





Abstract
Craniopharyngiomas, which are categorized as adamantinomatous (ACPs) or papillary (PCPs), have traditionally been treated with surgery and/or radiotherapy, although when the tumors progress or recur, therapeutic possibilities are very limited. Following recent advances in their molecular pathogenesis, new medical therapeutic options have emerged.
The search strategy that we selected to identify the appropriate evidence involved the following medical subject headings (MeSH) terms: (“Craniopharyngioma” [MeSH] AND “Craniopharyngioma/drug therapy” [MeSH]) NOT (“review” [Publication Type] OR “review literature as topic” [MeSH Terms] OR “review” [All Fields]) AND (“2009/05/01” [PDat]: “2019/04/28” [PDat]).
Mutations of β-catenin causing Wnt activation with alterations of the MEK/ERK pathway are encountered in the great majority of patients with ACPs; specific alterations also stratify patients to a more aggressive behavior. In most PCPs there is primary activation of the Ras/Raf/MEK/ERK pathway secondary to BRAF-V600E mutations. BRAF inhibitors, such as dabrafenib or vemurafenib, either alone or in combination with the MEK inhibitors trametinib and cobimetinib, have been administered to patients with PCPs producing clinically useful and, in some cases, sustained responses. In contrast to PCPs, drugs targeting β-catenin and its downstream MAPK pathway in ACPs have so far only been used in in vitro studies, but there appear to be promising new targets clinically.
The identification of specific genetic alterations in patients with craniopharyngiomas has expanded the therapeutic options, providing evidence for a customized approach using newer molecular agents. More studies including a larger number of carefully selected patients are required to evaluate the response to currently available and evolving agents alone and in combination.
The title of this review may sound like an oxymoron, as until very recently the mainstays of treatment of craniopharyngiomas (CPs) have been surgery and radiotherapy (RT). However, during the last few years there have been major developments in our understanding of the molecular pathology of these tumors that could set the scene for specific targeted therapy.
CPs develop as malformations of embryonic remnants along the original pathway of the craniopharyngeal duct. They are characterized as mostly benign epithelial tumors according to the World Health Organization Classification of Tumors of the Central Nervous System, namely, World Health Organization grade I tumors (1–3). They are relatively rare, with an incidence of 0.5 to 2.5 per 106 cases per year (2, 4–7), and are divided into two distinct subtypes, adamantinomatous CPs (ACPs) and papillary CPs (PCPs), differing in both histological features and genetic alterations (8). ACP is more prevalent and displays a bimodal age distribution, with peaks between the ages 5 to 15 years and 45 to 60 years, but it can occur at any age, even during the neonatal period (3, 9, 10); PCP has been classically considered as an adult entity peaking between 40 and 55 years of age (2, 4, 7, 11), although pediatric cases have occasionally been reported (12).
Harvey Cushing not only suggested the term “craniopharyngioma” but also recognized the substantial challenges in their management (13). Indeed, their anatomic vicinity to vital structures (hypothalamus, optic pathways, brain) and their intrinsic local infiltrative tendency render their growth pattern unpredictable and complete safe surgical removal problematic (14). Furthermore, malignant transformation may also rarely be seen and, overall, the long-term morbidities and mortality of patients with CPs are disappointedly high, particularly compared with patients with treated pituitary neuroendocrine tumors (15, 16).
Current therapeutic options are relatively limited: surgery, either with the intention to treat by gross total resection (GTR) or with the intention to reduce their mass by subtotal/partial resection (STR) (17, 18), with or without postoperative RT (19), is the primary therapeutic modality (3). In a systematic review, STR with adjuvant RT, and GTR, showed similar rates of long-term tumor control (20). Currently, STR followed by RT is recommended, unless there is a clear margin between the tumor and surrounding vital structures, allowing complete safe tumor removal. Whenever feasible, the tumors are approached by the transsphenoidal route (17, 18). The intracystic administration of sclerosing substances, mainly bleomycin and interferon (IFN)-α, has been suggested in the past to attenuate fluid formation and diminish cyst size (21–23). Bleomycin use is limited by its toxicity (21, 22), whereas IFN-α has shown some benefit with an acceptable safety profile (24); however, no prospective or randomized study has been performed, and there are isolated reports of relatively poor responses and major adverse effects following IFN-α instillation, limiting its use (21).
Recently, progress in molecular biology has unraveled novel potentially druggable molecular pathways in CPs that offer new promising therapeutic options. The use of such drugs is especially important to minimize the otherwise adverse effects associated with surgery and RT, particularly in recurrent tumors where there are few or no alternative options. The purpose of this mini-review is to present the available literature on clinical studies and case reports describing targeted treatments that are based on the recently identified molecular alterations in both ACPs and PCPs. The search strategy that we selected to identify the appropriate evidence involved the following MeSH terms: (“Craniopharyngioma” [MeSH] AND “Craniopharyngioma/drug therapy” [MeSH]) NOT (“review” [Publication Type] OR “review literature as topic” [MeSH Terms] OR “review” [All Fields]) AND (“2009/05/01” [PDat]: “2019/04/28” [PDat]) (Fig. 1).
![The MeSH terms used in the search strategy: (“Craniopharyngioma” [MeSH] AND “Craniopharyngioma/drug therapy” [MeSH]) NOT (“review” [Publication Type] OR “review literature as topic” [MeSH Terms] OR “review” [All Fields]) AND (“2009/05/01” [PDat]: “2019/04/28” [PDat]).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jcem/104/12/10.1210_jc.2019-01299/1/m_jc.2019-01299f1.jpeg?Expires=1747964157&Signature=gjKVXooroul1oZBWY7DFV9fmCwDuES3VM3rgHZ9xvGOloPX-TCNDCdqDMk9Vlsj18cuRxfTAE8ZJc7-bcdSJQ9Mm5ekuj~QqHDGFji85RDTdglfQxIsO9Xe-uO~3Dk-qCaF~QfRX7wrPsNulkE4K0Bz6FQRZALiffH6vk4Y-imioUSKn6fPVeic5Jhaz~su5gXQdsIrKp9xUinQF2My4tJG0hIrlzq2VWpkCVY~cfdse~yGIzJOgZoPxyspAdo5oTNS6vYn-lhcwuljaXfCb7txGKP1uUb4y2wqsoYYsVaLi8UV0uD1vH9ea7580KAfJ8ylzM9CHGzAQx~4BYsV5TA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The MeSH terms used in the search strategy: (“Craniopharyngioma” [MeSH] AND “Craniopharyngioma/drug therapy” [MeSH]) NOT (“review” [Publication Type] OR “review literature as topic” [MeSH Terms] OR “review” [All Fields]) AND (“2009/05/01” [PDat]: “2019/04/28” [PDat]).
