for ovulation induction. In contrast, patient 1 had regular menstrual cycles, and the evaluation of ovarian function showed an LH surge and progesterone levels comparable to those of the luteal phase, indicating that she had spontaneous ovulations. In patient 3, we also detected an LH surge. Therefore, these female patients with nonclassic LCAH had enough estrogen-producing capacity to provoke the LH surge and probably to induce ovulation.
Patients 1 and 2 had successful pregnancy outcomes without progesterone replacement therapy. During pregnancy, both the corpus luteum of maternal-derived tissue and the placenta of fetal-derived tissue can produce progesterone. Until 7 or 8 weeks’ gestation, the corpus luteum produces mainly progesterone. Subsequently, the main tissue of progesterone production shifts from the corpus luteum to the placenta (22). Therefore, the progesterone-producing capacity of the corpus luteum in the early stage of pregnancy in patients with LCAH is critical for the maintenance of the pregnancy (23). In previous reports, all three patients with classic LCAH with four successful deliveries received progesterone replacement therapy during the early stage of their pregnancies (10–12). In contrast, the serum progesterone levels during the early pregnancy of patient 1 were between the fifth percentile and the mean progesterone level in healthy pregnant women and then increased after the luteal-placental shift. Patients 1 and 2 were able to maintain three pregnancies without progesterone replacement therapy. Based on these data, patients with nonclassic LCAH could have sufficient ovarian progesterone-producing capacity to maintain their pregnancies.
We have demonstrated that female patients with nonclassic LCAH maintained their pregnancies without progesterone replacement therapy, with one patient achieving natural conception. Patients with nonclassic LCAH maintain ovarian function, which enables normal pubertal development and even a successful pregnancy outcome without progesterone replacement despite overt PAI.
Abbreviations:
- LCAH
lipoid congenital adrenal hyperplasia
- PAI
primary adrenal insufficiency
- StAR
steroidogenic acute regulatory protein
Acknowledgments
We thank Professor Takao Takahashi for fruitful discussion.
Financial Support: This work was supported by a grant from the Ministry of Health, Labour and Welfare, Japan [Jitsuyoka Nanbyo-Ippan-046 (to T.H. and T.I.)] and grants from the Japan Society for the Promotion of Science [16K09998 (to N.A.) and 16K09996 (to T.I.)].
Disclosure Summary: The authors have nothing to disclose.

{kind=link}