





Abstract
Thyroid hemiagenesis (THA) constitutes a rare, congenital disorder that is characterized by an absence of one thyroid lobe. Because the pathogenesis and clinical significance of this malformation remain undefined, specific clinical recommendations are lacking, especially for asymptomatic cases.
The PubMed database was searched (years 1970 to 2017), and the following terms were used to retrieve the results: “thyroid hemiagenesis,” “thyroid hemiaplasia,” “one thyroid lobe agenesis,” and “one thyroid lobe aplasia.” Subsequently, reference sections of the retrieved articles were searched.
There is a noticeable susceptibility of subjects with THA to develop additional thyroid and nonthyroidal pathologies. In pathogenesis of concomitant thyroid pathologies, a chronic elevation in thyroid-stimulating hormone values may play an important role. Thus far, genetic studies failed to find a common genetic background of the anomaly, and the potential underlying cause was identified in a minority of the cases.
Patients with THA are prone to develop additional thyroid pathologies and theoretically might benefit from l-thyroxine treatment to lower the thyrotropin levels to those observed in the normal population. However, further research should be done to ascertain whether such intervention early in life would prevent development of associated thyroid conditions. At least, increased vigilance should be maintained to reveal all of the concomitant disorders as soon as possible during follow-up examinations. Application of high-throughput technologies enabling a genome-wide search for novel factors involved in thyroid embryogenesis might be the next step to expand the knowledge on THA pathogenesis.
Thyroid hemiagenesis (THA) constitutes a rare, congenital disorder that is characterized by an absence of one thyroid lobe. The presence of this anomaly is usually unsuspected and detected incidentally (i.e., during screening tests or diagnostic procedures performed for other problems). Because the pathogenesis and clinical significance of this malformation remain undefined, specific clinical recommendations are lacking, especially for asymptomatic cases. The scope of this review is to summarize the genetic factors underlying its pathogenesis and embryogenesis and explore known epidemiologic factors and associated thyroidal and nonthyroidal disorders to develop an understanding of the potential clinical importance. To achieve this goal, the PubMed database was searched (years 1970 to 2017), and the following terms were used to retrieve the results: “thyroid hemiagenesis,” “thyroid hemiaplasia,” “one thyroid lobe agenesis,” and “one thyroid lobe aplasia.” Subsequently, reference sections of the retrieved articles were searched.
Thyroid Formation and Lobulation During Embryogenesis
Thyroid embryogenesis is a complex and still not fully defined process. The mechanisms governing the descent and lobulation of the thyroid primordium are yet to be elucidated. The thyroid gland is the first endocrine organ to develop during embryogenesis (1). The thyroid anlage can usually be visualized by the 20th day following conception (2). Thyroid precursor cells develop due to proliferation of endodermal epithelium, lining the floor of the primitive pharynx between the first and second pharyngeal arch (3). Experimentally, a potent trophic influence of the surrounding endoderm on the proliferation of thyrocytes has been demonstrated (4). Thus, a reduced mass of thyroid tissue could result from a lack of the stimulus from the adjacent endoderm, thereby depressing the process of proliferation of the thyroid primordium. The thyroid anlage subsequently migrates to its destination in the lower neck, gradually acquiring its classic bilobed configuration (5, 6). The lobes grow laterally, whereas the development of the median portion is restricted. During this developmental descent, the thyroid primordium stays connected to the base of the tongue by the thyroglossal duct, which apparently disappears by the end of the fifth week of pregnancy (7). By that time, the thyroid already possesses its bilobed shape with a distinct isthmus (6). Both lobes continue to expand laterally from the time of arrival of the primordium to its final locus until approximately the 50th day of pregnancy (5, 6, 8).
Whereas thyroid follicular cells of endodermal derivation dominate in the mature thyroid, parafollicular cells of neuroectodermal origin can be identified and are typically found in greatest abundance in the lower portion of the upper third of the lobes (9). Their precursors migrate from the neural crest bilaterally toward the fourth and fifth pharyngeal pouches and are deposited in the ultimobranchial bodies. The latter migrate medially and fuse with the descending thyroid primordium at about the 44th day of development. The parafollicular cells become deposited among the follicular cells, and the final histological structure of the gland is formed (10).
The Role of Vascular Factors in Thyroid Development
Development of the thyroid gland is strongly dependent on the precursory formation and continuing development of the cardiovascular system, which is realized by the proximate positioning of cardiac mesoderm to the thyroid bud (11). Experimental studies have demonstrated that as the thyroid anlage expands, cells are gradually recruited from the surrounding endoderm. In fact, the path of the thyroid primordium during embryogenesis is similar to that of the descent of the aortic sac. It has been proposed that the migration and final localization of the thyroid primordium is governed by its relation to developing arteries (4, 11). The fact that thyroid anlage stays connected to the aortic sac vessels may explain instances of ectopic thyroid tissue found in the pericardium (11, 12). The process of thyroid bilobation takes place in parallel with the development of the blood vessels of the third pharyngeal arch. These vessels are destined to form the internal carotid artery and a portion of the common carotid artery, with their final adult localization closely related to each mature thyroid lobe (11).
The process of thyroid anlage descent is thought to be rather passive, with the position of the thyroid primordium determined based on changes taking place in the local environment (12–14). The thyroid lobes migrate initially in the caudal direction and then move laterally (12). The weight of experimental data makes it highly probable that close contact with vessels or factors responsible for proper formation of vessels determine normal thyroid lobe formation. Conceivably then, maldevelopment or maldescent of vessels may be associated with, or be the cause of, anomalous thyroid development (11). Indeed, an association of heart and vessel defects to thyroid dysgenesis has been described (15, 16). One recent report (17) demonstrated agenesis of the thyroid artery on the agenetic side in patients with THA by computed tomography angiography of the neck, whereas the corresponding vessel on the side of the normal lobe was visualized. Therefore, one can speculate that at least one cause for the lack of thyroid lobe formation results from inadequate blood supply leading to aplasia of the lobe at a very early stage of embryogenesis (17).
Despite numerous reports supporting the fact that embryology of the thyroid appears to be connected to that of the heart, there is no evidence that this could be the reason why THA is most often left-sided. However, observations on a large cohort of subjects with THA led to the conclusion that most often, THA presents with isolated agenesis of the left lobe (the right lobe and isthmus are present), whereas the agenesis of the right lobe is often associated with simultaneous isthmus absence (18). That might suggest that the process of thyroid descent takes place not symmetrically in the midline, but occurs more to the left side. This is in accord with reports that the pyramidal lobe arises from the left side of the isthmus more often than from the midportion or the right side (19).