Molecular Biological Advances in ACPs and PCPs
It has recently been shown that each CP histological subtype (Figs. 2 and 3) is characterized by alterations in oncogenic molecular signaling pathways, harboring distinct mutational, transcriptomic, and epigenomic profiles as a result of different gene mutations, gene expression, and methylation patterns (25–28). These findings have shed light on the identification of tumor-specific signaling pathway activation that may lead to CP-targeted therapies. An additional role of the different molecular profiles may be to differentiate pathologically indeterminate cases of CPs (28, 29).
![Photomicrograph of a representative case of a PCP. Hematoxylin and eosin staining; original magnification, ×100 magnification. [Dr. A. Ramanathan and Prof. A. B. Grossman, unpublished data.]](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jcem/104/12/10.1210_jc.2019-01299/1/m_jc.2019-01299f2.jpeg?Expires=1747964157&Signature=H35t6hCMbU1Mb5QuJ77grL6tCY3s7N38qXYHhyNx3TH93pfQ~Ml1GE5a2KsbAqx4PBtNlz9SQJ8LBvLMz9WiZZJA-b9aQ1jTKGEkKTk4dP1x36L7eUzAc-f8oY7~uxv0bqZg3vsko0XHO1r1deCsHefr~syfNxYbcHNyTh4hQ4-2UUtA-ySKZrWoMxZP1IooqgCmCiMUtUcUGxEq9It-JucOOb0a1sGq4TRnV9wO5V7y73uhquqSaf4XZ6JxO5gGW-tKm9-TpUa2k9sQg1HEk-6n8qShOIg~OzcFxPCoxLY4z7bPaxOYYoYrCA~cYUbjcAIKHlizQ9ciJuqprMw2tg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Photomicrograph of a representative case of a PCP. Hematoxylin and eosin staining; original magnification, ×100 magnification. [Dr. A. Ramanathan and Prof. A. B. Grossman, unpublished data.]
![(A) Histology of ACP. Basal epithelium demonstrating the aberrant nuclear accumulation of β-catenin in nodular whorls (arrowheads) due to β-catenin mutation. Anti–β-catenin stain; original magnification, ×400. (B) Histology of PCP. Squamous epithelium showing membranous immunoreactivity of β-catenin, lacking clusters with aberrant nuclear accumulation. Original magnification, ×400. [Reproduced from Lithgow K, Pohl U, Karavitaki N. Craniopharyngiomas. In: Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-2019 Mar 14.]](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jcem/104/12/10.1210_jc.2019-01299/1/m_jc.2019-01299f3.jpeg?Expires=1747964157&Signature=ZfjU5kphIohyzhxIj6Vcft5tmOJBIA54AzECl4NULi61bJGaII7sX35kgJSJmJXFnM0pqDOO~~JoTyQHTlEMNTst4tXbU3Z~DDyW2t1yyzAjk6CCzdjdQUb1PClbdhweO2NudfYbHSGRLJ894uwUDRdH2OWBREIgz5rL4R7b0u92M41jQFdTbaSQcOQiEs4UtWLMSNhYAhENSPvlU56xRacRVnm76nydj3loMlOEydKVG52wi615~Z763JXGAwv1GJjE2bRFlBmwZaQZ1m9t3DWuOIJ8FcLXFQxBbvwFskCtlrJ9jecdqv-gQw4hjmf7h-ZYhdn3cLpn0PX9-u2upA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(A) Histology of ACP. Basal epithelium demonstrating the aberrant nuclear accumulation of β-catenin in nodular whorls (arrowheads) due to β-catenin mutation. Anti–β-catenin stain; original magnification, ×400. (B) Histology of PCP. Squamous epithelium showing membranous immunoreactivity of β-catenin, lacking clusters with aberrant nuclear accumulation. Original magnification, ×400. [Reproduced from Lithgow K, Pohl U, Karavitaki N. Craniopharyngiomas. In: Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-2019 Mar 14.]
Sekine et al. (30) first described activating mutations of the β-catenin encoding gene CTNNB1 in ACPs as early as 2002. Whole-exome sequencing revealed mutations in CTNNB1 in most of a small group of ACPs (11/12, 92%) (25). Additionally, targeted genotyping revealed CTNNB1 mutations in 96% of ACPs (51/53) (25). Next-generation panel sequencing did not identify any mutations other than those in CTNNB1 (27). In 76% of the ACPs, a mutation in exon 3 of CTNNB1 was found, and there was a trend toward a worse event-free survival in cases mutated at Thr41 (27). When primary and recurrent ACP tumor samples from the same patient were tested, the same CTNNB1 mutation (S45P) was found without additional ones (28). No substantial large chromosomal aberrations have been found, although a fraction of ACPs showed recurrent focal gains of chromosomal material, whereas in other cases there was loss of chromosomal region Xq28 (27).
