





Abstract
46,XY Gonadal dysgenesis (GD) is a heterogeneous group of disorders with a wide phenotypic spectrum, including embryonic testicular regression syndrome (ETRS).
To report a gene for 46,XY GD etiology, especially for ETRS.
Screening of familial cases of 46,XY GD using whole-exome sequencing and sporadic cases by target gene-panel sequencing.
Tertiary Referral Center for differences/disorders of sex development (DSD).
We selected 87 patients with 46,XY DSD (17 familial cases from 8 unrelated families and 70 sporadic cases); 55 patients had GD (among them, 10 patients from 5 families and 8 sporadic cases had ETRS), and 32 patients had 46,XY DSD of unknown etiology.
We identified four heterozygous missense rare variants, classified as pathogenic or likely pathogenic in the Asp-Glu-Ala-His-box (DHX) helicase 37 (DHX37) gene in five families (n = 11 patients) and in six sporadic cases. Two variants were recurrent: p.Arg308Gln (in two families and in three sporadic cases) and p.Arg674Trp (in two families and in two sporadic cases). The variants were specifically associated with ETRS (7/14 index cases; 50%). The frequency of rare, predicted-to-be-deleterious DHX37 variants in this cohort (14%) is significantly higher than that observed in the Genome Aggregation Database (0.4%; P < 0.001). Immunohistochemistry analysis in human testis showed that DHX37 is mainly expressed in germ cells at different stages of testis maturation, in Leydig cells, and rarely in Sertoli cells.
This strong genetic evidence identifies DHX37 as a player in the complex cascade of male gonadal differentiation and maintenance.
46,XY gonadal dysgenesis (GD) represents a heterogeneous group of disorders/differences of sex development (DSD), characterized by abnormal gonadal development, leading to a wide phenotypic spectrum. Variable degrees of external genitalia undervirilization are observed, ranging from micropenis to female-like genitalia and partially or fully developed Mullerian derivatives. The gonads from these patients display a wide spectrum of histological abnormalities, ranging from ovarian-like stroma with disorganized seminiferous tubules to complete absence of gonadal tissue (1). Embryonic testicular regression syndrome (ETRS) is considered a part of the clinical spectrum of 46,XY GD (2). Most individuals with ETRS present with micropenis or atypical genitalia and lack of gonadal tissue on one or both sides (2).
Partial or complete Mullerian duct regression associated with micropenis suggests an intrinsically functional testis in the first months of fetal life, with subsequent loss of testicular function before the last trimester of gestation, when the increase in penile length occurs.
Numerous genes are known to be involved in the process of gonadal determination (3). However, a genetic diagnosis is identified in <40% of patients with 46,XY GD (4). Moreover, few patients with ETRS were included in large cohorts of 46,XY DSD previously studied (4). However, the fact that some familial cases of ETRS were reported indicates a genetic etiology (5, 6).
In the present work, high-throughput, parallel-sequencing methods, including whole-exome sequencing (WES) and targeted DSD gene panels, were used to investigate the underlying genetic etiology in a large cohort of 46,XY patients with GD and 46,XY DSD patients with unknown etiological cause.
We identified recurrent, rare variants in Asp-Glu-Ala-His-box (DHX) helicase 37 (DHX37) in several affected individuals from distinct families, establishing a genetic cause for the 46,XY GD spectrum, including ETRS.
Methods
Ethics
This study was approved by the Ethics Committee of the Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, Institutional Review Board of the University of Michigan Medical School, Hospital de Garrahan Escuela de Medicina, Pontificia Universidad Católica de Chile, and Hospital Nacional Prof. Dr. A. Posadas. Written, informed consent was obtained from all patients, their parents, or legal guardians.
Subjects
We studied 87 46,XY DSD patients without previous molecular diagnosis, including 17 familial cases of 46,XY GD from 8 nonconsanguineous families and 70 sporadic cases (38 with GD and 32 with 46,XY DSD of unknown etiology). Out of the 55 patients with GD, 10 patients from 5 families and 8 sporadic cases had an ETRS phenotype. The patients had different nationalities: Brazilian (81 patients), Argentinian (3 siblings), Chilean (2 siblings), and Chinese-American (1 patient).
All patients have a normal Giemsa Trypsin G-banded metaphases 46,XY karyotype.
The 46,XY DSD patients were classified as having complete GD if they had female external genitalia, Mullerian derivatives, and streak gonads; partial GD (PGD) if they had atypical external genitalia, Mullerian derivatives, and at least one gonad with histopathological features of dysgenetic testis; ETRS if they had micropenis, partially developed Mullerian derivatives, and no gonadal tissue or small area of gonadal stroma; and 46,XY DSD of unknown etiology if the hormonal profile was not conclusive or not available as a result of previous gonadectomy. In this latter group, molecular defects of luteinizing hormone/choriogonadotropin and androgen receptor genes CYP17A1, HSD17B3, HSD3B2, and 5ARD2 were ruled out by DNA sequencing.
Genomic DNA
For molecular diagnosis, genomic DNA was extracted from peripheral blood leukocytes by the proteinase K–SDS salting-out method (7).
Genetic study
WES was performed in 14 familial cases from 7 families. In all but one family (F), the probands and their first-degree relatives and other affected family members were studied.
Sixty-eight sporadic cases were studied by targeted, massively parallel sequencing. DHX37 was studied by Sanger sequencing in two sporadic cases and in three patients from F2 (Fig. 1) (8).
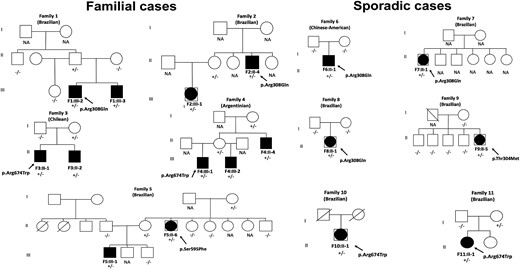
Pedigrees of the 11 families with potential disease-causing DHX37 variants. Closed symbols represent affected individuals. The affected males (46, XY males) are indicated by closed squares, and the affected individuals raised as females (46,XY females) are shown by large, closed circles within the squares. Symbols with a diagonal line represent deceased individuals. The DHX37 genotype is shown for the individuals whose DNA sample was available. Paternity and maternity were confirmed in Families 6 and 8. +/−, Heterozygous state; −/−, homozygous state for wild-type allele; NA, DNA not available.
Enrichment for massively parallel sequencing was performed with the Nextera Exome Enrichment Kit (Illumina, San Diego, CA), followed by paired-end sequencing on the HiSeq 2500 System (Illumina)
For target sequencing, we designed an amplicon-based capture panel against exonic regions of 63 genes, including 43 genes known to be associated with human DSDs and 20 candidate genes, including DHX37 (3, 9). Target sequences were captured using a custom Sure Select Target Enrichment System Kit (Agilent Technologies, Santa Clara, CA), and sequencing was performed on the MiSeq platform (Illumina).