Establishing the Diagnosis
THA tends to be discovered incidentally during imaging of the neck region. The condition may be suspected if no apparent thyroid tissue is detected on palpation on one side of the neck (Fig. 1). The diagnosis can be confirmed if any contralateral thyroid tissue is revealed by imaging techniques, mostly by the means of ultrasonography (Fig. 2) or thyroid scintiscan (Fig. 3). Less frequently, the anomaly may be incidentally detected on computed tomography or magnetic resonance imaging performed in the evaluation of other medical conditions (20). A combination of ultrasound examination and isotope scintigraphy is sufficient to establish and confirm the diagnosis of THA and most other thyroid developmental anomalies (21). Sonography can visualize lobe absence and the presence or absence of an isthmus and often demonstrate underlying pathology of the gland. The appearance of THA with an associated isthmus has been described as having the appearance of a hockey stick (“hockey stick” abnormality) (8). Unilateral uptake of isotope on a scintiscan can be misleading, whereas anatomic imaging (e.g., ultrasonography) will distinguish between true THA and other conditions that may present unilateral radionuclide uptake, such as unilateral thyroiditis or infiltrative disease (for example, amyloidosis) or the presence of a nonfunctional hypoplastic lobe (20, 22). The utility and popularity of thyroid ultrasonography has grown to almost gold-standard status due to its wide availability, noninvasiveness, and low cost (23, 24). Scintigraphy, in contrast, can identify possible ectopic thyroid tissue not seen by ultrasound by demonstrating functional tissue with parenchymal radionuclide uptake.
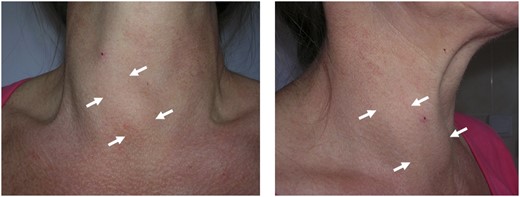
Compensatory enlargement of the right lobe in a patient with late-diagnosed left-sided THA.
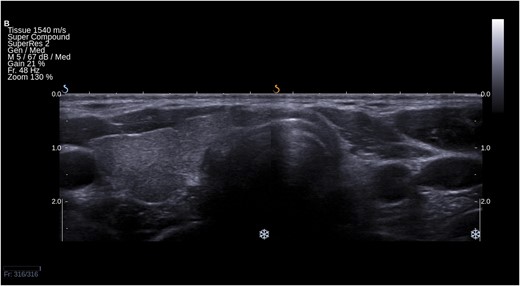
Ultrasound picture of left-sided THA in a transverse section of the neck. Right lobe and isthmus are present.

Different variants of THA visible on 131I scintiscan. From the left: (a) the most typical isolated left lobe agenesis (“hockey stick” sign), (b) agenesis of the left lobe and isthmus, (c) isolated agenesis of the right lobe, and (d) right lobe and isthmus agenesis. Left lobe and long pyramidal lobe are visible.
Epidemiology
The prevalence of THA is reported to vary from 0.05% to 0.5%, although the true frequency is uncertain (25–28). This uncertainty is likely due to widespread unawareness of its existence in affected asymptomatic, euthyroid individuals. In 2009, Duarte et al. (29) analyzed the thyroid volume, ultrasonographic abnormalities, and urinary iodine excretion in 964 schoolchildren living in an iodine-sufficient area in southern Brazil. Approximately 0.5% of children were affected by THA (29). Earlier autopsy studies have provided frequency estimates of THA, such as the report by Marshall in 1895 (30) that described only 1 patient with this malformation among 600 children. Because the remaining lobe is usually capable of sufficient hormonal synthesis and secretion to sustain clinical euthyroidism, the disorder rarely presents with hypothyroidism unless there is underlying Hashimoto thyroiditis. Rather, the abnormality is much more likely to be discovered incidentally during investigation of concomitant thyroid pathologies or during the course of imaging examinations. Nonetheless, juvenile subjects with overt hypothyroidism presumed to be due to the lack of one thyroid lobe have been reported (31). Calaciura et al. (32) stated that THA was responsible for 10.7% of congenital hyperthyrotropinemia identified during neonatal screening cases in 56 newborns. Other workers have reported a prevalence of THA varying from 0.02% to 5.7% in children with congenital hypothyroidism (33–35).
In the first systematic analysis of a large cohort of subjects with THA, our group observed a higher risk of THA development in female subjects, noting a 7:1 female to male ratio (18), confirming the conclusions drawn from previous studies (27, 28, 36). Perhaps the reported greater susceptibility of girls and women for THA may relate to both the generally higher frequency of thyroid diseases in females and the relatively greater number of female subjects presenting for evaluation to hospitals and other thyroid centers. Maiorana et al. (25) came to the opposite conclusion after an analysis of a systematic screening of healthy schoolchildren for thyroid developmental abnormalities that indicated a higher prevalence of THA in males, with a male to female ratio at the level of 1.4:1.0.
Remarkably, and for unknown reasons, left-sided agenesis is the considerably more frequent presentation, being present in up to ∼87.5% of the subjects studied (18). Other workers found the left lobe to be absent in 80% of the cases, with a left to right hemiagenesis ratio of 4:1 (8). The most typical patterns of THA are either an isolated left lobe agenesis or combined THA of the right lobe with concomitant isthmus absence (18).
Hormonal Status and Concomitant Thyroid Disorders
As indicated, these patients are most frequently clinically euthyroid and consequently will have normal circulating levels of thyroxine (T4) and triiodothyronine (T3). However, when compared with controls with an anatomically normal bilobate gland, the average serum thyroid-stimulating hormone (TSH) and free T3 (fT3) values were significantly higher in both children and adults (18, 25).
Twelve cases of THA were detected in the study by Maiorana et al. (25) of 24,032 unselected 11- to 14-year-old children who were examined. The study took place in southeastern Sicily, in the area where moderate iodine deficiency may be present; thus, the authors analyzed whether this abnormality was more frequent in endemic, iodine-deficient areas. However, no statistically notable difference was found. The mean TSH level value was 2.8 ± 0.6 mIU/L, significantly higher than in the control group (1.9 ± 0.5 mIU/L). Moreover, average fT3 serum value was significantly higher (5.5 ± 0.8 vs 4.5 ± 1.1 pmol/L). In most cases, a compensatory hypertrophy of the right lobe was described relative to half of the normal total thyroid volume. The results of a study of 40 adult patients living in Poland, currently regarded as an iodine-sufficient region (18), are of interest in this regard. The mean age of enrolled subjects was 37.4 ± 17.1 years. The average TSH and fT3 serum concentrations were significantly higher in the study group than in the bilobed controls (2.37 ± 1.4 mIU/L vs 1.23 ± 0.7 mIU/L and 6.26 ± 0.8 pmol/L vs. 4.88 ± 2.1 pmol/L, respectively). The observed noteworthy increase in fT3 level accompanied by a higher TSH level, without changes in free T4 level (resulting in higher fT3/free T4 ratio), could result from either increased thyroidal release of T3 due to the higher TSH levels or to augmented peripheral monodeiodination of T4. Because hormonal balance appears maintained in most patients, THA has been considered as a benign condition requiring no treatment. That belief and practice was based mostly on studies conducted in the pediatric population. However, studies in adult patients describe initial increments in TSH triggering an age-dependent increase in thyroid volume that is subsequently followed by gradual declines in serum TSH. Such a sequence of events is a proposed hypothesis based on cross-sectional data from large cohort studies on patients with THA. Given this scenario, one would expect a compensatory hypertrophy of the single lobe in a majority of cases as a direct result of chronic thyroidal overstimulation by an increased TSH serum level. Consistent with this hypothesis is one large case-control study that observed a significantly higher incidence of concomitant thyroid disorders in patients with THA than in a control group, the most frequent associated pathologies being thyroid nodules and autoimmune thyroid disease (18). Subjects with THA and underlying thyroid autoimmunity may present with either thyroid functional state. In general, hypothyroidism has been reported significantly more often among patients with THA in comparison with control subjects with normal thyroid function, whereas hyperthyroidism has occurred with a comparable frequency in both groups. Simple goiter and nonautoimmune subclinical hypothyroidism have also been observed, and of 40 subjects analyzed, 26 (∼65%) were euthyroid (18).