These CTNNB1 mutations are exclusively found in exon 3, which encodes the destruction/degradation complex of β-catenin: β-catenin is held in a multiprotein complex that can be phosphorylated at specific residues, and is then very quickly degraded. If such phosphorylation sites are mutated then there is aberrant nucleocytoplasmic accumulation of β-catenin (30) (Fig. 3A). This can also be demonstrated by immunocytochemistry (IHC), which is highly reliable in differentiating ACPs from other (para)sellar lesions (31, 32). β-Catenin is a dominant mediator of the classic Wnt molecular signaling pathway and is also part of the adherens complex (8). Normally, there are only low levels of β-catenin in the cytoplasm, as when phosphorylated it is rapidly degraded in a multiprotein destruction–regulator complex, facilitating its ubiquitination and degradation by the proteasome. Thus, absent Wnt ligands, such signaling is inactive, the default or “off” state (8, 33–35) (Fig. 4A). Other proteins held in this regulatory complex include the scaffold proteins Axin and Axin2, the tumor suppressor protein adenomatosis polyposis coli (APC), and the phosphokinases glycogen synthase kinase-3β (GSK-3β), casein kinase 1α (CK1), protein phosphatase 2A (PP2A) or, alternatively, by IκB kinase α (IKK-α) (36). The kinases phosphorylate APC, which in turn recruits β-catenin to the complex (34, 36); in this way the complex is marked to be degraded via phosphorylation of serine (Ser33, Ser37, Ser45) or threonine (Thr41) residues (of exon 3 on the CTNNB1) (34, 36–39). Additionally, recognition of such phosphorylation by multiple ubiquitin molecules, such as a component of E3 ubiquitin ligase, β-TrCP, allows ubiquitination of β-catenin and dissociation of α-catenin from β-catenin, resulting in loss of cadherin adhesion (34, 35, 37–41). In an alternative scenario, exogenous Wnt signaling ligands “switch on” (i.e., activate) Wnt signaling (41, 42), The Wnt protein family comprises ∼20 different proteins that bind to the Frizzled family of receptors. When activated, the Wnt pathway (34) leads to an intracellular signaling cascade that includes dimerization of Frizzled with LRP5/6 (34, 43), which in turn binds to Axin, GSK-3β, and CK1. These latter mediators are then sequestered at the cell membrane by Dishevelled (Dvl) distant to the β-catenin destruction complex, diminishing phosphorylation of β-catenin and thus stabilizing it by rendering it refractory to degradation (34–36, 43–45). The accumulation of β-catenin allows for its translocation to the nucleus, possibly mediated by its Armadillo-like sequences, and then leads to the transcription of target genes, including CyclinD1, c-Myc, CD44, Survivin, vascular endothelial growth factor (VEFG), and fascin (26, 46–49). These various factors acting alone or in combination stimulate cellular proliferation and other Wnt-regulated processes (50). Importantly, note that it is the nuclear accumulation of β-catenin that eventuates in Wnt-signaling target gene expression. It has been shown that the target genes Axin2, bone morphogenetic protein (BMP)4, and Fascin-1, stem cell markers (CD133 and CD44), and the cell cycle inhibitor p21Waf1/CIP1 are specifically concentrated in cells with nuclear β-catenin (8). One particular target of special interest is Fascin (48), which cross-links actin and is involved in filopodia formation, and thus mediates tumor cell migration and related tumoral invasion (36); β-catenin binds fascin by its armadillo-repeat sequence (as noted above, this is also involved in nuclear pore complex translocation of β-catenin) (51), leading to the relocation of β-catenin changing cell adhesion and decreasing β-catenin degradation (36). As a consequence, loss of either fascin or β-catenin expression has been shown to attenuate the ability of ACP tumor cells to migrate when cultured (36). The armadillo-repeat sequences are clearly significant in that they also bind to the TCP family transcription factors, Axin2, and APC (52). Furthermore, other growth factors pathways are also involved: β-catenin cell clusters demonstrate activation of epidermal growth factor (EGF) receptor (EGFR), or src-driven phosphorylation at the tyrosine of codon 654 (Y654) and sonic hedgehog (SHH) signaling pathways (8, 26, 28, 48, 52–55). Such activation reduces the binding affinity of β-catenin to E-cadherin such that the adherens complex is destabilized (40, 56) and may additionally aid in tumor stem cell maintenance (57–59). Additionally, Fyn, Fer, or c-Met promote phosphorylation of β-catenin at tyrosine residue 142, which also facilitates the loosened binding of α-catenin to β-catenin (Fig. 4A). RNA profiling (60) has confirmed that human ACP clusters express high levels of the fibroblast growth factor (FGF), BMP, and Wnt families (50); these are growth-related signaling mediators that have been shown to be involved in a variety of pathophysiological and embryological situations. These molecules may be useful in terms of predicting tumor recurrence: FGF-2 is only seen in recurrent ACPs, whereas platelet-derived growth factor (PDGF) receptor-α expression is significantly higher in recurrent ACP compared with nonrecurrent tumors. In contrast, VEGF and fibronectin are expressed in both ACP and PCP (61). BMP2 and BMP4 are downstream of the Wnt/β-catenin pathway and are present at high expression levels in ACP (52, 53, 62). Of particular importance from the therapeutic perspective, as discussed below, is that EGFR is a member of the human EGFR (HER) family, and its most significant signal transduction pathways are those involving the Ras-Raf MAPK (MAPK/ERK pathway) and the phosphatidylinositol 3-kinase (PI3K)/Akt pathways (8) (Fig. 4B).
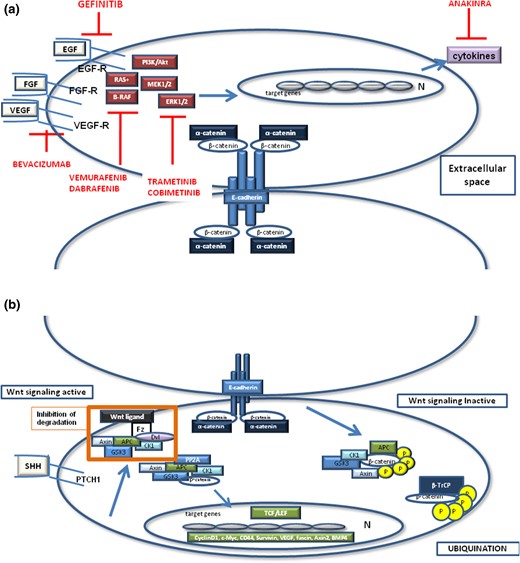
(a) Schematic presentation of adherens junction complex (including β- and α-catenin and E-cadherin) and the molecular signaling pathways that may crosstalk in both ACPs or PCPs. In ACPs, β-catenin cluster cells show an activation of the Wnt, SHH, and EGFR pathway, which are reported to crosstalk with each other. When Wnt signaling is inactive, axin recruits and binds to GSK-3β, CK1α, and PP2A. APC is phosphorylated by these kinases, which recruit β-catenin to the complex and tag it for degradation through phosphorylation of the serine (S33, S37, S45) or threonine (T41) residues of exon 3 on the CTNNB1 gene. This phosphorylation pattern is recognized by β-TrCP, which ubiquitinates β-catenin, When Wnt signaling is active, by the activation of a Wnt ligand or when extracellular SHH binds to the transmembrane receptor PTCH1, the protein degradation complex is inhibited and β-catenin is accumulated first in the cytoplasm and then in the nucleus. There, β-catenin represents a transcription cofactor by interacting with transcription factors of the0020TCF family TCF/LEF1, initiating expression of the target genes. (b) In ACPs, paracrine growth-related signaling molecules such as SOX2. FGFs, EGF, and PDGF bind to their receptors, activating the Ras/Raf/MAPK/ERK1/2 pathway and the PI3K/Akt pathway. Similarly, in PCPs the BRAF-V600E serine/threonine kinase is a critical component of the Ras-Raf-Mek-ERK1/2 pathway that has been mutated. Red lines show the inhibitors that may be used as molecular-targeted therapy for CPs. BMP, bone morphogenetic protein; Dvl, Dishevelled; FGF, fibroblast growth factor; Fz, Frizzled; IKK-α, IκB kinase α; LEF1, lymphocyte enhancer factor 1; PI3K, phosphatidylinositol 3-kinase; PP2A, protein phosphatase 2A; PTCH1, patched 1; TCF, T-cell factor; VEGF-R, vascular endothelial growth factor receptor.