Sanger sequencing was used to confirm the potentially pathogenic variants identified by massively parallel sequencing and for segregation analysis. PCR products were sequenced with a BigDye® Terminator version 3.1 cycle sequencing kit followed by automated sequencing on an ABI PRISM® 3130xl genetic analyzer (Applied Biosystems, Foster City, CA).
The identified variants were classified according to the American College of Medical Genetics criteria (10).
Data analysis
The exome and the targeted panel sequencing data were screened for rare variants [minor allele frequency <0.1% in the public databases: Genome Aggregation Database (gnomAD) (11), 1000 Genomes (12), and the Brazilian population database Arquivo Brasileiro Online de Mutações (ABraOM) (13)], located in exonic and consensus splice-site regions. Subsequently, the filtration pipeline prioritized potentially pathogenic candidate variants (loss-of-function variants and variants classified as pathogenic by multiple in silico programs). For variants identified by WES, we selected variants that fit an autosomal-dominant model.
The sequencing reads carrying candidate variants were visually confirmed using the Integrative Genomics Viewer (Broad Institute, Cambridge, MA). Candidate variants were segregated in the family members by the Sanger method.
The filtering of the variants is provided in da Silva et al.(8).
Histological analysis
Immunohistochemical staining
Eight formalin-fixed, paraffin-embedded testicular tissue samples from 46,XY individuals with different chronological ages (27 and 33 weeks gestational age; 1, 53, and 180 days of age; 13, 23, and 53 years of age) were collected during autopsy and used for DHX37 expression analysis by immunohistochemistry. All samples were sliced into 3 μm-thick sections using an automatic Leica RM2255 microtome (Leica Biosystems, Nussloch, Germany). The sections were briefly stretched in xylol at 60°C for 20 minutes, cooled in xylol, and dried in an incubator (Orion 515; Fanem, São Paulo, Brazil) at 60°C. Sections were subjected to hematoxylin-eosin staining for histological analysis. For the immunohistochemical study, slides were deparaffinized with xylene, hydrated in ethanol, washed in PBS (0.01 M/pH 7.4), and blocked using methanol and hydrogen peroxide. Epitope exposure was carried out by placing the slide in boiling 10 mM citric acid (pH 6) or 100 mM EDTA (pH 9), followed by blocking nonspecific protein. Rabbit polyclonal anti-DHX37 antibody (NB110-40581; Novus Biologicals, Centennial, CO) was added at a dilution factor of 1:50. Dilution was standardized after testing ovarian and skin tissues, where protein expression was identified in the cytoplasm of oocytes and nuclear membranes of ovarian stromal and squamous cells. The samples were incubated with universal secondary antibodies using the Novo Link Detection Systems kit (Leica Biosystems, Newcastle upon Tyne, United Kingdom), according to the manufacturer’s instruction.
Statistical analysis
To test the genetic evidence for the association between DHX37 and GD phenotype, we performed aggregate variant analyses comparing allele frequencies among our 46,XY DSD cohort and public databases (gnomAD and ABraOM).
Variants with similar characteristics of the DHX37 variants observed in our cohort [rare, nonsynonymous variants with a minor allele frequency of 1% and located in the two highly conserved proteins (ATP-binding and helicase C-terminal domains) that are predicted to be pathogenic by at least four in silico tools: Mutation Taster, Sorting Intolerant From Tolerant PolyPhen-2, Mutation Assessor, and Protein Variation Effect Analyzer were selected. Allele frequency differences among groups were analyzed by the χ2 test, and statistical significance was set at P < 0.05. Statistical analyses were performed using the SigmaStat statistical software package (Windows, version 3.5; SPSS Inc., San Rafael, CA).
Results
Patient phenotype and DHX37 variants
Firstly, WES identified the same DHX37 variant p.Arg308Gln (c.923G>A; GenBank NM_032656.3) in a heterozygous state in two unrelated Brazilian families with ETRS (Families 1 and 2). All of the affected individuals have the same phenotype (micropenis and absence or bilateral rudimentary gonadal tissue; Fig. 1 and Table 1). A founder effect for the p.Arg308Gln variant was ruled out in Families 1 and 2.
Phenotype of 46,XY DSD Patients With Familial ETRS With Rare and Predicted Pathogenic or Likely Pathogenic DHX37 Variants
| Nationality | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variables | Brazilian | Brazilian | Chilean | Argentinian | Brazilian | ||||||
| Patient | F1:III-2 | F1:III-3 | F2:II-4 | F2:III-1 | F3:II-1 | F3:II-2 | F4:III-1 | F4:III-2 | F4:II-4 | F5:II-6 | F5:III-1 |
| Sex of rearing | Male | Male | Male | Female | Male | Male | Male | Male | Male | Female | Male |
| Age at presentation, y | 2.2 | 1.8 | 14.0 | 1.8 | 0.6 | 24 Days | Newborn | 4 | 2 | 18 | 10 |
| Diagnosis | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | PGD | PGD | ETRS |
| External genitalia | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Atypical | Micropenis |
| Gonads | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Left testis–scrotum | Previous gonadectomy | Nonpalpable |
| Histologic analysis | Small bilateral dysgenetic gonads | Left gonad not found; right dysgenetic gonad | No gonadal tissue found | Left gonad not found; small right dysgenetic gonad | Small bilateral dysgenetic gonads | Small bilateral dysgenetic gonads | No gonadal tissue | No gonadal tissue | Right gonad not found; left dysgenetic testis with GCNISa | Bilateral dysgenetic gonads | No gonadal tissue |
| Wolffian derivatives | Present | Present | Present | Present | Present | Present | Present | Present | Present | Present | Present |
| Mullerian derivatives | |||||||||||
| Tubes | Presentb | Absent | Presentb | Presentb | Absent | Present | Presentb | Presentb | Absent | Absent | Absent |
| Uterus | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| LH, IU/L | 14.5 | 12 | 3.5 | 1.9 | <0.5 | <0.5 | <0.5 | <0.5 | 26 | NA | 19 |
| FSH, IU/L | 117 | 133 | 87 | 56 | 10.9 | 9.5 | NA | 9 | 112 | NA | 43 |
| Basal testosterone, ng/dL | <10 | <10 | <10 | NA | 16 | <10 | 38 | 27 | 16 | NA | 21 |
| Testosterone after hCG test, ng/dL | <10 | NA | 29 | 26 | 18 | <10 | 40 | 29 | NA | NA | NA |
| Allelic variant | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Ser595Phe | p.Ser595Phe |
| Variant state | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
