





Abstract
Hyperglucagonemia in the absence of glucagonomas is rare. Biallelic-inactivating mutations in the glucagon receptor gene (GCGR) cause glucagon cell hyperplasia and neoplasia (GCHN), also termed Mahvash syndrome. Here, we report the first case to our knowledge of GCHN presenting with hypercalcemia and demonstrate a unique relationship between calcium and α-cell hyperplasia.
A 47-year-old man presented with severe PTH-independent hypercalcemia, 13.95 mg/dL (3.48 mmol/L). Imaging and extensive pathology tests yielded no conclusive cause. Glucagon levels >300 times the upper limit of normal were discovered. Subtotal pancreatectomy identified α-cell hyperplasia and neoplasia with metastatic disease in lymph nodes. Genomic analysis confirmed a homozygous missense variant in GCGR (Asp63Asn). This is a previously described pathologic variant and has a known association with GCHN.
Inactivating mutations of the glucagon receptor gene lead to nonfunctional hyperglucagonemia and are associated with GCHN. Homozygous or compound heterozygous GCGR mutations are associated with α-cell hyperplasia, a known precursor to pancreatic neuroendocrine tumors that can metastasize. Hypercalcemia is an unreported consequence of GCHN with an unclear mechanism.
Hyperglucagonemia in the absence of glucagonoma(s) is very rare. Biallelic-inactivating mutations in the GCGR gene, which encodes the glucagon receptor, cause glucagon cell hyperplasia and neoplasia (GCHN), which was recently established as a distinct clinicopathological entity under the World Health Organization 2017 classification of endocrine neoplasia (1, 2). The typical presentation includes hyperglucagonemia together with α-cell hyperplasia, which may progress to multiple glucagon-producing tumors and then frankly malignant and metastatic disease. As the glucagon receptor is nonfunctional, these patients do not exhibit the characteristic rash and hyperglycemia classically associated with glucagonomas and in fact may experience hypoglycemia (1, 2).
Mahvash syndrome and glucagon cell adenomatosis are often used interchangeably with GCHN, with the term Mahvash syndrome sometimes restricted to the 50% of patients in whom biallelic mutations of GCGR are definitively identified (2). To date, nine pathogenic GCGR variants associated with GCHN have been reported, summarized in Table 1. The natural history of this disease involves high levels of glucagon associated with the transformation of hyperplastic islets into glucagon-producing microadenomas and, in time, pancreatic neuroendocrine tumors with metastatic potential.
Clinical Features of Reported Patients With GCGR Mutations
| Characteristic | Miller et al. ( 3 ) | Sipos et al. ( 4 ) (Patient 1) | Sipos et al. ( 4 ) (Patient 2) | Sipos et al. ( 4 ) (Patient 3) | Zhou et al. ( 5 ) | Present Case |
|---|---|---|---|---|---|---|
| Patient age, y/sex | 36/F | 25/M | 43/F | 58/M | 60/F | 48/M |
| Presenting symptoms | Abdominal pain | Abdominal pain | Abdominal pain | Fatigue, diarrhea, weight loss | Abdominal pain | Abdominal pain |
| Pancreatitis | Hypercalcemia | |||||
| Glucagon | 66 (NR <50) | 135× ULN | 25× ULN | Not performed | 20,000 | 100,000 |
| Pathology | ACH | ACH | ACH | Hyperplastic islets and microtumors | ACH | ACH |
| Hyperplastic islets | Microadenomas | P-NET | ||||