Based on a study of 65 patients with THA, it was concluded that subjects with THA constitute a unique in vivo model of long-term elevation of serum TSH with the chronic overstimulation resulting in thyroid tissue hyperplasia and goiter. It has also been suggested that constant TSH stimulation may be the trigger for development of autoimmune thyroid disease in the group described (37). Similarly, in a recent literature summary on THA, the most frequent disorders accompanying THA were hypothyroidism, hyperthyroidism, and goiter (38). The TSH response to thyrotropin-releasing hormone administration was seen to be exaggerated in patients with THA, suggesting underlying subclinical hypothyroidism (39). However, other studies suggest that the higher prevalence of thyroid disorders in subjects with THA is overestimated. Gursoy et al. (28) have suggested that this may be related to a selection bias, because all subjects included would have been referred to thyroid clinics for some reason. Although patients with THA may be more prone to developing thyroid diseases than the population at large, coexisting thyroid pathology is not necessarily an obligate finding (25, 28). Selection bias aside, we remain impressed by the volume of published case reports describing coexistence of THA with other thyroid disorders (Table 1) (18, 24, 28, 33, 36, 37, 40–90).
Thyroid Disorders Associated With THA
| Type of Associated Thyroidal Disorders | Associated Disorder | Total Number of Patients Reported |
|---|---|---|
| Autoimmune thyroid diseases or thyroiditis | Graves disease (18, 28, 40–58) | 38 |
| Hashimoto thyroiditis (18, 24, 43, 49, 55, 59–62) | 30 | |
| Postpartum silent thyroiditis/subacute thyroiditis (63, 64) | 2 | |
| Goiter | Toxic nodular goiter (18, 28, 50, 65) | 5 |
| Simple goiter (40, 66) | 3 | |
| Multinodular nontoxic goiter (18, 28, 37, 49, 67–74) | 111 | |
| Differentiated thyroid cancer | Papillary thyroid carcinoma (36, 41, 75–80) | 11 |
| Follicular carcinoma (40) | 1 | |
| Mixed papillary and follicular carcinoma (81) | 1 | |
| Other thyroid cancers | Medullary thyroid carcinoma (80) | 1 |
| Developmental and congenital disorders | Thyroglossal duct cyst (82, 83) | 2 |
| Ectopic thyroid (84–88) | 5 | |
| Congenital hypothyroidism (33, 89, 90) | 11 |
| Type of Associated Thyroidal Disorders | Associated Disorder | Total Number of Patients Reported |
|---|---|---|
| Autoimmune thyroid diseases or thyroiditis | Graves disease (18, 28, 40–58) | 38 |
| Hashimoto thyroiditis (18, 24, 43, 49, 55, 59–62) | 30 | |
| Postpartum silent thyroiditis/subacute thyroiditis (63, 64) | 2 | |
| Goiter | Toxic nodular goiter (18, 28, 50, 65) | 5 |
| Simple goiter (40, 66) | 3 | |
| Multinodular nontoxic goiter (18, 28, 37, 49, 67–74) | 111 | |
| Differentiated thyroid cancer | Papillary thyroid carcinoma (36, 41, 75–80) | 11 |
| Follicular carcinoma (40) | 1 | |
| Mixed papillary and follicular carcinoma (81) | 1 | |
| Other thyroid cancers | Medullary thyroid carcinoma (80) | 1 |
| Developmental and congenital disorders | Thyroglossal duct cyst (82, 83) | 2 |
| Ectopic thyroid (84–88) | 5 | |
| Congenital hypothyroidism (33, 89, 90) | 11 |
Thyroid Disorders Associated With THA
| Type of Associated Thyroidal Disorders | Associated Disorder | Total Number of Patients Reported |
|---|---|---|
| Autoimmune thyroid diseases or thyroiditis | Graves disease (18, 28, 40–58) | 38 |
| Hashimoto thyroiditis (18, 24, 43, 49, 55, 59–62) | 30 | |
| Postpartum silent thyroiditis/subacute thyroiditis (63, 64) | 2 | |
| Goiter | Toxic nodular goiter (18, 28, 50, 65) | 5 |
| Simple goiter (40, 66) | 3 | |
| Multinodular nontoxic goiter (18, 28, 37, 49, 67–74) | 111 | |
| Differentiated thyroid cancer | Papillary thyroid carcinoma (36, 41, 75–80) | 11 |
| Follicular carcinoma (40) | 1 | |
| Mixed papillary and follicular carcinoma (81) | 1 | |
| Other thyroid cancers | Medullary thyroid carcinoma (80) | 1 |
| Developmental and congenital disorders | Thyroglossal duct cyst (82, 83) | 2 |
| Ectopic thyroid (84–88) | 5 | |
| Congenital hypothyroidism (33, 89, 90) | 11 |
| Type of Associated Thyroidal Disorders | Associated Disorder | Total Number of Patients Reported |
|---|---|---|
| Autoimmune thyroid diseases or thyroiditis | Graves disease (18, 28, 40–58) | 38 |
| Hashimoto thyroiditis (18, 24, 43, 49, 55, 59–62) | 30 | |
| Postpartum silent thyroiditis/subacute thyroiditis (63, 64) | 2 | |
| Goiter | Toxic nodular goiter (18, 28, 50, 65) | 5 |
| Simple goiter (40, 66) | 3 | |
| Multinodular nontoxic goiter (18, 28, 37, 49, 67–74) | 111 | |
| Differentiated thyroid cancer | Papillary thyroid carcinoma (36, 41, 75–80) | 11 |
| Follicular carcinoma (40) | 1 | |
| Mixed papillary and follicular carcinoma (81) | 1 | |
| Other thyroid cancers | Medullary thyroid carcinoma (80) | 1 |
| Developmental and congenital disorders | Thyroglossal duct cyst (82, 83) | 2 |
| Ectopic thyroid (84–88) | 5 | |
| Congenital hypothyroidism (33, 89, 90) | 11 |
Thus, once a diagnosis of THA is made, the next step is to assess the thyroid functional state as well as ruling out associated neoplasia. In the case of underlying autoimmune thyroid disease, measurement of antithyroid autoantibodies (antithyroperoxidase and antithyroglobulin) are indicated in hypothyroid or goitrous subjects, and elevated titers will be found frequently. In the setting of either thyroidal hyperfunction (based upon elevated T4 and T3 and suppressed TSH) or clinical stigmata of Graves disease, measurement of anti-TSH receptor antibodies should be considered. In either case, there is a need for careful follow-up of patients affected by THA, because of higher risk of development of thyroid dysfunction due to limited functional reserve of the unilobate thyroid and/or presumably increased incidence of antithyroid autoantibodies.