Of the pathways disrupted in human cancers, Ras-Raf-Mek-ERK1/2 is one of the most important (63, 64). Extracellular growth factors (FGFs, EGF, PDGF) bind to and activate receptor tyrosine kinases, causing a downstream phosphorylation cascade that eventually leads to transcription of target genes regulating cellular proliferation, differentiation, apoptosis, and senescence (63). Downstream activation of the MAPK/ERK pathway, as evidenced by phosphorylation of ERK1/2, was identified by IHC around the clusters with particularly prominent staining at the forefront, known as the leading edge, of the tumor (50). Phospholipase Cγ (PLCγ), the signal transducer and activator of transcription (STAT), and src/FAK cascades are also stimulated (8, 65, 66). SHH signaling pathway gene products, such as Gli2 and patched (Ptch)1, are overexpressed in ACP, and their cleaved active forms are especially highly expressed (8, 26, 28, 53). Upregulation of certain matrix metallopeptidases (MMPs) has also been reported in ACP transcriptome studies (23, 50). A variable degree of colocalization between Ki-67 and pERK1/2 expression has also been also shown (60). In ACP, whole-exome sequencing showed additional mutations in genes listed in the Cancer Gene Census (67), although many of these may be “passenger” mutations (25); there is also evidence for recurrent copy number changes in some tumors (27). Thus, mutation-produced loss of phosphorylation sites on β-catenin leads to a plethora of downstream changes initiating and augmenting tumor growth.
Considering PCPs, a BRAF-V600E mutation was documented (25) by targeted genotyping in 95% of PCPs (36/39), whereas whole-exome sequencing revealed recurrent mutations in BRAF-V600E in all PCPs (3/3) (25). In a parallel study, an analysis of known oncogenes revealed that BRAF-V600E mutations were seen in 81% (17/21) of PCPs by targeted Sanger sequencing and in 86% (18/21) of PCPs by IHC (68). Agreement between methods was seen in 95% (20/21) of cases; however, interpretation of anti–BRAF-V600E staining was challenging due to occasional nonspecific reactivity (68). It is well established that the gain-of-function mutation BRAF-V600E, a critical serine/threonine kinase in the Ras-Raf-Mek-ERK1/2 pathway, renders it a potent oncogene, leading to increased cell proliferation and survival, resulting in cell transformation and tumorigenesis (69, 70) (Fig. 4B). Such mutations render the BRAF kinase constitutively active. The MAPK pathway also plays a role in controlling stem-cell specification during development and perhaps during stem-cell homeostasis in the postnatal period (70), modulating the balance between the proliferation and differentiation of Sox2+ cells, implying that persistent proliferative capacity of Sox2+ cells may underlie the pathogenesis of PCP (70). Sox2+ progenitor cells appear to show tumor-inducing potential, driving tumor formation in a paracrine manner by inducing tumorigenic events in adjacent cells but without gains or losses of genetic material (53, 54, 71). In PCPs (25), whole-exome sequencing with a recently described novel technique—MuTect—also showed isolated mutations of cancer-related genes encoding chromatin remodeling factors (CHD5, CHD6) and cell adhesion molecules (CDH26, PTPRT), and one (KIAA1549) that is fused to BRAF in most cases of pilocytic astrocytoma (72, 73). However, it is presently unclear whether these mutations are pathogenetic or merely passenger mutations (25).
CTNNB1 and BRAF mutations were mutually exclusive and clonal in ACP and PCP, according to an original observation by Brastianos et al. (25), and there were no other recurrent mutations or genomic aberrations in either subtype (Fig. 3A and 3B). Thus, the specific oncogenic change appears to be Wnt activation in ACP, as well as activation of the Ras/Raf/MEK/ERK pathway by BRAF-V600E mutations in PCP (27). It was then suggested that one should classify CPs as ACP CTNNB1 mutated, ACP CTNNB1 wild type, PCP BRAF-V600E mutated, and PCP BRAF wild type (74). Comparison microarray analysis of 18 ACP and 10 PCP samples revealed significant upregulation of several direct targets of the Wnt/β-catenin signaling pathway in ACPs compared with PCPs, including LEF1 and AXIN, whereas components of the SHH such as GLI2, PTCH1, and SHH were also overexpressed specifically in ACPs in comparison with PCPs (8, 36, 48, 52, 71, 75, 76). Unsupervised consensus clustering of the gene expression values of the 5000 most variable genes resulted in two distinct and stable clusters that perfectly separated ACP and PCP samples. Gene expression data showed increased expression of microtubule-associated protein 2 (MAP2), tenascin C (TNC), and the stem cell marker CD133 (PROM1) in ACPs. CD44 and claudin-1 (CLDN1) were significantly downregulated in ACPs, but showed higher expression in PCPs (28, 52, 75). Moreover, reduction of claudin-1 protein levels point toward an invasive growth pattern in ACPs (75). Downregulation of miRNA miR-132 was identified as a marker of aggressiveness, playing a role in epithelial–mesenchymal transition, in a study of 754 miRNAs from childhood CPs (77). However, in contrast to the view of a complete molecular separation between these two tumor subtypes, Larkin et al. (68), in a small cohort of ACPs, reported a dual aberration of CTNNB1 mutated at Thr41 together with BRAF-V600E mutation in two tumors. This finding was validated by review of morphology and comparison with IHC findings. Sequencing in forward and reverse directions from two DNA samples extracted on different occasions, confirmed these findings. Whether this represents the rare occurrence of a collision tumor needs to be established in future larger studies (68). The expression of additional stem cell markers SOX9, KLF4, and Okt4 was also identified by IHC in both ACPs and PCPs (8, 78).
Other transcriptional molecular analyses of ACPs have revealed the expression of immune system gene expression, including IL-1β, IL-6, IL-8 (CXCL-8), IL-10, IL-18. and TNF-α. In particular, there is a characteristic inflammatory cytokine/chemokine reaction in both ACP cyst fluid and solid tumor components. Cytokines and chemokines with elevated concentrations in ACPs included IL-6, CXCL-1 (GRO), CXCL-8, and the immunosuppressive cytokine IL-10, and their relevant receptors are also expressed in ACPs, as were IL-10 as well as the immunosuppressive factor IDO-1 at both mRNA and protein levels (79). This interaction between tumor cells and chemokines is not dissimilar to that seen in some pituitary neuroendocrine tumors (80).
In summary, in the era of molecular-targeted treatments, the molecular pathways that characterize either ACPs or PCPs are under vigorous investigation. Currently, ACPs are characterized and identified by activating mutations of the β-catenin–encoding gene CTNNB1, whereas PCPs are characterized by the activating BRAF-V600E mutation. In both cases, there is upregulation of the MAPK pathway, which is critically involved in cell proliferation.