| GCGR status | K349_G359 | HomozygousW83Lfs*35 | Exon 2: R8* (heterozygous) Exon 11: Q327* (heterozygous) | Exon 8: R225H (homozygous) Exon 12: V368 M (homozygous) | Homozygous | Homozygous c.187G>A, Asp63Asn |
| Deletion | c.256C>T, p.P86S |
| Characteristic | Miller et al. ( 3 ) | Sipos et al. ( 4 ) (Patient 1) | Sipos et al. ( 4 ) (Patient 2) | Sipos et al. ( 4 ) (Patient 3) | Zhou et al. ( 5 ) | Present Case |
|---|---|---|---|---|---|---|
| Patient age, y/sex | 36/F | 25/M | 43/F | 58/M | 60/F | 48/M |
| Presenting symptoms | Abdominal pain | Abdominal pain | Abdominal pain | Fatigue, diarrhea, weight loss | Abdominal pain | Abdominal pain |
| Pancreatitis | Hypercalcemia | |||||
| Glucagon | 66 (NR <50) | 135× ULN | 25× ULN | Not performed | 20,000 | 100,000 |
| Pathology | ACH | ACH | ACH | Hyperplastic islets and microtumors | ACH | ACH |
| Hyperplastic islets | Microadenomas | P-NET | ||||
| GCGR status | K349_G359 | HomozygousW83Lfs*35 | Exon 2: R8* (heterozygous) Exon 11: Q327* (heterozygous) | Exon 8: R225H (homozygous) Exon 12: V368 M (homozygous) | Homozygous | Homozygous c.187G>A, Asp63Asn |
| Deletion | c.256C>T, p.P86S |
Abbreviations: ACH, alpha cell hyperplasia; F, female; M, male; NR, normal range; P-NET, pancreatic neuroendocrine tumor; ULN, upper limit of normal.
Clinical Features of Reported Patients With GCGR Mutations
| Characteristic | Miller et al. ( 3 ) | Sipos et al. ( 4 ) (Patient 1) | Sipos et al. ( 4 ) (Patient 2) | Sipos et al. ( 4 ) (Patient 3) | Zhou et al. ( 5 ) | Present Case |
|---|---|---|---|---|---|---|
| Patient age, y/sex | 36/F | 25/M | 43/F | 58/M | 60/F | 48/M |
| Presenting symptoms | Abdominal pain | Abdominal pain | Abdominal pain | Fatigue, diarrhea, weight loss | Abdominal pain | Abdominal pain |
| Pancreatitis | Hypercalcemia | |||||
| Glucagon | 66 (NR <50) | 135× ULN | 25× ULN | Not performed | 20,000 | 100,000 |
| Pathology | ACH | ACH | ACH | Hyperplastic islets and microtumors | ACH | ACH |
| Hyperplastic islets | Microadenomas | P-NET | ||||
| GCGR status | K349_G359 | HomozygousW83Lfs*35 | Exon 2: R8* (heterozygous) Exon 11: Q327* (heterozygous) | Exon 8: R225H (homozygous) Exon 12: V368 M (homozygous) | Homozygous | Homozygous c.187G>A, Asp63Asn |
| Deletion | c.256C>T, p.P86S |
| Characteristic | Miller et al. ( 3 ) | Sipos et al. ( 4 ) (Patient 1) | Sipos et al. ( 4 ) (Patient 2) | Sipos et al. ( 4 ) (Patient 3) | Zhou et al. ( 5 ) | Present Case |
|---|---|---|---|---|---|---|
| Patient age, y/sex | 36/F | 25/M | 43/F | 58/M | 60/F | 48/M |
| Presenting symptoms | Abdominal pain | Abdominal pain | Abdominal pain | Fatigue, diarrhea, weight loss | Abdominal pain | Abdominal pain |
| Pancreatitis | Hypercalcemia | |||||
| Glucagon | 66 (NR <50) | 135× ULN | 25× ULN | Not performed | 20,000 | 100,000 |
| Pathology | ACH | ACH | ACH | Hyperplastic islets and microtumors | ACH | ACH |
| Hyperplastic islets | Microadenomas | P-NET | ||||
| GCGR status | K349_G359 | HomozygousW83Lfs*35 | Exon 2: R8* (heterozygous) Exon 11: Q327* (heterozygous) | Exon 8: R225H (homozygous) Exon 12: V368 M (homozygous) | Homozygous | Homozygous c.187G>A, Asp63Asn |
| Deletion | c.256C>T, p.P86S |
Abbreviations: ACH, alpha cell hyperplasia; F, female; M, male; NR, normal range; P-NET, pancreatic neuroendocrine tumor; ULN, upper limit of normal.