Coexisting Extrathyroidal Pathologies
As is apparent from Table 1, disorders associated with THA largely have been related to the thyroid gland, but some cases of superimposed parathyroid abnormalities have also been reported. These are mostly coexisting parathyroid adenomas on the ipsilateral side of THA and have been single or even double. Primary hyperparathyroidism due to parathyroid hyperplasia has also been described (Table 2) (89, 91–108). One unusual presentation involved both adenoma and hyperplasia of the parathyroid glands accompanied by Hashimoto thyroiditis. The patient was a 66-year-old female who presented with elevated serum levels of parathormone as well as antithyroglobulin antibodies and antithyroperoxidase antibodies. Scintigraphy with technetium 99mTc–methoxyisobutyl isonitrile confirmed the diagnosis of right THA, and a left lower hyperplastic parathyroid and a right upper parathyroid adenoma were subsequently identified when she underwent thyroidectomy for a tumor observed on ultrasound examination (97).
Extrathyroidal Disorders Reported To Be Associated With THA
| Type of Extrathyroidal Disorders | Associated Disorder | Total Number of Patients Reported |
|---|---|---|
| Parathyroid disorders | Ipsilateral parathyroid adenoma (91–94) | 4 |
| Contralateral parathyroid adenoma (95, 96) | 2 | |
| Primary parathyroid hyperplasia (97, 98) | 3 | |
| Congenital disorders | Right aortic arch (99) | 1 |
| Down syndrome (89) | 1 | |
| Marfan syndrome (100) | 1 | |
| Williams syndrome (101) | 1 | |
| Dysmorphic face with short stature (102) | 1 | |
| Fourth branchial cleft cyst (103) | 1 | |
| Brain cavernoma and pituitary Rathke cleft cyst (104) | 1 | |
| Familial dilated cardiomyopathy and hypergonadotrophic hypogonadism (105) | 1 | |
| Other | Neonatal lingual choristoma (106) | 1 |
| Pituitary adenoma (107) | 1 | |
| Autoimmune polyglandular syndrome type III (108) | 1 |
| Type of Extrathyroidal Disorders | Associated Disorder | Total Number of Patients Reported |
|---|---|---|
| Parathyroid disorders | Ipsilateral parathyroid adenoma (91–94) | 4 |
| Contralateral parathyroid adenoma (95, 96) | 2 | |
| Primary parathyroid hyperplasia (97, 98) | 3 | |
| Congenital disorders | Right aortic arch (99) | 1 |
| Down syndrome (89) | 1 | |
| Marfan syndrome (100) | 1 | |
| Williams syndrome (101) | 1 | |
| Dysmorphic face with short stature (102) | 1 | |
| Fourth branchial cleft cyst (103) | 1 | |
| Brain cavernoma and pituitary Rathke cleft cyst (104) | 1 | |
| Familial dilated cardiomyopathy and hypergonadotrophic hypogonadism (105) | 1 | |
| Other | Neonatal lingual choristoma (106) | 1 |
| Pituitary adenoma (107) | 1 | |
| Autoimmune polyglandular syndrome type III (108) | 1 |
Extrathyroidal Disorders Reported To Be Associated With THA
| Type of Extrathyroidal Disorders | Associated Disorder | Total Number of Patients Reported |
|---|---|---|
| Parathyroid disorders | Ipsilateral parathyroid adenoma (91–94) | 4 |
| Contralateral parathyroid adenoma (95, 96) | 2 | |
| Primary parathyroid hyperplasia (97, 98) | 3 | |
| Congenital disorders | Right aortic arch (99) | 1 |
| Down syndrome (89) | 1 | |
| Marfan syndrome (100) | 1 | |
| Williams syndrome (101) | 1 | |
| Dysmorphic face with short stature (102) | 1 | |
| Fourth branchial cleft cyst (103) | 1 | |
| Brain cavernoma and pituitary Rathke cleft cyst (104) | 1 | |
| Familial dilated cardiomyopathy and hypergonadotrophic hypogonadism (105) | 1 | |
| Other | Neonatal lingual choristoma (106) | 1 |
| Pituitary adenoma (107) | 1 | |
| Autoimmune polyglandular syndrome type III (108) | 1 |
| Type of Extrathyroidal Disorders | Associated Disorder | Total Number of Patients Reported |
|---|---|---|
| Parathyroid disorders | Ipsilateral parathyroid adenoma (91–94) | 4 |
| Contralateral parathyroid adenoma (95, 96) | 2 | |
| Primary parathyroid hyperplasia (97, 98) | 3 | |
| Congenital disorders | Right aortic arch (99) | 1 |
| Down syndrome (89) | 1 | |
| Marfan syndrome (100) | 1 | |
| Williams syndrome (101) | 1 | |
| Dysmorphic face with short stature (102) | 1 | |
| Fourth branchial cleft cyst (103) | 1 | |
| Brain cavernoma and pituitary Rathke cleft cyst (104) | 1 | |
| Familial dilated cardiomyopathy and hypergonadotrophic hypogonadism (105) | 1 | |
| Other | Neonatal lingual choristoma (106) | 1 |
| Pituitary adenoma (107) | 1 | |
| Autoimmune polyglandular syndrome type III (108) | 1 |
Apart from these unusual cases with concomitant parathyroid disease, the calcium-phosphate balance in THA subjects is not in any way disturbed. The biochemical parameters (total calcium, parathormone, and calcitonin) do not differ significantly between patients with THA and controls with a normal thyroid. However, abundance of the thyroid C cells was observed in patients with THA in comparison with controls. Conceivably, an increased proportion of parafollicular cells might be observed in case of a reduced number of follicular cells in a patient with THA (40, 109). It remains uncertain whether thyroid lobe agenesis influences ipsilateral parathyroid development, but support for this thesis arose from descriptions of missing parathyroid glands and reduced thyroid artery development ipsilateral to THA (95, 99, 110). On the contrary, the normal presence of two parathyroid glands ipsilateral to THA has been reported (97, 111). Based on an analysis of nine previously reported cases of THA accompanied by hyperparathyroidism, and because the embryologic pathways of the thyroid and parathyroid glands differ, it is suggested that parathyroid exploration should follow standard surgical recommendations for primary hyperparathyroidism (96).