Targeting Molecular Aberrations in CPs
Currently, there are no approved targeted or cytotoxic therapies available for the systemic treatment of CPs. The development of molecular-targeted treatments in oncology has opened new horizons in the pharmacological treatment of tumors caused by genetic alterations. Recently, in cancers harboring BRAF-V600E alterations, V600E mutation–specific BRAF inhibitors, such as dabrafenib or vemurafenib (81, 82), have been shown to be effective antitumor agents. Additionally, the MEK inhibitors trametinib and cobimetinib have been administered in combination with BRAF inhibitors, because these compounds override any resistance to BRAF inhibition (81, 83). These data have led to the use of BRAF inhibitors in patients with aggressive PCPs (see Table 1 for summary). Aylwin et al. (83) described the beneficial effect of the off-label administration of vemurafenib (960 mg twice daily) in a 41-year-old female with a 16-year history of a recurrent PCP (carrying a BRAF-V600E mutation) with progressive visual failure following three transsphenoidal operations. MRI 2 weeks after starting treatment showed marked reduction in the size of the tumor with resolution of the surrounding edema, and 3 months later there was evidence of radiological improvement. The CP recurred 6 weeks later and vemuranefib was restarted with reduction of tumor size followed by stabilization for 7 months, but this was followed by further growth. In the same year, Brastianos et al. (84) reported on the use of dabrafenib (150 mg twice daily) in a 39-year-old male with a PCP harboring a BRAF-V600E mutation. This patient had undergone four craniotomies along with cyst decompression during a period of 11 months: as early as 4 days after treatment, the tumor showed a 23% size reduction, with the cyst volume having decreased by 32%. The beneficial effect was even more obvious 17 days after treatment, achieving a 52% reduction in tumor size and 70% decrease in the cyst volume. The MEK inhibitor trametinib (2 mg, orally, twice daily) was added to the therapeutic scheme on day 21 aiming to enhance the effect. Fourteen days after this combined therapy, there was an 85% reduction in tumor size and 81% decrease in the cyst volume. On day 38 of treatment, the patient underwent endoscopic transsphenoidal surgery: 2 weeks later the combined treatment was discontinued, and a week later RT was offered (50.4 Gy in 28 fractions). Seven months later, the patient has remained free of symptomatic recurrence. In the following year, Roque and Odia (29) described a 47-year-old female with acute visual loss and a 4-month history of amenorrhea, cold intolerance, and headache. A 2.7-cm cystic lesion was identified in the suprasellar area harboring a BRAF-V600E mutation (29). Subsequently, she underwent right frontal craniotomy, but the tumor regrew, causing bilateral visual impairment necessitating an Ommaya catheter placement into the cyst and then RT (54 Gy in 30 fractions). Seven months later, the tumor regrew and dabrafenib (150 mg twice daily) and trametinib (2 mg daily) were administered. After 2 months, a 52% reduction in tumor volume was documented, and after 5 months, the tumor size was reduced by 75%. The patient was continuing to improve on this treatment both radiologically and clinically 7 months posttreatment. Subsequently, Rostami et al. (85) reported a 65-year-old male with visual deficits who underwent a partial transsphenoidal resection of a PCP harboring a BRAF-V600E mutation. After rapid tumor growth, dabrafenib (150 mg twice daily) was initiated, followed by trametinib (2 mg daily) 3 weeks later for a total of 7 weeks. An 11% tumor reduction was seen at 4 weeks and 91% by the 8th week.
Case Reports and Current Clinical Trials of Patients With PCPs and BRAF-V600E Alterations Treated With V600E Mutation-Specific BRAF Inhibitors (Dabrafenib or Vemurafenib) With or Without Combined Treatment With MEK Inhibitors (Trametinib or Cobimetinib)
| Case Series/Reports | CP | Previous Treatment | Histopathological And Molecular Genetic Analysis | Treatment | Duration Of Treatment | Outcome (Mean Of Measurement) | Comments |
|---|---|---|---|---|---|---|---|
| Aylwin et al., 2015 (83) | PCP | TSS ×2, RT | Pyrosequencing analysis indicated the presence of BRAF mutation c.1799T>A (p.Val600Glu). | Vemurafenib 960 mg bd | 3 mo | Near complete resolution (MRI) | Recurrence 6 wk off treatment; restart of treatment and stabilization for 7 mo with a regrowth |
| Brastianos et al., 2015 (84) | PCP | Craniotomy ×3 | Uniform staining of the BRAFV600E protein (VE1 antibody) by IHC; V600E mutation confirmed by allele-specific genetic testing. Mutation detectable in peripheral blood during treatment. Absence of nuclear staining for β-catenin by IHC. | Dabrafenib 150 mg bd + after 3 wk trametinib 2 mg od | 35 d (followed by other treatments) | 85% reduction in tumor volume and 81% reduction in tumor-associated | |
| cyst (MRI) | |||||||
| Roque et al., 2016 (29) | PCP | Craniotomy, Ommaya catheter with percutaneous aspiration of cyst fluid, RT 54 Gy in 30 fractions | Positive staining for BRAFV600E by IHC; next-generation sequencing confirmed BRAFV600E mutation and FGFR3 splice site (437_445 + 3del12) with two variants of unknown significance (BRCA2-G1771D and FGFR4-S551F). | Dabrafenib 150 mg orally bd and trametinib 2 mg orally od | 7 mo | Almost disappearance of 2.6 × 2.3 × 3.2-cm tumor (MRI); reduced volume and intensity of FDG uptake on PET | |
| Rostami et al., 2017 (85) | PCP | TSS | Weak staining of mutated BRAFV600E (VE1 antibody) by IHC; BRAFV600E genotype confirmed by pyrosequencing mutational analysis. | Dabrafenib 150 mg bd plus after 3 wk trametinib 2 mg od | 7 wk | 91% reduction tumor size (MRI) | Combined BRAF and MEK-targeted therapy |
| Himes et al., 2019 (86) | PCP | Craniotomy, RT 36 Gy in 12 fractions | BRAF V600E mutation confirmed in specimens from original resection. | Dabrafenib 150 mg bd (shortly) → od (several weeks) → 225 mg od | 1 y under treatment and follow-up for 1 y off treatment. | Minimal residual tumor | |
| Juratli et al., 2019 (87) | PCP | No treatment | BRAF V600E confirmed by biopsy | Dabrafenib 150 mg bd plus trametinib 2 mg od | 6 mo | >80% reduction of tumor size (MRI) | Characterized as neoadjuvant therapy |