Hypercalcemia is not a recognized complication of this syndrome, and we believe this is the first report of this association. Although malignant hypercalcemia is relatively frequent in metastatic neuroendocrine tumors, the mechanism is related to PTH-related protein, which was absent in our case. Furthermore, the trajectory of the calcium in our patient mirrors the clinical activity of the disease.
Case Illustration
A 47-year-old man presented with refractory PTH-independent hypercalcemia (13.95 mg/dL) and abdominal pain. Initial investigations, including angiotensin converting enzyme level (low, 87 U/L), myeloma screen, PTH-related protein (<2 pmol/L), and 1,25 dihydroxyvitamin D (low, 16 pmol/L), yielded no clear cause. History was unremarkable; there were no relevant mediations and family history was only relevant for parental consanguinity. Imaging demonstrated multiple punctate foci of calcification in the pancreas (Fig. 1A). Fluorodeoxyglucose–positron emission tomography (PET) showed no evidence of fluorodeoxyglucose-avid disease or lymphoma. Further investigation with magnetic resonance cholangiopancreatography showed no solid neoplasms in the pancreas, and endoscopic ultrasound showed lobulation and stranding heterogeneity of the entire pancreas, without a dominant lesion. Pancreatic hormones were assessed for any functional derangement. Plasma glucagon was striking at 43,547 pg/mL (range, 40 to 140 pg/mL). Close clinical review revealed no evidence of necrolytic migratory erythema or hyperglycemia. 68Ga-DOTATATE-PET showed intense diffusely increased tracer uptake throughout the pancreas, with no abnormal uptake elsewhere. Concern for a diffuse neuroendocrine cell hyperplastic process was raised, and he was given a provisional diagnosis of GCHN. Glucagon sampling was performed to assess for a localized area of glucagon secretion. The area of highest production was found around the pancreatic neck. The patient underwent splenectomy and subtotal distal pancreatectomy with 65% of his large calcified pancreas being excised (Fig. 1B). Serum calcium initially improved (Fig. 2). Pathology confirmed diffuse α-cell hyperplasia (Fig. 1C) characterized by multiple enlarged islets with a striking preponderance of glucagon-producing cells progressing to hundreds of coalescing glucagon-producing tumors up to 10 mm in diameter. Micrometastatic glucagon-producing neuroendocrine tumor was found in 11 of 30 lymph nodes. Immunohistochemistry was diffusely positive for glucagon (Fig. 1D) and pancreatic polypeptide and negative for insulin and other hormones. The Ki-67 proliferative index was no more than 0.5% in both primary and metastatic disease.
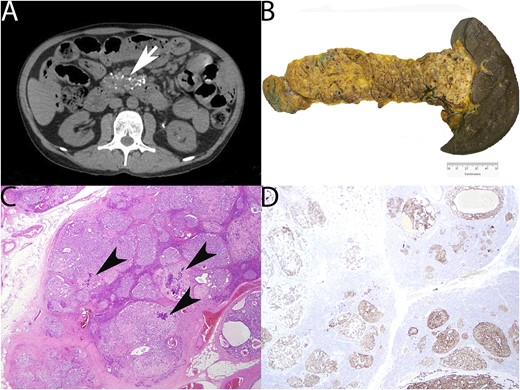
Radiology and surgical pathology. (A) CT abdomen demonstrating pancreatic enlargement and calcification (arrow). (B) Gross appearance of distal pancreatectomy and splenectomy demonstrating brawny induration and nodularity of the pancreas corresponding to myriad coalescing tumors (weight of distal pancreas removed, 264 g). (C) Photomicrograph of pancreas showing multiple hyperplastic islets coalescing into small neuroendocrine tumors. Dystrophic calcification (arrowheads) is noted (hematoxylin and eosin, ×100). (D) Glucagon immunohistochemistry demonstrating diffuse expression in hyperplastic islets and small tumors (glucagon immunohistochemistry, ×100).