Familial Clustering of THA and Other Thyroid Developmental Anomalies
Although the majority of reported cases of THA have been sporadic, familial clustering of the anomaly has also been reported. Six of 40 (15%) patients with THA described by Szczepanek et al. (112) belonged to three families; THA of the same side was detected in a mother and daughter, brother and sister, as well as in two sisters out of five siblings. Rajmil et al. (39) diagnosed THA in two sisters, whereas Kizys et al. (113) described the anomaly in a father and his son. These familial cases have been taken to suggest at least a partial genetic basis for THA. Moreover, Castanet et al. (67) observed that THA might be associated with an increased incidence of other thyroid developmental abnormalities. In 9 out of 22 (41%) patients with THA, their first-degree relatives presented different forms of thyroid dysgenesis, including: thyroid ectopy (four times), thyroid agenesis (once), THA (two times), and thyroglossal duct cyst in two subjects (67). The exact prevalence of thyroid developmental anomalies in relatives of subjects with THA is likely to be underestimated due to the asymptomatic nature of these abnormalities and would not be diagnosed unless ultrasound screening was performed. Other observations of interest include the report of three cases of THA in a group of 241 first-degree relatives of 84 patients diagnosed with congenital hypothyroidism due to thyroid dysgenesis (114) and a report of five siblings, of whom two had thyroid ectopy, and one had THA (115). Although the coincidence of THA and other thyroid dysgenesis in families suggests a genetic background of the anomaly, we cannot rule out a role of other unknown factors, which may modulate expression of the final phenotype. One of the proposed mechanisms of this discordance is postzygotic mosaicism (116). Alternatively, THA might be one of the manifestations of a broader spectrum of thyroid developmental anomalies, which share common background. Based on reports like that of monozygotic twins, one of whom had thyroid ectopy and the other had THA (117), one could infer that not only genes but also some epigenetic factors or stochastic events during embryogenesis might contribute to the final phenotype of a hemiagenetic thyroid.
Genetic Factors Potentially Involved in Pathogenesis of THA
Common genetic factors are suggested by the occurrence in children with thyroid dysgenesis of an increased risk for cardiac anomalies. A similar rationale holds for the reported coincidence of THA with several genetic conditions, including familial dilated cardiomyopathy and hypergonadotropic hypogonadism, Williams syndrome, and short stature with facial dysmorphia as well as Down syndrome, all of which tend to support the concept of genetic factors contributing to the development of THA (89, 101, 102, 105). Table 3 summarizes the putative genetic alterations thus far analyzed concerning the development of THA in the studies on human and mice (3, 4, 41, 67, 112, 113, 118–124).
Genes Analyzed and Mutations Found Thus Far in Subjects Presenting With the Phenotype of THA
| Author | No. of Subjects Studied | Material | Gene | Potential Role in Thyroid Development | Mutation Found |
|---|---|---|---|---|---|
| Amendola et al. (118) | — | Knockout mice model | TTF1 and PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. | Simultaneous heterozygous deletion of TTF1 and PAX8 |
| Castanet et al. (67) | 22 patients | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | No mutations |
| Tonacchera et al. (119) | 6 patients | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | No mutations |
| Macchia et al. (120) | 1 patient | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | Heterozygous mutation |
| Szczepanek et al. (112) | 36 sporadic and 4 familial cases | Human DNA from peripheral blood | FOXE1 (TTF2) | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | Longer variants (≥16 codons) of FOXE1-polyAla were significantly higher in patients with the familial form of THA in comparison with those with sporadic THA (P = 0.003) and controls (P = 0.005) |
| Budny et al. (121) | 34 sporadic and 2 familial cases | Human DNA from peripheral blood | PAX8, TTF1, TTF2 | No mutations | |
| PAX8, NKX2-1 (TTF1), FOXE (TTF2) | No structural and copy number variations in MLPA analysis | ||||
| HHEX | Encodes transcription factor expressed in the thyroid. Necessary to maintain TTF-1, PAX8, and TTF-2 expression in the developing thyroid. | No mutations | |||
| PSMA1, PSMA3, PSMD3 | Involved in the degradation of ubiquitinated proteins | Duplication (one sporadic patient) and deletion (three sporadic patients) | |||
| PSMD2 | Involved in the degradation of ubiquitinated proteins | Splice site mutation (c.612T>C cDNA.1170T>C, g.3271T>C) in mother and daughter with THA | |||
| VPS13C | Involved in the degradation of ubiquitinated proteins | Deletion (2 sporadic patients) | |||
| RAD23B | Involved in the degradation of ubiquitinated proteins | Deletion (1 sporadic patient) | |||
| Fagman et al. (4, 122) | — | Knockout mice model | Tbx1 or Shh | Genes not expressed in the thyroid bud. Involved is governing symmetric formation of organs | Mice with knockout of Tbx1 or Shh present abnormal development of follicular cells. Their thyroid lacks parafollicular cells probably due to impaired development of the ultimobranchial bodies |
| Kim et al. (123) | 1 patient | Human DNA from peripheral blood | Chromosome region 22q11.2 (contains TBX1 gene) | The same region is deleted in DiGeorge syndrome (associated with multiple organ malformations including parathyroids and thyroid gland; may present with thyroid hypoplasia or hemiagenesis) | Microduplication |
| Kizys et al. (113) | 4 (2 familial and 2 sporadic) | HOXA3, HOXB3, HOXD3 | Encode a class of transcription factors that regulate the regionalization of the embryo along its major axes during development and morphogenesis of several structures. Expressed in the foregut in the early embryo and have been related to thyroid organogenesis. | Polymorphisms in the HOXB3 (rs2229304), HOXD3 (rs34729309, rs1051929, c.543-199G>T and c.543-34G>A; and a new synonymous variant, NP_008829.3:p.314;C>G) but no deleterious mutations | |
| PITX2 | Plays essential role in embryogenic development and organogenesis. Involved in left-right patterning and the determination of internal organ positioning and asymmetry. | Polymorphism (c.45+76C>T) but no deleterious mutations | |||
| Manley and Capecchi (3) | — | Knockout mice model | Hoxa3 | Encode a class of transcription factors that regulate the regionalization of the embryo along its major axes during development and morphogenesis of several structures. Expressed in the foregut in the early embryo and have been related to thyroid organogenesis. | Mice with knockout of Hoxa3 present with fewer follicular cells as well as absence or hypoplasia of one thyroid lobe |
| Szczepanek-Parulska et al. (124) | 1 patient | Somatic DNA from the thyroid tissue obtained from a patient with THA coexisting with nontoxic multinodular goiter | PAX8 | Transcription factor coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. | A novel variant of PAX8 mRNA was identified, which characterizes with an extension of the 5′ untranslated region of the second exon by 97 nucleotides |
| Campenni et al. (41) | 1 patient | Somatic DNA from the thyroid tissue obtained from a patient with THA coexisting with Graves disease and papillary thyroid cancer | PAX8 and TSHR genes | PAX8 is a transcription factor coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. TSHR is expressed in thyroid follicular cells, crucial for TSH-mediated thyroid cells proliferation | No mutations detected |
| Author | No. of Subjects Studied | Material | Gene | Potential Role in Thyroid Development | Mutation Found |
|---|---|---|---|---|---|
| Amendola et al. (118) | — | Knockout mice model | TTF1 and PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. | Simultaneous heterozygous deletion of TTF1 and PAX8 |