| Nationality | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variables | Brazilian | Brazilian | Chilean | Argentinian | Brazilian | ||||||
| Patient | F1:III-2 | F1:III-3 | F2:II-4 | F2:III-1 | F3:II-1 | F3:II-2 | F4:III-1 | F4:III-2 | F4:II-4 | F5:II-6 | F5:III-1 |
| Sex of rearing | Male | Male | Male | Female | Male | Male | Male | Male | Male | Female | Male |
| Age at presentation, y | 2.2 | 1.8 | 14.0 | 1.8 | 0.6 | 24 Days | Newborn | 4 | 2 | 18 | 10 |
| Diagnosis | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | PGD | PGD | ETRS |
| External genitalia | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Atypical | Micropenis |
| Gonads | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Left testis–scrotum | Previous gonadectomy | Nonpalpable |
| Histologic analysis | Small bilateral dysgenetic gonads | Left gonad not found; right dysgenetic gonad | No gonadal tissue found | Left gonad not found; small right dysgenetic gonad | Small bilateral dysgenetic gonads | Small bilateral dysgenetic gonads | No gonadal tissue | No gonadal tissue | Right gonad not found; left dysgenetic testis with GCNISa | Bilateral dysgenetic gonads | No gonadal tissue |
| Wolffian derivatives | Present | Present | Present | Present | Present | Present | Present | Present | Present | Present | Present |
| Mullerian derivatives | |||||||||||
| Tubes | Presentb | Absent | Presentb | Presentb | Absent | Present | Presentb | Presentb | Absent | Absent | Absent |
| Uterus | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| LH, IU/L | 14.5 | 12 | 3.5 | 1.9 | <0.5 | <0.5 | <0.5 | <0.5 | 26 | NA | 19 |
| FSH, IU/L | 117 | 133 | 87 | 56 | 10.9 | 9.5 | NA | 9 | 112 | NA | 43 |
| Basal testosterone, ng/dL | <10 | <10 | <10 | NA | 16 | <10 | 38 | 27 | 16 | NA | 21 |
| Testosterone after hCG test, ng/dL | <10 | NA | 29 | 26 | 18 | <10 | 40 | 29 | NA | NA | NA |
| Allelic variant | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Ser595Phe | p.Ser595Phe |
| Variant state | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
Conversion factors to International System of Units: T, ng/dL to nM, multiply by 0.0347.
Abbreviations: GCNIS, germ-cell neoplasia in situ; hCG, human chorionic gonadotropin; NA, not available.
Testicular biopsy.
Rudimentary fallopian tubes.
Phenotype of 46,XY DSD Patients With Familial ETRS With Rare and Predicted Pathogenic or Likely Pathogenic DHX37 Variants
| Nationality | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variables | Brazilian | Brazilian | Chilean | Argentinian | Brazilian | ||||||
| Patient | F1:III-2 | F1:III-3 | F2:II-4 | F2:III-1 | F3:II-1 | F3:II-2 | F4:III-1 | F4:III-2 | F4:II-4 | F5:II-6 | F5:III-1 |
| Sex of rearing | Male | Male | Male | Female | Male | Male | Male | Male | Male | Female | Male |
| Age at presentation, y | 2.2 | 1.8 | 14.0 | 1.8 | 0.6 | 24 Days | Newborn | 4 | 2 | 18 | 10 |
| Diagnosis | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | PGD | PGD | ETRS |
| External genitalia | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Atypical | Micropenis |
| Gonads | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Left testis–scrotum | Previous gonadectomy | Nonpalpable |
| Histologic analysis | Small bilateral dysgenetic gonads | Left gonad not found; right dysgenetic gonad | No gonadal tissue found | Left gonad not found; small right dysgenetic gonad | Small bilateral dysgenetic gonads | Small bilateral dysgenetic gonads | No gonadal tissue | No gonadal tissue | Right gonad not found; left dysgenetic testis with GCNISa | Bilateral dysgenetic gonads | No gonadal tissue |
| Wolffian derivatives | Present | Present | Present | Present | Present | Present | Present | Present | Present | Present | Present |
| Mullerian derivatives | |||||||||||
| Tubes | Presentb | Absent | Presentb | Presentb | Absent | Present | Presentb | Presentb | Absent | Absent | Absent |
| Uterus | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| LH, IU/L | 14.5 | 12 | 3.5 | 1.9 | <0.5 | <0.5 | <0.5 | <0.5 | 26 | NA | 19 |
| FSH, IU/L | 117 | 133 | 87 | 56 | 10.9 | 9.5 | NA | 9 | 112 | NA | 43 |
| Basal testosterone, ng/dL | <10 | <10 | <10 | NA | 16 | <10 | 38 | 27 | 16 | NA | 21 |
| Testosterone after hCG test, ng/dL | <10 | NA | 29 | 26 | 18 | <10 | 40 | 29 | NA | NA | NA |
| Allelic variant | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Ser595Phe | p.Ser595Phe |
| Variant state | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
| Nationality | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variables | Brazilian | Brazilian | Chilean | Argentinian | Brazilian | ||||||
| Patient | F1:III-2 | F1:III-3 | F2:II-4 | F2:III-1 | F3:II-1 | F3:II-2 | F4:III-1 | F4:III-2 | F4:II-4 | F5:II-6 | F5:III-1 |
| Sex of rearing | Male | Male | Male | Female | Male | Male | Male | Male | Male | Female | Male |
| Age at presentation, y | 2.2 | 1.8 | 14.0 | 1.8 | 0.6 | 24 Days | Newborn | 4 | 2 | 18 | 10 |
| Diagnosis | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | ETRS | PGD | PGD | ETRS |
| External genitalia | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Micropenis | Atypical | Micropenis |
| Gonads | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Nonpalpable | Left testis–scrotum | Previous gonadectomy | Nonpalpable |
| Histologic analysis | Small bilateral dysgenetic gonads | Left gonad not found; right dysgenetic gonad | No gonadal tissue found | Left gonad not found; small right dysgenetic gonad | Small bilateral dysgenetic gonads | Small bilateral dysgenetic gonads | No gonadal tissue | No gonadal tissue | Right gonad not found; left dysgenetic testis with GCNISa | Bilateral dysgenetic gonads | No gonadal tissue |
| Wolffian derivatives | Present | Present | Present | Present | Present | Present | Present | Present | Present | Present | Present |
| Mullerian derivatives | |||||||||||
| Tubes | Presentb | Absent | Presentb | Presentb | Absent | Present | Presentb | Presentb | Absent | Absent | Absent |
| Uterus | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| LH, IU/L | 14.5 | 12 | 3.5 | 1.9 | <0.5 | <0.5 | <0.5 | <0.5 | 26 | NA | 19 |
| FSH, IU/L | 117 | 133 | 87 | 56 | 10.9 | 9.5 | NA | 9 | 112 | NA | 43 |
| Basal testosterone, ng/dL | <10 | <10 | <10 | NA | 16 | <10 | 38 | 27 | 16 | NA | 21 |
| Testosterone after hCG test, ng/dL | <10 | NA | 29 | 26 | 18 | <10 | 40 | 29 | NA | NA | NA |
| Allelic variant | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Arg674Trp | p.Ser595Phe | p.Ser595Phe |
| Variant state | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
Conversion factors to International System of Units: T, ng/dL to nM, multiply by 0.0347.