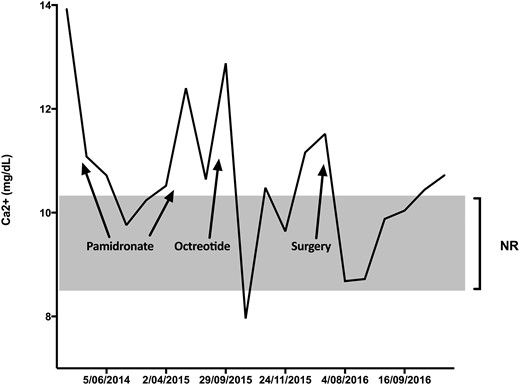
Graph of calcium levels over time. NR, normal range.
Germline whole-genome sequencing demonstrated a homozygous pathogenic missense variant c.187G>A (Asp63Asn) in exon 4 of the GCGR gene, which was confirmed by Sanger sequencing.
Serum calcium levels were labile during investigation and treatment with recurrent administration of pamidronate tempering them for short periods (Fig. 2). The most significant reductions in serum calcium occurred following initial administration of octreotide (10.5 to 7.98 mg/dL) and then after pancreatectomy (11.54 to 8.77 mg/dL), with both reductions corresponding to a fall in serum glucagon levels. After the pancreatectomy, serum calcium has continued to remain just above the upper limit of normal with one episode of severe hypercalcemia (14.43 mg/dL) following a 9-month period of noncompliance to lanreotide. As pamidronate was no longer being tolerated due to abdominal pain, denosumab was given and serum calcium subsequently returned to normal. 68Ga-DOTATATE-PET scans have remained stable and no distant metastases have been identified.
During his latest assessment, we performed an oral glucose tolerance test (75g glucose) and measured changes in serum concentrations of pancreatic hormones and other analytes of interest. As shown in Table 2, we found an expected increase in C-peptide and insulin, a reduction in glucagon, and stable glucagon-like peptide-1 (slightly raised at baseline), calcium, and PTH levels. Interestingly, the urinary amino acids citrulline, arginine, and glycine were raised at baseline and remained so after the glucose load.
Change in Parameters Following Oral Glucose Tolerance Test
| Characteristic | 0 min | 120 min | Normal Range |
|---|---|---|---|
| Calcium, mg/dL, mmol/L | 10.74 (2.68) | 10.46 (2.61) | 8.5–10.2 |
| Phosphate, mg/dL, mmol/L | 4.15 (1.34) | 3.72 (1.20) | 2.4–4.5 |
| Glucose, mg/dL, mmol/L | 77.4 (4.3) | 145.8 (8.1) | 70–140 |
| Insulin, mIU/L | 2.6 | 17.5 | <10 |
| C-peptide, ng/mL, pmol/L | 1.62 (537) | 8.12 (2690) | 260–1739 |
| PTH, ng/L | 4.0 | <4.0 | 15–68 |
| P1NP, μg/L | 24 | 21 | 14–80 |
| CTX, ng/L | 482 | 454 | <580 |
| TRAP 5b, U/L | 3.1 | 2.6 | <4.8 |
| Glucagon, pg/mL | 5380 | 4795 | 40–140 |
| Glucagon-like peptide-1, pM | 17.9 | 17.9 | 5–10 |
| Abnormal amino acids | |||
| Arginine | 150 | 126 | 34–118 |
| Citrulline | 102 | 80 | 10–45 |
| Cystine | 86 | 75 | 24–67 |
| Glycine | 733 | 680 | 119–368 |
| Aromatic amino acids | |||
| Phenylalanine | 52 | 40 | 30–75 |
| Tyrosine | 43 | 31 | 32–114 |
| Characteristic | 0 min | 120 min | Normal Range |
|---|---|---|---|
| Calcium, mg/dL, mmol/L | 10.74 (2.68) | 10.46 (2.61) | 8.5–10.2 |