| NCT03224767 (88, 89) | PCP | Cohort A: with/without surgery | IHC for BRAF V600E mutation (VE1) and β-catenin IHC (membranous, nonnuclear pattern) | Vemurafenib and cobimetinib | Vemurafenib bd days 1–28 plus cobimetinib | Every 28 d for up to five courses in the absence of disease progression or unacceptable toxicity (MRI) | Patients may then receive RT, surgery, or continued treatment |
| Cohort B: RT with/without other treatment (except for BRAF or MEK inhibitors) | od on days 1–21 |
| Case Series/Reports | CP | Previous Treatment | Histopathological And Molecular Genetic Analysis | Treatment | Duration Of Treatment | Outcome (Mean Of Measurement) | Comments |
|---|---|---|---|---|---|---|---|
| Aylwin et al., 2015 (83) | PCP | TSS ×2, RT | Pyrosequencing analysis indicated the presence of BRAF mutation c.1799T>A (p.Val600Glu). | Vemurafenib 960 mg bd | 3 mo | Near complete resolution (MRI) | Recurrence 6 wk off treatment; restart of treatment and stabilization for 7 mo with a regrowth |
| Brastianos et al., 2015 (84) | PCP | Craniotomy ×3 | Uniform staining of the BRAFV600E protein (VE1 antibody) by IHC; V600E mutation confirmed by allele-specific genetic testing. Mutation detectable in peripheral blood during treatment. Absence of nuclear staining for β-catenin by IHC. | Dabrafenib 150 mg bd + after 3 wk trametinib 2 mg od | 35 d (followed by other treatments) | 85% reduction in tumor volume and 81% reduction in tumor-associated | |
| cyst (MRI) | |||||||
| Roque et al., 2016 (29) | PCP | Craniotomy, Ommaya catheter with percutaneous aspiration of cyst fluid, RT 54 Gy in 30 fractions | Positive staining for BRAFV600E by IHC; next-generation sequencing confirmed BRAFV600E mutation and FGFR3 splice site (437_445 + 3del12) with two variants of unknown significance (BRCA2-G1771D and FGFR4-S551F). | Dabrafenib 150 mg orally bd and trametinib 2 mg orally od | 7 mo | Almost disappearance of 2.6 × 2.3 × 3.2-cm tumor (MRI); reduced volume and intensity of FDG uptake on PET | |
| Rostami et al., 2017 (85) | PCP | TSS | Weak staining of mutated BRAFV600E (VE1 antibody) by IHC; BRAFV600E genotype confirmed by pyrosequencing mutational analysis. | Dabrafenib 150 mg bd plus after 3 wk trametinib 2 mg od | 7 wk | 91% reduction tumor size (MRI) | Combined BRAF and MEK-targeted therapy |
| Himes et al., 2019 (86) | PCP | Craniotomy, RT 36 Gy in 12 fractions | BRAF V600E mutation confirmed in specimens from original resection. | Dabrafenib 150 mg bd (shortly) → od (several weeks) → 225 mg od | 1 y under treatment and follow-up for 1 y off treatment. | Minimal residual tumor | |
| Juratli et al., 2019 (87) | PCP | No treatment | BRAF V600E confirmed by biopsy | Dabrafenib 150 mg bd plus trametinib 2 mg od | 6 mo | >80% reduction of tumor size (MRI) | Characterized as neoadjuvant therapy |
| NCT03224767 (88, 89) | PCP | Cohort A: with/without surgery | IHC for BRAF V600E mutation (VE1) and β-catenin IHC (membranous, nonnuclear pattern) | Vemurafenib and cobimetinib | Vemurafenib bd days 1–28 plus cobimetinib | Every 28 d for up to five courses in the absence of disease progression or unacceptable toxicity (MRI) | Patients may then receive RT, surgery, or continued treatment |
| Cohort B: RT with/without other treatment (except for BRAF or MEK inhibitors) | od on days 1–21 |
Abbreviations: bd, twice a day; FDG, 18fluoro-d-glucose; od, once a day; PET, positron emission tomography; TSS, transsphenoidal surgery.
Case Reports and Current Clinical Trials of Patients With PCPs and BRAF-V600E Alterations Treated With V600E Mutation-Specific BRAF Inhibitors (Dabrafenib or Vemurafenib) With or Without Combined Treatment With MEK Inhibitors (Trametinib or Cobimetinib)
| Case Series/Reports | CP | Previous Treatment | Histopathological And Molecular Genetic Analysis | Treatment | Duration Of Treatment | Outcome (Mean Of Measurement) | Comments |
|---|---|---|---|---|---|---|---|
| Aylwin et al., 2015 (83) | PCP | TSS ×2, RT | Pyrosequencing analysis indicated the presence of BRAF mutation c.1799T>A (p.Val600Glu). | Vemurafenib 960 mg bd | 3 mo | Near complete resolution (MRI) | Recurrence 6 wk off treatment; restart of treatment and stabilization for 7 mo with a regrowth |
| Brastianos et al., 2015 (84) | PCP | Craniotomy ×3 | Uniform staining of the BRAFV600E protein (VE1 antibody) by IHC; V600E mutation confirmed by allele-specific genetic testing. Mutation detectable in peripheral blood during treatment. Absence of nuclear staining for β-catenin by IHC. | Dabrafenib 150 mg bd + after 3 wk trametinib 2 mg od | 35 d (followed by other treatments) | 85% reduction in tumor volume and 81% reduction in tumor-associated | |
| cyst (MRI) | |||||||
| Roque et al., 2016 (29) | PCP | Craniotomy, Ommaya catheter with percutaneous aspiration of cyst fluid, RT 54 Gy in 30 fractions | Positive staining for BRAFV600E by IHC; next-generation sequencing confirmed BRAFV600E mutation and FGFR3 splice site (437_445 + 3del12) with two variants of unknown significance (BRCA2-G1771D and FGFR4-S551F). | Dabrafenib 150 mg orally bd and trametinib 2 mg orally od | 7 mo | Almost disappearance of 2.6 × 2.3 × 3.2-cm tumor (MRI); reduced volume and intensity of FDG uptake on PET | |
| Rostami et al., 2017 (85) | PCP | TSS | Weak staining of mutated BRAFV600E (VE1 antibody) by IHC; BRAFV600E genotype confirmed by pyrosequencing mutational analysis. | Dabrafenib 150 mg bd plus after 3 wk trametinib 2 mg od | 7 wk | 91% reduction tumor size (MRI) | Combined BRAF and MEK-targeted therapy |
| Himes et al., 2019 (86) | PCP | Craniotomy, RT 36 Gy in 12 fractions | BRAF V600E mutation confirmed in specimens from original resection. | Dabrafenib 150 mg bd (shortly) → od (several weeks) → 225 mg od | 1 y under treatment and follow-up for 1 y off treatment. | Minimal residual tumor | |
| Juratli et al., 2019 (87) | PCP | No treatment | BRAF V600E confirmed by biopsy | Dabrafenib 150 mg bd plus trametinib 2 mg od | 6 mo | >80% reduction of tumor size (MRI) | Characterized as neoadjuvant therapy |
| NCT03224767 (88, 89) | PCP | Cohort A: with/without surgery | IHC for BRAF V600E mutation (VE1) and β-catenin IHC (membranous, nonnuclear pattern) | Vemurafenib and cobimetinib | Vemurafenib bd days 1–28 plus cobimetinib | Every 28 d for up to five courses in the absence of disease progression or unacceptable toxicity (MRI) | Patients may then receive RT, surgery, or continued treatment |