Abbreviations: GCNIS, germ-cell neoplasia in situ; hCG, human chorionic gonadotropin; NA, not available.
Testicular biopsy.
Rudimentary fallopian tubes.
The p.Arg308Gln variant was also identified by WES in a Chinese-American sporadic case of ETRS from the University of Michigan performed in the laboratory of E.V. (sporadic case F6:II-1; Fig. 1 and Table 2).
Phenotype of 46,XY DSD Patients With a Sporadic GD Spectrum and Heterozygous Rare Pathogenic or Likely Pathogenic DHX37 Variants
| Nationality | ||||||
|---|---|---|---|---|---|---|
| Variables | Chinese-American | Brazilian | ||||
| Patient | F6:II-1 | F7:II-1 | F8:II-1 | F9:II-5 | F10:II-1 | F11:II-1 |
| Social sex | Male | Male to Female | Female | Female | Male to female | Female |
| Age at presentation, y | 0.18 | 30 | 7.7 | 35 | 19 | 3.7 |
| Diagnosis | ETRS | ETRS | PGD | Previous gonadectomy | ETRS | PGD |
| External genitalia | Micropenis | Micropenis | Female | Previous genitoplasty | Micropenis | Atypical |
| Gonads | Nonpalpable | Nonpalpable | Nonpalpable | NA | Nonpalpable | Nonpalpable |
| Histological analysis | No gonadal tissue | No gonadal tissue | Bilateral dysgenetic gonads | NA | No gonadal tissue | Bilateral dysgenetic gonads |
| Wolff derivatives | Present | Present | Present | NA | NA | Present |
| Mullerian derivatives | ||||||
| Tubes | Absent | Present | Absent | NA | Absent | Present |
| Uterus | Absent | Absent | Absent | Absent | Absent | Absent |
| LH, IU/L | 0.1 | 10 | 0.1 | NA | 23 | NA |
| FSH, IU/L | 0.4 | 40 | 4.9 | NA | 62 | NA |
| Basal testosterone, ng/dL | <10 | NA | <10 | NA | 21 | 25 |
| Testosterone after hCG test, ng/dL | <10 | <10 | NA | NA | NA | 33 |
| Allelic variant | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Thr304Met | p.Arg674Trp | p.Arg674Trp |
| Variant state | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
| Nationality | ||||||
|---|---|---|---|---|---|---|
| Variables | Chinese-American | Brazilian | ||||
| Patient | F6:II-1 | F7:II-1 | F8:II-1 | F9:II-5 | F10:II-1 | F11:II-1 |
| Social sex | Male | Male to Female | Female | Female | Male to female | Female |
| Age at presentation, y | 0.18 | 30 | 7.7 | 35 | 19 | 3.7 |
| Diagnosis | ETRS | ETRS | PGD | Previous gonadectomy | ETRS | PGD |
| External genitalia | Micropenis | Micropenis | Female | Previous genitoplasty | Micropenis | Atypical |
| Gonads | Nonpalpable | Nonpalpable | Nonpalpable | NA | Nonpalpable | Nonpalpable |
| Histological analysis | No gonadal tissue | No gonadal tissue | Bilateral dysgenetic gonads | NA | No gonadal tissue | Bilateral dysgenetic gonads |
| Wolff derivatives | Present | Present | Present | NA | NA | Present |
| Mullerian derivatives | ||||||
| Tubes | Absent | Present | Absent | NA | Absent | Present |
| Uterus | Absent | Absent | Absent | Absent | Absent | Absent |
| LH, IU/L | 0.1 | 10 | 0.1 | NA | 23 | NA |
| FSH, IU/L | 0.4 | 40 | 4.9 | NA | 62 | NA |
| Basal testosterone, ng/dL | <10 | NA | <10 | NA | 21 | 25 |
| Testosterone after hCG test, ng/dL | <10 | <10 | NA | NA | NA | 33 |
| Allelic variant | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Thr304Met | p.Arg674Trp | p.Arg674Trp |
| Variant state | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
Conversion factors to International System of Units: T, ng/dL to nM, multiply by 0.0347.
Abbreviation: NA, not available.
Phenotype of 46,XY DSD Patients With a Sporadic GD Spectrum and Heterozygous Rare Pathogenic or Likely Pathogenic DHX37 Variants
| Nationality | ||||||
|---|---|---|---|---|---|---|
| Variables | Chinese-American | Brazilian | ||||
| Patient | F6:II-1 | F7:II-1 | F8:II-1 | F9:II-5 | F10:II-1 | F11:II-1 |
| Social sex | Male | Male to Female | Female | Female | Male to female | Female |
| Age at presentation, y | 0.18 | 30 | 7.7 | 35 | 19 | 3.7 |
| Diagnosis | ETRS | ETRS | PGD | Previous gonadectomy | ETRS | PGD |
| External genitalia | Micropenis | Micropenis | Female | Previous genitoplasty | Micropenis | Atypical |
| Gonads | Nonpalpable | Nonpalpable | Nonpalpable | NA | Nonpalpable | Nonpalpable |
| Histological analysis | No gonadal tissue | No gonadal tissue | Bilateral dysgenetic gonads | NA | No gonadal tissue | Bilateral dysgenetic gonads |
| Wolff derivatives | Present | Present | Present | NA | NA | Present |
| Mullerian derivatives | ||||||
| Tubes | Absent | Present | Absent | NA | Absent | Present |
| Uterus | Absent | Absent | Absent | Absent | Absent | Absent |
| LH, IU/L | 0.1 | 10 | 0.1 | NA | 23 | NA |
| FSH, IU/L | 0.4 | 40 | 4.9 | NA | 62 | NA |
| Basal testosterone, ng/dL | <10 | NA | <10 | NA | 21 | 25 |
| Testosterone after hCG test, ng/dL | <10 | <10 | NA | NA | NA | 33 |
| Allelic variant | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Thr304Met | p.Arg674Trp | p.Arg674Trp |
| Variant state | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
| Nationality | ||||||
|---|---|---|---|---|---|---|
| Variables | Chinese-American | Brazilian | ||||
| Patient | F6:II-1 | F7:II-1 | F8:II-1 | F9:II-5 | F10:II-1 | F11:II-1 |
| Social sex | Male | Male to Female | Female | Female | Male to female | Female |
| Age at presentation, y | 0.18 | 30 | 7.7 | 35 | 19 | 3.7 |
| Diagnosis | ETRS | ETRS | PGD | Previous gonadectomy | ETRS | PGD |
| External genitalia | Micropenis | Micropenis | Female | Previous genitoplasty | Micropenis | Atypical |
| Gonads | Nonpalpable | Nonpalpable | Nonpalpable | NA | Nonpalpable | Nonpalpable |
| Histological analysis | No gonadal tissue | No gonadal tissue | Bilateral dysgenetic gonads | NA | No gonadal tissue | Bilateral dysgenetic gonads |
| Wolff derivatives | Present | Present | Present | NA | NA | Present |
| Mullerian derivatives | ||||||
| Tubes | Absent | Present | Absent | NA | Absent | Present |
| Uterus | Absent | Absent | Absent | Absent | Absent | Absent |
| LH, IU/L | 0.1 | 10 | 0.1 | NA | 23 | NA |
| FSH, IU/L | 0.4 | 40 | 4.9 | NA | 62 | NA |
| Basal testosterone, ng/dL | <10 | NA | <10 | NA | 21 | 25 |
| Testosterone after hCG test, ng/dL | <10 | <10 | NA | NA | NA | 33 |
| Allelic variant | p.Arg308Gln | p.Arg308Gln | p.Arg308Gln | p.Thr304Met | p.Arg674Trp | p.Arg674Trp |
| Variant state | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
Conversion factors to International System of Units: T, ng/dL to nM, multiply by 0.0347.