| Castanet et al. (67) | 22 patients | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | No mutations |
| Tonacchera et al. (119) | 6 patients | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | No mutations |
| Macchia et al. (120) | 1 patient | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | Heterozygous mutation |
| Szczepanek et al. (112) | 36 sporadic and 4 familial cases | Human DNA from peripheral blood | FOXE1 (TTF2) | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | Longer variants (≥16 codons) of FOXE1-polyAla were significantly higher in patients with the familial form of THA in comparison with those with sporadic THA (P = 0.003) and controls (P = 0.005) |
| Budny et al. (121) | 34 sporadic and 2 familial cases | Human DNA from peripheral blood | PAX8, TTF1, TTF2 | No mutations | |
| PAX8, NKX2-1 (TTF1), FOXE (TTF2) | No structural and copy number variations in MLPA analysis | ||||
| HHEX | Encodes transcription factor expressed in the thyroid. Necessary to maintain TTF-1, PAX8, and TTF-2 expression in the developing thyroid. | No mutations | |||
| PSMA1, PSMA3, PSMD3 | Involved in the degradation of ubiquitinated proteins | Duplication (one sporadic patient) and deletion (three sporadic patients) | |||
| PSMD2 | Involved in the degradation of ubiquitinated proteins | Splice site mutation (c.612T>C cDNA.1170T>C, g.3271T>C) in mother and daughter with THA | |||
| VPS13C | Involved in the degradation of ubiquitinated proteins | Deletion (2 sporadic patients) | |||
| RAD23B | Involved in the degradation of ubiquitinated proteins | Deletion (1 sporadic patient) | |||
| Fagman et al. (4, 122) | — | Knockout mice model | Tbx1 or Shh | Genes not expressed in the thyroid bud. Involved is governing symmetric formation of organs | Mice with knockout of Tbx1 or Shh present abnormal development of follicular cells. Their thyroid lacks parafollicular cells probably due to impaired development of the ultimobranchial bodies |
| Kim et al. (123) | 1 patient | Human DNA from peripheral blood | Chromosome region 22q11.2 (contains TBX1 gene) | The same region is deleted in DiGeorge syndrome (associated with multiple organ malformations including parathyroids and thyroid gland; may present with thyroid hypoplasia or hemiagenesis) | Microduplication |
| Kizys et al. (113) | 4 (2 familial and 2 sporadic) | HOXA3, HOXB3, HOXD3 | Encode a class of transcription factors that regulate the regionalization of the embryo along its major axes during development and morphogenesis of several structures. Expressed in the foregut in the early embryo and have been related to thyroid organogenesis. | Polymorphisms in the HOXB3 (rs2229304), HOXD3 (rs34729309, rs1051929, c.543-199G>T and c.543-34G>A; and a new synonymous variant, NP_008829.3:p.314;C>G) but no deleterious mutations | |
| PITX2 | Plays essential role in embryogenic development and organogenesis. Involved in left-right patterning and the determination of internal organ positioning and asymmetry. | Polymorphism (c.45+76C>T) but no deleterious mutations | |||
| Manley and Capecchi (3) | — | Knockout mice model | Hoxa3 | Encode a class of transcription factors that regulate the regionalization of the embryo along its major axes during development and morphogenesis of several structures. Expressed in the foregut in the early embryo and have been related to thyroid organogenesis. | Mice with knockout of Hoxa3 present with fewer follicular cells as well as absence or hypoplasia of one thyroid lobe |
| Szczepanek-Parulska et al. (124) | 1 patient | Somatic DNA from the thyroid tissue obtained from a patient with THA coexisting with nontoxic multinodular goiter | PAX8 | Transcription factor coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. | A novel variant of PAX8 mRNA was identified, which characterizes with an extension of the 5′ untranslated region of the second exon by 97 nucleotides |
| Campenni et al. (41) | 1 patient | Somatic DNA from the thyroid tissue obtained from a patient with THA coexisting with Graves disease and papillary thyroid cancer | PAX8 and TSHR genes | PAX8 is a transcription factor coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. TSHR is expressed in thyroid follicular cells, crucial for TSH-mediated thyroid cells proliferation | No mutations detected |
Abbreviation: MLPA, multiplex ligation-dependent probe amplification.
Genes Analyzed and Mutations Found Thus Far in Subjects Presenting With the Phenotype of THA
| Author | No. of Subjects Studied | Material | Gene | Potential Role in Thyroid Development | Mutation Found |
|---|---|---|---|---|---|
| Amendola et al. (118) | — | Knockout mice model | TTF1 and PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. | Simultaneous heterozygous deletion of TTF1 and PAX8 |
| Castanet et al. (67) | 22 patients | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | No mutations |
| Tonacchera et al. (119) | 6 patients | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | No mutations |
| Macchia et al. (120) | 1 patient | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | Heterozygous mutation |
| Szczepanek et al. (112) | 36 sporadic and 4 familial cases | Human DNA from peripheral blood | FOXE1 (TTF2) | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | Longer variants (≥16 codons) of FOXE1-polyAla were significantly higher in patients with the familial form of THA in comparison with those with sporadic THA (P = 0.003) and controls (P = 0.005) |
| Budny et al. (121) | 34 sporadic and 2 familial cases | Human DNA from peripheral blood | PAX8, TTF1, TTF2 | No mutations | |
| PAX8, NKX2-1 (TTF1), FOXE (TTF2) | No structural and copy number variations in MLPA analysis | ||||
| HHEX | Encodes transcription factor expressed in the thyroid. Necessary to maintain TTF-1, PAX8, and TTF-2 expression in the developing thyroid. | No mutations | |||
| PSMA1, PSMA3, PSMD3 | Involved in the degradation of ubiquitinated proteins | Duplication (one sporadic patient) and deletion (three sporadic patients) | |||
| PSMD2 | Involved in the degradation of ubiquitinated proteins | Splice site mutation (c.612T>C cDNA.1170T>C, g.3271T>C) in mother and daughter with THA | |||
| VPS13C | Involved in the degradation of ubiquitinated proteins | Deletion (2 sporadic patients) | |||
| RAD23B | Involved in the degradation of ubiquitinated proteins | Deletion (1 sporadic patient) | |||
| Fagman et al. (4, 122) | — | Knockout mice model | Tbx1 or Shh | Genes not expressed in the thyroid bud. Involved is governing symmetric formation of organs | Mice with knockout of Tbx1 or Shh present abnormal development of follicular cells. Their thyroid lacks parafollicular cells probably due to impaired development of the ultimobranchial bodies |
| Kim et al. (123) | 1 patient | Human DNA from peripheral blood | Chromosome region 22q11.2 (contains TBX1 gene) | The same region is deleted in DiGeorge syndrome (associated with multiple organ malformations including parathyroids and thyroid gland; may present with thyroid hypoplasia or hemiagenesis) | Microduplication |
| Kizys et al. (113) | 4 (2 familial and 2 sporadic) | HOXA3, HOXB3, HOXD3 | Encode a class of transcription factors that regulate the regionalization of the embryo along its major axes during development and morphogenesis of several structures. Expressed in the foregut in the early embryo and have been related to thyroid organogenesis. | Polymorphisms in the HOXB3 (rs2229304), HOXD3 (rs34729309, rs1051929, c.543-199G>T and c.543-34G>A; and a new synonymous variant, NP_008829.3:p.314;C>G) but no deleterious mutations | |
| PITX2 | Plays essential role in embryogenic development and organogenesis. Involved in left-right patterning and the determination of internal organ positioning and asymmetry. | Polymorphism (c.45+76C>T) but no deleterious mutations | |||
| Manley and Capecchi (3) | — | Knockout mice model | Hoxa3 | Encode a class of transcription factors that regulate the regionalization of the embryo along its major axes during development and morphogenesis of several structures. Expressed in the foregut in the early embryo and have been related to thyroid organogenesis. | Mice with knockout of Hoxa3 present with fewer follicular cells as well as absence or hypoplasia of one thyroid lobe |