| Cohort B: RT with/without other treatment (except for BRAF or MEK inhibitors) | od on days 1–21 |
| Case Series/Reports | CP | Previous Treatment | Histopathological And Molecular Genetic Analysis | Treatment | Duration Of Treatment | Outcome (Mean Of Measurement) | Comments |
|---|---|---|---|---|---|---|---|
| Aylwin et al., 2015 (83) | PCP | TSS ×2, RT | Pyrosequencing analysis indicated the presence of BRAF mutation c.1799T>A (p.Val600Glu). | Vemurafenib 960 mg bd | 3 mo | Near complete resolution (MRI) | Recurrence 6 wk off treatment; restart of treatment and stabilization for 7 mo with a regrowth |
| Brastianos et al., 2015 (84) | PCP | Craniotomy ×3 | Uniform staining of the BRAFV600E protein (VE1 antibody) by IHC; V600E mutation confirmed by allele-specific genetic testing. Mutation detectable in peripheral blood during treatment. Absence of nuclear staining for β-catenin by IHC. | Dabrafenib 150 mg bd + after 3 wk trametinib 2 mg od | 35 d (followed by other treatments) | 85% reduction in tumor volume and 81% reduction in tumor-associated | |
| cyst (MRI) | |||||||
| Roque et al., 2016 (29) | PCP | Craniotomy, Ommaya catheter with percutaneous aspiration of cyst fluid, RT 54 Gy in 30 fractions | Positive staining for BRAFV600E by IHC; next-generation sequencing confirmed BRAFV600E mutation and FGFR3 splice site (437_445 + 3del12) with two variants of unknown significance (BRCA2-G1771D and FGFR4-S551F). | Dabrafenib 150 mg orally bd and trametinib 2 mg orally od | 7 mo | Almost disappearance of 2.6 × 2.3 × 3.2-cm tumor (MRI); reduced volume and intensity of FDG uptake on PET | |
| Rostami et al., 2017 (85) | PCP | TSS | Weak staining of mutated BRAFV600E (VE1 antibody) by IHC; BRAFV600E genotype confirmed by pyrosequencing mutational analysis. | Dabrafenib 150 mg bd plus after 3 wk trametinib 2 mg od | 7 wk | 91% reduction tumor size (MRI) | Combined BRAF and MEK-targeted therapy |
| Himes et al., 2019 (86) | PCP | Craniotomy, RT 36 Gy in 12 fractions | BRAF V600E mutation confirmed in specimens from original resection. | Dabrafenib 150 mg bd (shortly) → od (several weeks) → 225 mg od | 1 y under treatment and follow-up for 1 y off treatment. | Minimal residual tumor | |
| Juratli et al., 2019 (87) | PCP | No treatment | BRAF V600E confirmed by biopsy | Dabrafenib 150 mg bd plus trametinib 2 mg od | 6 mo | >80% reduction of tumor size (MRI) | Characterized as neoadjuvant therapy |
| NCT03224767 (88, 89) | PCP | Cohort A: with/without surgery | IHC for BRAF V600E mutation (VE1) and β-catenin IHC (membranous, nonnuclear pattern) | Vemurafenib and cobimetinib | Vemurafenib bd days 1–28 plus cobimetinib | Every 28 d for up to five courses in the absence of disease progression or unacceptable toxicity (MRI) | Patients may then receive RT, surgery, or continued treatment |
| Cohort B: RT with/without other treatment (except for BRAF or MEK inhibitors) | od on days 1–21 |
Abbreviations: bd, twice a day; FDG, 18fluoro-d-glucose; od, once a day; PET, positron emission tomography; TSS, transsphenoidal surgery.
Very recently, Himes et al. (86) presented the longer-term effects of monotherapy with dabrafenib in a patient with a history of non-Hodgkin lymphoma and colon cancer who had a PCP with the BRAF-V600E mutation treated by cranial surgery and RT. Three years after the RT he developed tumor recurrence, and dabrafenib was initiated (150 mg twice daily, reduced to 150 mg daily due to joint pains, but then increased to 225 mg daily). Two months following treatment, an enlargement of the cystic component was documented in parallel with a reduction of the solid component. However, at 6 months posttreatment substantial reduction in the size of both components was identified, and by 9 months only minimal residual tumor was evident and this remained stable until 12 months; treatment was discontinued, and the tumor remained stable for further 12 months. A sixth case was recently reported at the European Congress of Endocrinology 2019 by Jurtali (87), who administered dabrafenib in a neoadjuvant setting to a 21-year-old male prior to surgery and found a >80% reduction of tumor size. All of these cases support the potential benefits of BRAF inhibitors, at least in achieving transient tumor responses, either as neoadjuvant treatment or following recurrence (Table 1). What is unclear from these single reports relates to the efficacy of different inhibitors, the mechanisms of tumor resistance, and the means to overcome such resistance. An interesting observation from one of these case reports was the documentation of a circulating BRAF-V600E mutation in the peripheral blood of the patient during treatment with the BRAF inhibitor (84). The authors stated that their finding of detectable BRAF-V600E in peripheral blood was unexpected, because there was not any previously reported case of circulating tumor cells or cell-free DNA in patients with intracranial benign tumors. However, if this is not a result of the surgical procedures or drug treatment, and the presence of BRAF-V600E can be detected in the blood before any treatment, then this assessment may be of value as a selection criterion of the patients who may benefit from BRAF-inhibitor neoadjuvant therapy (84). Following these observations, an ongoing cooperative group trial (Alliance A071601) is testing the combination of vemurafenib and cobimetinib in an open-label, phase 2 study in patients with BRAF-V600E–mutant CPs (88) to determine the frequency, durability, and extent of BRAF treatment responses in patients with PCPs. In this study, vemurafenib is administered twice daily on days 1 through 28, and cobimetinib is administered four times daily on days 1 through 21; treatment is repeated every 28 days to a maximum of five cycles as long as the disease shows a failure to progress and there is no unacceptable toxicity. Patients may then be treated with RT, surgery, or continue the combination of medical treatment. In another published series, a patient in whom GTR was achieved had a recurrence of a cystic tumor component after 2 years and has been referred to a phase 2 trial of combined BRAF/MEK inhibitors (NCT03224767) (88, 89).