Abbreviation: NA, not available.
As a candidate gene for 46,XY DSD, DHX37 was included in our target DSD panel. The same p.Arg308Gln variant was identified in another two sporadic cases: one had ETRS (sporadic case F7:II-1), and the other had PGD (sporadic case F8:II-1; Fig. 1 and Table 2).
A further three different heterozygous DHX37 missense variants (p.Arg674Trp, p.Ser595Phe, and p.Thr304Met) were identified in seven affected members from three families and in three sporadic cases (Fig. 2).

The identified variants are localized within conserved helicase domains of DHX37. (Top) Schematic protein structure of DHX37, showing conserved motifs of the helicase core region, helicase-associated domain (HA2), and oligonucleotide/oligosaccharide-binding fold. (Middle) Nucleotide-interacting motifs (I, II, and VI); nucleic acid-binding motifs (Ia, Ib, and IV); motif V, which binds nucleic acid and interacts with nucleotides; and motif III, which couples ATP hydrolysis to RNA unwinding. (Bottom) Amino acids (AAs) within conserved motifs of the helicase core region. The position of the first and last AA within each motif is denoted below left and right, respectively. The positions of the allelic variants identified in this study are indicated with vertical arrows and shown in bold in the different species sequences. N-, N terminus; -C, C terminus.
All of these four variants are predicted to be pathogenic by at least four in silico prediction tools (Table 3) and are absent in genomic population databases, except for p.Arg308Gln, which has a very low allele frequency (0.003%) in the gnomAD database (Tables 4 and 5).
In Silico Prediction Analysis of DHX37 Allelic Variants Identified in 46,XY DSD Patients
| Families | Nucleotide Changed | AA Changed | Functional Domain | In Silico Prediction Tools | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutation Taster | Mutation Assessor | SIFT | Polyphen-2 | PROVEAN | CADD | GERP | ||||
| F1, F2, F6, F7, F8 | c.923G>A | p.Arg308Gln | Helicase ATP binding | Disease cause (score: 0.999) | High functional impact (score: 4.38) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −3.93) | 35 | 5.3 |
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | Helicase superfamily C-terminal domain | Disease cause (score 1.000) | Middle functional impact (score: 4.83) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −7.42) | 33 | 4.2 |
| F5 | c.1784C>T | p.Ser595Phe | Helicase superfamily C-terminal domain | Disease cause (score: 1.000) | High functional impact (score: 4.26) | Protein function affected (score 0.001) | Benign (score 0.24) | Deleterious (score −5.57) | 24.4 | 4.13 |
| F9 | c.911C>T | p.Thr304Met | Helicase ATP binding | Disease cause (score 1.000) | High functional impact (score: 4.45) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −5.89) | 29.8 | 5.3 |
| Families | Nucleotide Changed | AA Changed | Functional Domain | In Silico Prediction Tools | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutation Taster | Mutation Assessor | SIFT | Polyphen-2 | PROVEAN | CADD | GERP | ||||
| F1, F2, F6, F7, F8 | c.923G>A | p.Arg308Gln | Helicase ATP binding | Disease cause (score: 0.999) | High functional impact (score: 4.38) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −3.93) | 35 | 5.3 |
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | Helicase superfamily C-terminal domain | Disease cause (score 1.000) | Middle functional impact (score: 4.83) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −7.42) | 33 | 4.2 |
| F5 | c.1784C>T | p.Ser595Phe | Helicase superfamily C-terminal domain | Disease cause (score: 1.000) | High functional impact (score: 4.26) | Protein function affected (score 0.001) | Benign (score 0.24) | Deleterious (score −5.57) | 24.4 | 4.13 |
| F9 | c.911C>T | p.Thr304Met | Helicase ATP binding | Disease cause (score 1.000) | High functional impact (score: 4.45) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −5.89) | 29.8 | 5.3 |
Abbreviations: CADD, Combined Annotation Dependent Depletion; GERP, Genomic Evolutionary Rate Profiling; PROVEAN, Protein Variation Effect Analyzer; SIFT, Sorting Intolerant From Tolerant.
In Silico Prediction Analysis of DHX37 Allelic Variants Identified in 46,XY DSD Patients
| Families | Nucleotide Changed | AA Changed | Functional Domain | In Silico Prediction Tools | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutation Taster | Mutation Assessor | SIFT | Polyphen-2 | PROVEAN | CADD | GERP | ||||
| F1, F2, F6, F7, F8 | c.923G>A | p.Arg308Gln | Helicase ATP binding | Disease cause (score: 0.999) | High functional impact (score: 4.38) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −3.93) | 35 | 5.3 |
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | Helicase superfamily C-terminal domain | Disease cause (score 1.000) | Middle functional impact (score: 4.83) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −7.42) | 33 | 4.2 |
| F5 | c.1784C>T | p.Ser595Phe | Helicase superfamily C-terminal domain | Disease cause (score: 1.000) | High functional impact (score: 4.26) | Protein function affected (score 0.001) | Benign (score 0.24) | Deleterious (score −5.57) | 24.4 | 4.13 |
| F9 | c.911C>T | p.Thr304Met | Helicase ATP binding | Disease cause (score 1.000) | High functional impact (score: 4.45) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −5.89) | 29.8 | 5.3 |
| Families | Nucleotide Changed | AA Changed | Functional Domain | In Silico Prediction Tools | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutation Taster | Mutation Assessor | SIFT | Polyphen-2 | PROVEAN | CADD | GERP | ||||
| F1, F2, F6, F7, F8 | c.923G>A | p.Arg308Gln | Helicase ATP binding | Disease cause (score: 0.999) | High functional impact (score: 4.38) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −3.93) | 35 | 5.3 |
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | Helicase superfamily C-terminal domain | Disease cause (score 1.000) | Middle functional impact (score: 4.83) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −7.42) | 33 | 4.2 |
| F5 | c.1784C>T | p.Ser595Phe | Helicase superfamily C-terminal domain | Disease cause (score: 1.000) | High functional impact (score: 4.26) | Protein function affected (score 0.001) | Benign (score 0.24) | Deleterious (score −5.57) | 24.4 | 4.13 |
| F9 | c.911C>T | p.Thr304Met | Helicase ATP binding | Disease cause (score 1.000) | High functional impact (score: 4.45) | Protein function affected (score 0.001) | Probably damaging (score 1.000) | Deleterious (score −5.89) | 29.8 | 5.3 |
Abbreviations: CADD, Combined Annotation Dependent Depletion; GERP, Genomic Evolutionary Rate Profiling; PROVEAN, Protein Variation Effect Analyzer; SIFT, Sorting Intolerant From Tolerant.