| Szczepanek-Parulska et al. (124) | 1 patient | Somatic DNA from the thyroid tissue obtained from a patient with THA coexisting with nontoxic multinodular goiter | PAX8 | Transcription factor coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. | A novel variant of PAX8 mRNA was identified, which characterizes with an extension of the 5′ untranslated region of the second exon by 97 nucleotides |
| Campenni et al. (41) | 1 patient | Somatic DNA from the thyroid tissue obtained from a patient with THA coexisting with Graves disease and papillary thyroid cancer | PAX8 and TSHR genes | PAX8 is a transcription factor coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. TSHR is expressed in thyroid follicular cells, crucial for TSH-mediated thyroid cells proliferation | No mutations detected |
| Author | No. of Subjects Studied | Material | Gene | Potential Role in Thyroid Development | Mutation Found |
|---|---|---|---|---|---|
| Amendola et al. (118) | — | Knockout mice model | TTF1 and PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. | Simultaneous heterozygous deletion of TTF1 and PAX8 |
| Castanet et al. (67) | 22 patients | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | No mutations |
| Tonacchera et al. (119) | 6 patients | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | No mutations |
| Macchia et al. (120) | 1 patient | Human DNA from peripheral blood | PAX8 | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | Heterozygous mutation |
| Szczepanek et al. (112) | 36 sporadic and 4 familial cases | Human DNA from peripheral blood | FOXE1 (TTF2) | Transcription factors coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function | Longer variants (≥16 codons) of FOXE1-polyAla were significantly higher in patients with the familial form of THA in comparison with those with sporadic THA (P = 0.003) and controls (P = 0.005) |
| Budny et al. (121) | 34 sporadic and 2 familial cases | Human DNA from peripheral blood | PAX8, TTF1, TTF2 | No mutations | |
| PAX8, NKX2-1 (TTF1), FOXE (TTF2) | No structural and copy number variations in MLPA analysis | ||||
| HHEX | Encodes transcription factor expressed in the thyroid. Necessary to maintain TTF-1, PAX8, and TTF-2 expression in the developing thyroid. | No mutations | |||
| PSMA1, PSMA3, PSMD3 | Involved in the degradation of ubiquitinated proteins | Duplication (one sporadic patient) and deletion (three sporadic patients) | |||
| PSMD2 | Involved in the degradation of ubiquitinated proteins | Splice site mutation (c.612T>C cDNA.1170T>C, g.3271T>C) in mother and daughter with THA | |||
| VPS13C | Involved in the degradation of ubiquitinated proteins | Deletion (2 sporadic patients) | |||
| RAD23B | Involved in the degradation of ubiquitinated proteins | Deletion (1 sporadic patient) | |||
| Fagman et al. (4, 122) | — | Knockout mice model | Tbx1 or Shh | Genes not expressed in the thyroid bud. Involved is governing symmetric formation of organs | Mice with knockout of Tbx1 or Shh present abnormal development of follicular cells. Their thyroid lacks parafollicular cells probably due to impaired development of the ultimobranchial bodies |
| Kim et al. (123) | 1 patient | Human DNA from peripheral blood | Chromosome region 22q11.2 (contains TBX1 gene) | The same region is deleted in DiGeorge syndrome (associated with multiple organ malformations including parathyroids and thyroid gland; may present with thyroid hypoplasia or hemiagenesis) | Microduplication |
| Kizys et al. (113) | 4 (2 familial and 2 sporadic) | HOXA3, HOXB3, HOXD3 | Encode a class of transcription factors that regulate the regionalization of the embryo along its major axes during development and morphogenesis of several structures. Expressed in the foregut in the early embryo and have been related to thyroid organogenesis. | Polymorphisms in the HOXB3 (rs2229304), HOXD3 (rs34729309, rs1051929, c.543-199G>T and c.543-34G>A; and a new synonymous variant, NP_008829.3:p.314;C>G) but no deleterious mutations | |
| PITX2 | Plays essential role in embryogenic development and organogenesis. Involved in left-right patterning and the determination of internal organ positioning and asymmetry. | Polymorphism (c.45+76C>T) but no deleterious mutations | |||
| Manley and Capecchi (3) | — | Knockout mice model | Hoxa3 | Encode a class of transcription factors that regulate the regionalization of the embryo along its major axes during development and morphogenesis of several structures. Expressed in the foregut in the early embryo and have been related to thyroid organogenesis. | Mice with knockout of Hoxa3 present with fewer follicular cells as well as absence or hypoplasia of one thyroid lobe |
| Szczepanek-Parulska et al. (124) | 1 patient | Somatic DNA from the thyroid tissue obtained from a patient with THA coexisting with nontoxic multinodular goiter | PAX8 | Transcription factor coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. | A novel variant of PAX8 mRNA was identified, which characterizes with an extension of the 5′ untranslated region of the second exon by 97 nucleotides |
| Campenni et al. (41) | 1 patient | Somatic DNA from the thyroid tissue obtained from a patient with THA coexisting with Graves disease and papillary thyroid cancer | PAX8 and TSHR genes | PAX8 is a transcription factor coexpressed in the precursors of thyroid follicular cells. Crucial role in the process of differentiation into thyroid cells as well as maintaining proper thyroid function. TSHR is expressed in thyroid follicular cells, crucial for TSH-mediated thyroid cells proliferation | No mutations detected |
Abbreviation: MLPA, multiplex ligation-dependent probe amplification.
Thyroid transcription factors
The first genes suspected to be related to the development of THA were thyroid transcription factors, including TTF1 (NKX2-1), TTF2 (FOXE1), and PAX8. They are expressed in different tissues, but coexpression relates exclusively to thyroid cells. These genes were found to be expressed in the precursors of follicular thyroid cells, and they play a crucial role in the process of cell differentiation (125). Mutations of these genes were found to cause syndromic cases of thyroid dysgenesis, mainly agenesis or ectopy. Mutations in PAX8 and NKX2-5 account for only a minority of patients with severe thyroid developmental abnormalities such as thyroid agenesis, ectopy, or severe hypoplasia (126). Homozygotic deletion of either of these genes in mice leads to thyroid dysgenesis (agenesis, ectopy, or severe hypoplasia). Amendola et al. (118) demonstrated that simultaneous heterozygous deletion of TTF1 and PAX8 also leads to thyroid developmental anomalies. What is interesting is, in the abovementioned study, a particularly high incidence of THA was observed that might suggest a multigenic etiology of the anomaly. Castanet et al. (67) searched for mutations in PAX8 gene and found no potential causative abnormalities in 22 patients with THA. Similar negative results were reported in a study of another group of six patients with THA (119). In contrast, Macchia et al. reported one familial form of THA caused by a heterozygous mutation in PAX8 gene (120). Also, the more frequent occurrence of longer variants (at least 16 codons) of polyalanine tract in FOXE1 gene in patients with the familial form of THA has been pointed out by Szczepanek et al. (112). The tract was longer at a significantly greater frequency in patients with the familial form of THA than in sporadic THA or healthy controls (112). In a recent study by Budny et al. (121), genetic screening of thyroid transcription factors, including PAX8, TTF1, TTF2, and HHEX, revealed no potential causative mutations in a cohort of 36 patients with THA. Additionally, multiplex ligation-dependent probe amplification analysis for structural and copy-number variations in PAX8, NKX2-1, and FOXE1 genes failed to reveal any abnormalities (121).