| Phosphate, mg/dL, mmol/L | 4.15 (1.34) | 3.72 (1.20) | 2.4–4.5 |
| Glucose, mg/dL, mmol/L | 77.4 (4.3) | 145.8 (8.1) | 70–140 |
| Insulin, mIU/L | 2.6 | 17.5 | <10 |
| C-peptide, ng/mL, pmol/L | 1.62 (537) | 8.12 (2690) | 260–1739 |
| PTH, ng/L | 4.0 | <4.0 | 15–68 |
| P1NP, μg/L | 24 | 21 | 14–80 |
| CTX, ng/L | 482 | 454 | <580 |
| TRAP 5b, U/L | 3.1 | 2.6 | <4.8 |
| Glucagon, pg/mL | 5380 | 4795 | 40–140 |
| Glucagon-like peptide-1, pM | 17.9 | 17.9 | 5–10 |
| Abnormal amino acids | |||
| Arginine | 150 | 126 | 34–118 |
| Citrulline | 102 | 80 | 10–45 |
| Cystine | 86 | 75 | 24–67 |
| Glycine | 733 | 680 | 119–368 |
| Aromatic amino acids | |||
| Phenylalanine | 52 | 40 | 30–75 |
| Tyrosine | 43 | 31 | 32–114 |
Abnormal results in bold.
Abbreviations: CTX, c-terminal telopeptide; GLP-1, glucagon-like peptide-1; P1NP, procollagen type 1 N propeptide; TRAP 5b, tartrate-resistant acid phosphatase 5b.
Change in Parameters Following Oral Glucose Tolerance Test
| Characteristic | 0 min | 120 min | Normal Range |
|---|---|---|---|
| Calcium, mg/dL, mmol/L | 10.74 (2.68) | 10.46 (2.61) | 8.5–10.2 |
| Phosphate, mg/dL, mmol/L | 4.15 (1.34) | 3.72 (1.20) | 2.4–4.5 |
| Glucose, mg/dL, mmol/L | 77.4 (4.3) | 145.8 (8.1) | 70–140 |
| Insulin, mIU/L | 2.6 | 17.5 | <10 |
| C-peptide, ng/mL, pmol/L | 1.62 (537) | 8.12 (2690) | 260–1739 |
| PTH, ng/L | 4.0 | <4.0 | 15–68 |
| P1NP, μg/L | 24 | 21 | 14–80 |
| CTX, ng/L | 482 | 454 | <580 |
| TRAP 5b, U/L | 3.1 | 2.6 | <4.8 |
| Glucagon, pg/mL | 5380 | 4795 | 40–140 |
| Glucagon-like peptide-1, pM | 17.9 | 17.9 | 5–10 |
| Abnormal amino acids | |||
| Arginine | 150 | 126 | 34–118 |
| Citrulline | 102 | 80 | 10–45 |
| Cystine | 86 | 75 | 24–67 |
| Glycine | 733 | 680 | 119–368 |
| Aromatic amino acids | |||
| Phenylalanine | 52 | 40 | 30–75 |
| Tyrosine | 43 | 31 | 32–114 |
| Characteristic | 0 min | 120 min | Normal Range |
|---|---|---|---|
| Calcium, mg/dL, mmol/L | 10.74 (2.68) | 10.46 (2.61) | 8.5–10.2 |
| Phosphate, mg/dL, mmol/L | 4.15 (1.34) | 3.72 (1.20) | 2.4–4.5 |
| Glucose, mg/dL, mmol/L | 77.4 (4.3) | 145.8 (8.1) | 70–140 |
| Insulin, mIU/L | 2.6 | 17.5 | <10 |
| C-peptide, ng/mL, pmol/L | 1.62 (537) | 8.12 (2690) | 260–1739 |
| PTH, ng/L | 4.0 | <4.0 | 15–68 |
| P1NP, μg/L | 24 | 21 | 14–80 |
| CTX, ng/L | 482 | 454 | <580 |
| TRAP 5b, U/L | 3.1 | 2.6 | <4.8 |
| Glucagon, pg/mL | 5380 | 4795 | 40–140 |
| Glucagon-like peptide-1, pM | 17.9 | 17.9 | 5–10 |
| Abnormal amino acids | |||
| Arginine | 150 | 126 | 34–118 |
| Citrulline | 102 | 80 | 10–45 |
| Cystine | 86 | 75 | 24–67 |
| Glycine | 733 | 680 | 119–368 |
| Aromatic amino acids | |||
| Phenylalanine | 52 | 40 | 30–75 |
| Tyrosine | 43 | 31 | 32–114 |
Abnormal results in bold.