DHX37 Missense Allelic Variants Identified in 46,XY DSD Patients and Their Frequency in Population Databases
| Families | cDNA Position | AA Changed | Phylogenetic Conservation | State | dbSNP | MAFs in Population Databases | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1000 Genomes | ExAC | gnomAD | ABraOM | ESP6500 | ||||||
| F1, F2, F6, F7, F8 | c.923 G>A | p.Arg308Gln | Highly conserved | Heterozygous | Not available | Absent | Absent | 0.00006677 Non-Finnish European | Absent | Absent |
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | Highly conserved | Heterozygous | Not available | Absent | Absent | Absent | Absent | Absent |
| F5 | c.1784C>T | p.Ser595Phe | Highly conserved | Heterozygous | Not available | Absent South Asian | Absent | Absent | Absent | Absent |
| F9 | c.911C>T | p.Thr304Met | Highly conserved | Heterozygous | Not available | Absent | Absent | Absent | Absent | Absent |
| Families | cDNA Position | AA Changed | Phylogenetic Conservation | State | dbSNP | MAFs in Population Databases | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1000 Genomes | ExAC | gnomAD | ABraOM | ESP6500 | ||||||
| F1, F2, F6, F7, F8 | c.923 G>A | p.Arg308Gln | Highly conserved | Heterozygous | Not available | Absent | Absent | 0.00006677 Non-Finnish European | Absent | Absent |
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | Highly conserved | Heterozygous | Not available | Absent | Absent | Absent | Absent | Absent |
| F5 | c.1784C>T | p.Ser595Phe | Highly conserved | Heterozygous | Not available | Absent South Asian | Absent | Absent | Absent | Absent |
| F9 | c.911C>T | p.Thr304Met | Highly conserved | Heterozygous | Not available | Absent | Absent | Absent | Absent | Absent |
Abbreviations: dbSNP, Single Nucleotide Polymorphism Database; ESP6500, Exome Sequencing Project; ExAC, Exome Aggregation Consortium; MAFs, minor allele frequencies.
DHX37 Missense Allelic Variants Identified in 46,XY DSD Patients and Their Frequency in Population Databases
| Families | cDNA Position | AA Changed | Phylogenetic Conservation | State | dbSNP | MAFs in Population Databases | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1000 Genomes | ExAC | gnomAD | ABraOM | ESP6500 | ||||||
| F1, F2, F6, F7, F8 | c.923 G>A | p.Arg308Gln | Highly conserved | Heterozygous | Not available | Absent | Absent | 0.00006677 Non-Finnish European | Absent | Absent |
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | Highly conserved | Heterozygous | Not available | Absent | Absent | Absent | Absent | Absent |
| F5 | c.1784C>T | p.Ser595Phe | Highly conserved | Heterozygous | Not available | Absent South Asian | Absent | Absent | Absent | Absent |
| F9 | c.911C>T | p.Thr304Met | Highly conserved | Heterozygous | Not available | Absent | Absent | Absent | Absent | Absent |
| Families | cDNA Position | AA Changed | Phylogenetic Conservation | State | dbSNP | MAFs in Population Databases | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1000 Genomes | ExAC | gnomAD | ABraOM | ESP6500 | ||||||
| F1, F2, F6, F7, F8 | c.923 G>A | p.Arg308Gln | Highly conserved | Heterozygous | Not available | Absent | Absent | 0.00006677 Non-Finnish European | Absent | Absent |
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | Highly conserved | Heterozygous | Not available | Absent | Absent | Absent | Absent | Absent |
| F5 | c.1784C>T | p.Ser595Phe | Highly conserved | Heterozygous | Not available | Absent South Asian | Absent | Absent | Absent | Absent |
| F9 | c.911C>T | p.Thr304Met | Highly conserved | Heterozygous | Not available | Absent | Absent | Absent | Absent | Absent |
Abbreviations: dbSNP, Single Nucleotide Polymorphism Database; ESP6500, Exome Sequencing Project; ExAC, Exome Aggregation Consortium; MAFs, minor allele frequencies.
Pathogenicity Classification of DHX37 Variants According to the American College of Medical Genetics and Genomics Guidelines
| Families | Nucleotide Changed | AA Changed | Population Data | Computational and Prediction Data | De novo Data | Other Data | Classification |
|---|---|---|---|---|---|---|---|
| F1, F2, F6, F7, F8 | c.923G>A | p.Arg308Gln | PM2a | PP2b | PS2c | PM1d | Pathogenic |
| PP3e | |||||||
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic | ||||||
| F5 | c.1784C>T | p.Ser595Phe | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic | ||||||
| F9 | c.911C>T | p.Thr304Met | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic |
| Families | Nucleotide Changed | AA Changed | Population Data | Computational and Prediction Data | De novo Data | Other Data | Classification |
|---|---|---|---|---|---|---|---|
| F1, F2, F6, F7, F8 | c.923G>A | p.Arg308Gln | PM2a | PP2b | PS2c | PM1d | Pathogenic |
| PP3e | |||||||
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic | ||||||
| F5 | c.1784C>T | p.Ser595Phe | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic | ||||||
| F9 | c.911C>T | p.Thr304Met | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic |
Abbreviations: PM1, pathogenic moderate; PM2, moderate piece of evidence for pathogenicity; PP2, supporting evidence using phenotype; PP3, supporting evidence for pathogenicity by computational (in silico) data; PS2, strong support for pathogenicity when the variants are de novo.
Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium.
Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease.
De novo (both maternity and paternity confirmed) in a patient with the disease and no family history.
Located in a mutational hot spot and/or critical and well-established functional domain without benign variation.
Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.).