Adverse effects of compounds currently used in the management of CPs
Data on the adverse effects profile of these treatments in patients with CPs are limited. The phenomenon of “pseudo-progression” with dabrafenib, defined as early initial cystic enlargement together with solid component reduction, followed by lesion shrinkage, has been observed, similar to that seen in gliomas after RT or chemotherapy (86). Additionally, transient fever has been described with BRAF inhibitors (29, 84, 85). Finally, the addition of the MEK inhibitor to BRAF inhibition may play a role in reducing the incidence of secondary squamous cell skin carcinomas, as previously shown in patients with melanoma (90).
Other potential treatments targeting CPs
Despite the identification of the deranged molecular pathways in ACP pathogenesis, drugs targeting β-catenin and its downstream MAPK pathway have been used only in in vitro studies. A transcriptome study used mRNA microarray gene expression analysis and unraveled several “druggable” pharmaceutical targets that were consistently overexpressed in a panel of 15 ACPs, compared with other brain tumors and normal brain tissue (26). Lymphocyte-specific protein tyrosine kinase (LCK), ephrin type-A2 (EPHA2), and SRC were identified as targets of the tyrosine kinase inhibitor dasatinib. AREG, EGFR, and ERBB-3 in the EGFR pathway may respond to inhibitors such as cetuximab, erlotinib, and lapatinib, and MMP9 and MMP12 could potentially respond to the oral MMP9/12 inhibitor AZD1236 (26, 50). Recurrence in ACPs has been associated with increased VEGF expression, suggesting a possible therapeutic role for the VEGF inhibitor bevacizumab (49, 61). Indeed, some of these targets can be shown to be expressed in the primary ACP tumor using Western blot analysis (26). Alternatively, although the SHH pathway may be implicated in the pathogenesis of ACPs, inhibition of this pathway may be tumorigenic (91). This relies on data showing that vismodegib, a well-established inhibitor of SHH, results in premature tumor formation and increased tumor cell proliferation in both genetically engineered and patient-derived xenograft mouse models (91). Inhibition of EGFR signaling by the tyrosine kinase inhibitor gefitinib reduced ACP cell migration in vitro by decreasing Fascin expression; this was attributed to the finding of nuclear colocalization of activated EGFR, β-catenin, and Fascin in ACP cell migration in vitro (48), implying crosstalk of these pathways in ACPs. Because annexin A2 expression, a Ca2+-regulated binding protein, correlates with gefitinib sensitivity in vitro, this may serve as a biomarker of a response to treatment (92). Small samples of human ACP tumors were cultured with and without trametinib, a specific MEK inhibitor; the inhibition of the MAPK/ERK pathway resulted in reduced proliferation along with a dose-dependent significant decrease in the Ki-67 proliferation index in the trametinib-treated tumors, suggesting a possible role in the management of not only PCPs but also ACPs (91). Notably, the well-established synergistic effects of combined targeted therapies in other neuroendocrine tumor models provide support for the potential use of combinatorial targeted therapy in CPs (93, 94).
Given the potential role of inflammation in the pathogenesis of CPs and previous studies on intratumor administration of IFN-α in patients with a CP (22), an open-label, phase 2 study is underway aiming to estimate the 1-year disease stabilization rate associated with the use of Peg–IFN-α2b in patients with progressive unresectable or recurrent CPs after surgery alone (without adjuvant RT). The study will also estimate the sustained objective response rate (partial and complete response) to Peg–IFN-α2b in patients with CPs that progress or recur following RT (95). Finally, noting the immune microenvironment in ACPs, IL-1R inhibitors, such as anakinra, may have a place in our therapeutic armamentarium (60).
Conclusions
The discovery of distinct oncogenic molecular signaling pathways in CPs has opened new avenues for the personalized treatment of patients with these challenging tumors. Τhe key pathogenetic event in ACPs appears to be Wnt activation along with alterations of the MEK/ERK pathway, whereas in PCPs there is primary activation of the Ras/Raf/MEK/ERK pathway by BRAF-V600E mutations. Current literature on PCPs includes single case reports, with both BRAF and MAPK inhibitors, in patients with V600E mutations. Their results are promising, but well-designed studies are needed that aim to provide robust data on treatment regimens and protocols, as well as on efficacy and safety. It also remains unclear whether single or combination therapies will be necessary, although in vitro data from other endocrine tumors have highlighted the use of multiple simultaneous combination treatments. The use of MAPK inhibitors for the more common ACPs also requires assessment in clinical trials, particularly in view of the risk of acquired drug resistance. The value of combination therapies aiming to target multiple molecular mediators implicated in the pathogenesis of ACP and possibly minimizing drug toxicities also requires further investigation.
Nevertheless, the recently expanding data on the pathogenesis of CPs offer substantial ground for the translation of molecular insights into practical therapies for primary, recurrent, or very aggressive tumors that will, hopefully, improve the prognosis of the patients. The eventual use of neoadjuvant medical treatment or even an entirely nonoperative treatment strategy for CPs should improve the quality of life of these patients, minimizing the neurologic and endocrinological sequelae caused either by the tumor extension or by the neurosurgical or radiotherapeutic complications. Combination therapy strategies could be also be effective in cases of ACPs to target the multiple molecular mediators that are implicated in ACP pathogenesis and to mimimize any adverse treatment effects.
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability: All data generated or analyzed during this study are included in this published article or in the data repositories listed in References.
Abbreviations:
- ACP
adamantinomatous craniopharyngioma
- APC
adenomatosis polyposis coli
- BMP
bone morphogenetic protein
- CK1
casein kinase 1α
- CP
craniopharyngioma
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- FGF
fibroblast growth factor
- GSK-3β
glycogen synthase kinase-3β
- GTR
gross total resection
- IFN
interferon
- IHC
immunocytochemistry
- MeSH
medical subject headings
- MMP
matrix metallopeptidase
- PCP
papillary craniopharyngioma
- PDGF
platelet-derived growth factor
- RT
radiotherapy
- SHH
sonic hedgehog
- STR
subtotal/partial resection
- VEGF
vascular endothelial growth factor
References and Notes
Author notes
N.K. and A.B.G. are joint senior authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}