22q11.21 region including SHH and TBX1 genes
THA has been reported occasionally in patients with DiGeorge or Williams syndrome (101). Of relevance to both of these syndromes is that the affected regions contain genes SHH and TBX1, knockouts of which in experimental studies were shown to cause a THA-alike phenotype in affected mice (4, 122, 127, 128). Animals with the Tbx1 knockout are characterized by abnormal development of follicular cells lacking parafollicular cells, probably due to impaired development of the ultimobranchial bodies (4). However, the presence of parafollicular cells was demonstrated, and the expression of calcitonin and other genes specific for C cells was documented by Ruchala et al. (109) in surgical thyroidectomy specimens. SHH is one of the genes governing symmetric formation of organs. SHH mutations in humans were associated with severe syndromic cases including holoprosencephaly, and no milder forms are known (129). In contrast, Kim et al. (123) reported a case of THA in a patient with microduplication of the region 22q11.2. In contrast, microdeletion of this region was found in 14 out of 30 (47%) of patients diagnosed with thyroid hypoplasia described by Stagi et al. (130); 10 of them presented additionally with congenital heart anomaly.
One of the mechanisms suggested to be potentially involved in the development of a unilobate thyroid might be a deleted TBX1 gene, localized in a 22q11.21 position. Fagman et al. (4) demonstrated that mice with a homozygotic deletion of this gene or Shh present with disturbed formation of a bilobed structure of the thyroid gland. The mice were proposed to be an animal model of THA, although the expression of neither of these genes at any stage of thyroid development was documented (4).
Proteasome genes
The role of this group of genes is degradation of ubiquitinated proteins. Impairment of this process may lead to accumulation of undegraded proteins, leading to toxicity to the cell. Mutations in proteasome genes can be linked to several conditions, including developmental abnormalities as well as neurologic and autoimmune diseases. It is conceivable that even subtle alterations in protein degradation may influence the action of thyroid-specific genes and thereby affect the process of thyroid primordium migration and lobulation. Budny et al. (121) recently applied advanced genomic techniques, including high-resolution microarrays as well as whole-exome sequencing, to further explore the underlying mechanism of THA development. Four genetic defects (one duplication and three deletions) affecting proteasome genes PSMA1, PSMA3, and PSMD3 were detected in four sporadic patients with THA. In addition, in a family in which THA was found to be an inherited disorder, a splice site mutation in the PSMD2 gene (c.612T>C cDNA.1170T>C, g.3271T>C) was found in an affected mother and daughter. The mutation introduces an exonic splicing silencer sequence. Moreover, the proteasome-related pathomechanism potentially contributing to the development of THA is further supported by occurrence of a deletion of VPS13C, as found in two patients with THA and RAD23B in another one patient with THA as both these genes are related to the ubiquitin-proteasome degradation process. These workers speculated that the detected variations influenced the highly dynamic thyroid bilobation process. However, functional analysis is required to confirm those findings. Although recurrent, the detected mutations in proteasome-related genes account for only a small portion of the studied patients with THA, whereas the mechanism remains unknown in the majority (121).
Human homeobox genes
It was recently demonstrated that mice with the knockout of Hoxa3 present with fewer follicular cells as well as absence or hypoplasia of one thyroid lobe (3). Kizys et al. (113) demonstrated lack of mutations in HOXA3, HOXB3, HOXC3, and PITX2 genes in four patients with THA, both sporadic and familial. However, some polymorphic variants were described.
Somatic mutation analysis
There have been few published reports on the somatic genetic alterations found in a hemiagenetic thyroid obtained during thyroidectomy. In the study by Szczepanek-Parulska et al. (124), surgical thyroid specimens obtained from three patients with confirmed THA who were thyroidectomized for unrelated medical reasons were subjected to genetic analysis. A novel variant of PAX8 mRNA was identified, characterized by an extension of the 5′ untranslated region of the second exon by 97 nucleotides. Genomic DNA analysis revealed that an intronic sequence is localized between exon 1 and 2, and 97 bp of its 3′ site were not spliced out but incorporated into a PAX8 transcript. Conceivably, an alternative 3′ acceptor site in the second exon of PAX8 could be designated. The authors speculate that the presence of such an insert may potentially impair binding of mRNA to the ribosome and consequently significantly decrease expression of the PAX8 protein (124). In another study, Campenni et al. (41) performed an extraction of somatic DNA from the thyroid tissue obtained from a patient with THA, in whom THA coexisting with Graves disease and papillary thyroid cancer were diagnosed. However, no mutations in PAX8 and TSH-receptor (TSHR) genes were detected.
Conclusions and Perspectives
To sum up, despite all of the available data, including a host of recent publications, our understanding of THA remains incomplete. Being unable to assess its true population frequency and actual influence on health in general, we are still unaware of the correct clinical approach to affected patients. Patients with THA are prone to develop additional thyroid pathologies and theoretically might benefit from l-thyroxine treatment to lower the thyrotropin levels to that observed in the normal population. However, further research should be done to ascertain whether such intervention early in life would prevent development of associated thyroid conditions. At least, increased vigilance should be maintained to reveal all of the concomitant or associated disorders as soon as possible during follow-up examinations. This is particularly the case concerning the Williams and DiGeorge syndromes for which screening for thyroid gland disorders is justified.
In spite of growing and convincing evidence on the potential genetic background of THA, our knowledge of the factors responsible for the development of a hemiagenetic thyroid is incomplete, and further studies are needed. Having failed to find a common genetic background of the anomaly and discordance of homozygotic twins with thyroid dysgenesis, including THA, it was suggested that non-Mendelian inheritance might contribute to the development of THA. Epigenetic changes, somatic mutations, or even stochastic events occurring at the early steps of embryogenesis might apply (131). The failure to identify mutations in thyroid-specific genes has led investigators to turn to a search for mutations in extrathyroid genes potentially involved in the process of formation of thyroid primordium, such as vascular factors or adhesion molecules (132), and proteasome genes (121). Application of high-throughput technologies (i.e., whole-exome sequencing) enabling a genome-wide search for novel factors involved in thyroid embryogenesis might be the next step that will allow the nature of this complex process to be revealed.
Abbreviations:
- fT3
free triiodothyronine
- T3
triiodothyronine
- T4
thyroxine
- THA
thyroid hemiagenesis
- TSH
thyroid-stimulating hormone.
Acknowledgments
Disclosure Summary: The authors have nothing to disclose.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}