Pathogenicity Classification of DHX37 Variants According to the American College of Medical Genetics and Genomics Guidelines
| Families | Nucleotide Changed | AA Changed | Population Data | Computational and Prediction Data | De novo Data | Other Data | Classification |
|---|---|---|---|---|---|---|---|
| F1, F2, F6, F7, F8 | c.923G>A | p.Arg308Gln | PM2a | PP2b | PS2c | PM1d | Pathogenic |
| PP3e | |||||||
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic | ||||||
| F5 | c.1784C>T | p.Ser595Phe | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic | ||||||
| F9 | c.911C>T | p.Thr304Met | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic |
| Families | Nucleotide Changed | AA Changed | Population Data | Computational and Prediction Data | De novo Data | Other Data | Classification |
|---|---|---|---|---|---|---|---|
| F1, F2, F6, F7, F8 | c.923G>A | p.Arg308Gln | PM2a | PP2b | PS2c | PM1d | Pathogenic |
| PP3e | |||||||
| F3, F4, F10, F11 | c.2020C>T | p.Arg674Trp | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic | ||||||
| F5 | c.1784C>T | p.Ser595Phe | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic | ||||||
| F9 | c.911C>T | p.Thr304Met | PM2a | PP2b | PM1d | Likely | |
| PP3e | Pathogenic |
Abbreviations: PM1, pathogenic moderate; PM2, moderate piece of evidence for pathogenicity; PP2, supporting evidence using phenotype; PP3, supporting evidence for pathogenicity by computational (in silico) data; PS2, strong support for pathogenicity when the variants are de novo.
Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium.
Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease.
De novo (both maternity and paternity confirmed) in a patient with the disease and no family history.
Located in a mutational hot spot and/or critical and well-established functional domain without benign variation.
Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.).
The p.Arg674Trp (c.2020C>T) variant was identified in the two Chilean brothers, both with ETRS (cases F3:II-1 and F3:II-2, F3), and also in the three Argentinian-affected members (two brothers with ETRS and their uncle with PGD; cases F4:III-1, F4:III-2, and F4:II-4, respectively, F4; Fig. 1 and Table 1). In addition, the p.Arg674Trp variant was identified in another two Brazilian sporadic cases: one patient with ETRS (sporadic case F10:II-1) and the other with PGD (sporadic case F11:II-1; Fig. 1 and Table 2).
The p.Ser595Phe (c.1784C>T) variant was identified in two affected individuals from the same Brazilian family (F5). The proband had PGD, and her nephew had ETRS (F5:II-6 and F5:III-1, respectively; Fig. 1 and Table 1).
The p.Thr304Met (c.911C>T) was identified in a Brazilian female (sporadic case F9:II-5), who had previously undergone bilateral gonadectomy and genitoplasty (Fig. 1 and Table 2).
The p.Arg308Gln variant is classified as pathogenic, and the other three variants (p.Arg674Trp, p.Ser595Phe, and p.Thr304Met) are classified as likely pathogenic, according to the American College of Medical Genetics criteria (Tables 4 and 5).
Segregation analysis of DHX37 variants
Segregation analysis of the DHX37 variants in eight families displayed a sex-limited, autosomal-dominant pattern, maternally inherited in five families (F2, F3, F4, F5, and F11). In F1, the presence of the p.Arg308Gln variant in the asymptomatic father suggests an autosomal-dominant pattern of inheritance with incomplete penetrance (Fig. 1). In two sporadic cases (F6:II-1 and F8:II-1), the confirmed paternity displayed a de novo status of the p.Arg308Gln DHX37 variant.
DHX37 gene and its protein structure
DHX37 is located in the 12q24.31 region. It is a member of the large Asp-Glu-Ala-His family of proteins and encodes an RNA helicase (14). The DHX37 protein (NP_116045) comprises 1157 amino acids (AAs) and four main domains. The conserved motifs of the helicase core region contain the helicase ATP-binding domain (position 262–429) and the helicase superfamily C-terminal domain (position 585–674); the two other domains are the helicase-associated domain (position 768–859) and the oligonucleotide/oligosaccharide-binding-fold domain (position 894–1011; Fig. 2). All of the identified variants are located in the helicase core region (Fig. 2).
DHX37 protein was identified in different testicular cells
DHX37 expression was characterized in testes from newborns, children, and adults using immunohistochemistry. DHX37 was expressed in fibroblasts, endothelial cells, and epithelial cells of epididymis. These cells were used as internal positive controls for immunohistochemistry. We found DHX37 expression in Leydig cell cytoplasm and in germ cells at different stages of maturation. Our analysis indicates that DHX37 expression in spermatogonia is characterized by a regular perinuclear halo pattern in both newborns (five samples) and adults (three samples). This pattern of staining differs from that seen in Leydig cells (granular cytoplasmatic) and during other stages of maturation of germ cells. A progressive condensation of protein around the nucleus was observed, as cells differentiate from spermatocytes to spermatids, generating a localized paranuclear dot-like pattern. There was no staining in spermatozoa. Rare Sertoli cells displayed a weak and focal cytoplasmatic stain (Fig. 3).
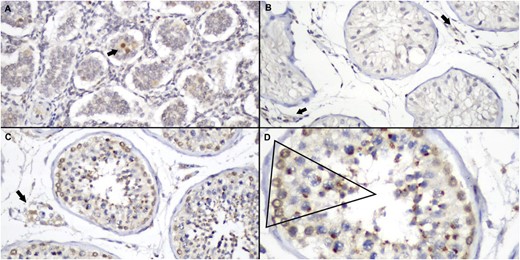
Immunoexpression patterns of DHX37 in testis tissues. (A) Newborn testis showing strongly positive staining in occasional spermatogonia (arrow) among numerous Sertoli cells, some of which show weak cytoplasmic staining (original magnification ×100). (B) Seminiferous tubules of a 13-year-old boy, demonstrating tubules with predominance of Sertoli cells, all of them negative for DHX37. Note some positive stromal cells (arrow; original magnification ×100). (C) Adult testis of a 54-year-old man, showing positive Leydig (arrow) and germ-cell staining. (original magnification ×100) (D) Detail of C, showing the different pattern of stain in different stages of germ cells. Note strong perinuclear halo in spermatogonia, progressive paranuclear condensation in spermatocytes and spermatids, and absence of DHX37 expression in spermatozoa (original magnification, ×20).
Frequency of the DHX37 variants in our 46,XY DSD cohort
The allele frequency of rare and predicted-to-be-deleterious DHX37 variants identified in our cohort of 46,XY DSD patients [11/78 index cases (0.14)] was markedly higher than that observed in individuals from gnomAD [568/141,456 individuals (0.4%; P < 0.001)] and from ABraOM [1/609 individuals (0.2%); P < 0.001)].