Abbreviations: CTX, c-terminal telopeptide; GLP-1, glucagon-like peptide-1; P1NP, procollagen type 1 N propeptide; TRAP 5b, tartrate-resistant acid phosphatase 5b.
Discussion
To our knowledge, this is the first time hypercalcemia has been reported in GCHN. Improvement of hypercalcemia after subtotal pancreatectomy suggests that this is a direct action of glucagon, presumably independent of the glucagon receptor. However, the exact mechanism of hypercalcemia in this case remains unclear.
The calcium-sensing receptor is activated not only by extracellular calcium ions but also by aromatic amino acids (6). Whether the calcium-sensing receptor is activated by the hyperaminoacidemia secondary to hyperglucagonemia is not known, but in this patient, we did not observe an increase in aromatic amino acids at baseline or following the glucose load to support this. We also explored whether increased bone turnover was associated with hypercalcemia. However, the patient had previously had an unremarkable bone mineral density and normal bone turnover markers P1NP, TRAP5b, and CTX (Table 2). Interestingly, in addition to glucagon, glucagon-like peptide-1 levels were also raised and could potentially contribute to hypercalcemia albeit through an undefined mechanism.
Gcgr−/− mice are a functional model of GCHN and also exhibit α-cell hyperplasia and hyperglucagonemia. These mice develop well-differentiated neuroendocrine neoplasms that occasionally metastasize to the liver (7), and it is assumed that a loss of negative feedback results in the progression of α-cell hyperplasia to true neoplasia (8). Notably, the phenotype of these mice also includes fasting hypoglycemia. β-Cells as well as α-cells have glucagon receptors that inhibit insulin release. Dysregulation of β-cells causes mild fasting hypoglycemia, which was also reported by our patient as a young adult.
Amino acids may play a role in both the α-cell hyperplasia and progression to neoplasia in this disease. Glucagon secretion is stimulated by amino acids acting on α-cells. Glucagon is ureogenic and also affects amino acid production in the liver, and patients with glucagonomas are hypoaminoacidemic (9). GCGR mutations are also associated with high aminoacidemia. In Gcgr−/− mice, amino acid catabolism genes are downregulated (8). A lack of functional glucagon leads to increased production of amino acids and an attempt to reinstitute normal levels of glucagon. That is, it appears that the high amino acids may be the mechanism driving the α-cell hyperplasia as α-cells sense amino acids and promote α-cell replication possibly through mammalian target of rapamycin activation (10). Given the success of mammalian target of rapamycin inhibitors in some pancreatic neuroendocrine tumors (11), these drugs may be a rational therapeutic option in GCHN and warrant further investigation.
In conclusion, we report a new case of GCHN and its first association, to our knowledge, with hypercalcemia. Further study is required to estimate the frequency of this association and understand the pathophysiology of the unexplained hypercalcemia.
Abbreviations:
Acknowledgments
Disclosure Summary: The authors have nothing to disclose.

{kind=link}
{kind=link}