Discussion
The current study analyzed a large cohort of 46,XY DSD patients without a molecular diagnosis, most of whom had a GD phenotype, including a large number of familial and sporadic cases with ETRS.
Pathogenic or likely pathogenic allelic variants in the DHX37 were identified in 11 familial cases from 5 unrelated families and in six sporadic cases. Deleterious variants are recurrent in familial and sporadic cases of 46,XY GD in patients of different nationalities.
The DHX37 gene has never been directly associated with gonadal development, but deletions or rearrangements of the 12q24 chromosomal region, which contains DHX37 gene, have been associated with atypical genital development (15). Four syndromic patients with micropenis or perineal hypospadias and/or hypergonadotropic hypogonadism are reported to have deletions or rearrangements involving the 12q24 region (15–17).
The DHX37 gene encodes an RNA helicase protein that is involved in RNA-related processes, including transcription, splicing, ribosome biogenesis (18), translation, and degradation (14, 19). DHX37 is required for maturation of the small ribosomal subunit in human cells through its catalytic activity, required for dissociation of the U3 small nucleolar RNA from preribosomal complexes (20). Disturbance of human ribosome production is associated with cancer and genetic diseases known as ribosomopathies (21).
Disease-causing variants in the DHX30 were previously described in syndromic patients with global developmental delay, intellectual disability, severe speech impairment, and gait abnormalities. Functional studies of allelic variants in DHX30 demonstrated that they affect protein folding or stability interfering with RNA binding (mutations located in motif Ia) or with ATPase activity (mutations located in motif II and VI) (19, 20). Two DHX37 allelic variants found in the current study are located in the same motifs.
Despite lack of experimental evidence to demonstrate formally the deleterious effects of the four variants identified in the current study, they are located in the highly conserved helicase core region of the DHX37 protein.
The spontaneous p.Leu489Pro Dhx37 pathogenic variant was identified in Zebrafish in association with behavior escape defects (22). This study demonstrated that Dhx37 is involved in pre-mRNA splicing, reinforcing the role of Dhx37 in RNA-related processes.
Although there is no direct evidence of DHX37 being involved in mRNA processing during gonadal development, DEAD-box and DEAH-box RNA helicase genes are differentially expressed between males and females during the critical period of male sex differentiation in channel catfish (23).
Furthermore, we show population evidence that the DHX37 variants are enriched among the 46,XY GD patients compared with the population database. The statistical analysis confirmed that the predicted deleterious DHX37 variants located in the helicase core region are more frequently identified in our 46,XY DSD cohort than in the public databases, emphasizing that this finding was not by chance (P < 0.01).
Therefore, in vitro and in vivo studies on the DHX37 mechanism have demonstrated a role of DHX37 in ribosome biogenesis (23). Based on this knowledge, 46,XY GD could be classified as a ribosomopathy, expanding the etiological mechanisms of the dysgenetic 46,XY DSD spectrum.
Since the discovery of the sex-determining region Y variants in patients with GD in 1990 (24), several genes have been associated with the molecular etiology of this disorder. The nuclear receptor subfamily 5 group A member 1 (NR5A1) and MAPK kinase kinase 1 variants are the most frequent causes of 46,XY GD identified to date (25–28).
In this study, we found pathogenic/likely pathogenic variants in DHX37 in patients with 46,XY GD at a frequency of 14%, which is slightly higher than the frequency of NR5A1 defects (11%) in our whole cohort (26, 29). With the consideration of only the ETRS phenotype (micropenis and absence of uni- or bilateral testicular tissue), this frequency increases to 50% (7/14 families).
In the literature, different inheritance patterns have already been described in 46,XY GD kindreds (30), including the description of asymptomatic male carriers of proven pathogenic variants of genes involved in testicular determination, such as sex-determining region Y and NR5A1 genes (31, 32). Uncertain mechanisms might prevent the appearance of the phenotype in asymptomatic 46,XY carriers.
Maternal inheritance was observed in all familial cases with pathogenic/likely pathogenic variants in DHX37, with the exception of F1, where the variant was inherited from a seemingly unaffected father carrier.
In adult humans, the DHX37 protein is expressed in the ovarian stroma and in the cells within seminiferous tubules (Human Protein Atlas database) (33–35). In our study, the immunohistochemistry analysis of normal testicular tissue from newborn, pubertal, and adult males revealed that DHX37 is expressed during specific stages of germ-cell maturation, in Leydig cells, and rarely in Sertoli cells.
An elaborate paracrine cell–cell network, transporting signaling molecules between germ cells and Sertoli cells, has been described (36). Indeed, in vitro studies have shown that there is a bidirectional trafficking between Sertoli and germ cells and that each cell type regulates the function of the other (37–40). In addition, RNA expression profiles of DHX37 in human testicular cancer cells are higher than in other tissues (The Human Protein Atlas–Pathology), suggesting that DHX37 may be involved in the regulatory process of the cell proliferation in the testis (33–35).
The current study provides several lines of genetic evidence to indicate that defects in DHX37 are associated with the 46,XY GD spectrum, mainly with ETRS. First, we observed that the variants segregate with the DSD phenotype in a dominant inheritance pattern in most of the families and that two de novo variants were identified. Second, we provide statistical evidence that rare DHX37 variants are enriched in the analyzed 46,XY DSD cohort compared with public databases involving a large number of individuals not selected by this phenotype.
In conclusion, our findings indicate that DHX37 is a player in the complex cascade of male gonadal differentiation and maintenance, thus establishing a frequent molecular etiology for the 46,XY GD spectrum, which includes a high proportion of individuals with ETRS.
Acknowledgments
The authors are very grateful to Dr. Frederico Moraes Ferreira for his technical assistance with the in silico prediction analysis, Drs. Beverly M. Yashar and John Park for medical care of patient F6:II-1, and Dr. Ivo Jorge Prado Arnhold for suggestions and significant review of the manuscript.
Financial Support: This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; Grant 305743/2011-2), Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; Grants 05/04726-0, 07/512156, 10/51102-0, 2013/02162-8, and 2014/50137-5), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES/PNPD; institutional fellowship grant).
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability: All data generated or analyzed during this study are included in this published article or in the data repositories listed in References.
Abbreviations:
- AA
amino acid
- ABraOM
Arquivo Brasileiro Online de Mutações
- DHX
Asp-Glu-Ala-His-box
- DSD
differences/disorders of sex development
- ETRS
embryonic testicular regression syndrome
- F
family
- GD
gonadal dysgenesis
- gnomAD
Genome Aggregation Database
- NR5A1
nuclear receptor subfamily 5 group A member 1
- PGD
partial gonadal dysgenesis
- WES
whole-exome sequencing
References and Notes
Author notes
T.E.d.S. and N.L.G. (first authors) and B.B.M. and S.D. (last authors) contributed equally to this study.

{kind=link}
{kind=link}
{kind=link}