





Abstract
Gastrointestinal stromal tumors (GISTs) are mesenchymal tumors of the gastrointestinal tract arising from the interstitial cells of Cajal. Succinate dehydrogenase (SDH)–deficient GISTs are a unique class of GIST defined by loss of immunohistochemical expression of SDHB, indicating dysfunction of the mitochondrial complex 2; lack of driver mutations in KIT and PDGFRA; and distinctive morphologic features and natural history. To date, all reported SDH-deficient GISTs have arisen in the stomach. We report an SDH-deficient GIST arising in the gastrointestinal tract outside the stomach.
A 29-year-old man with a germline SDHB mutation (p.Arg90*) presented with acute upper gastrointestinal hemorrhage. Endoscopy identified a lesion in the second part of the duodenum, close to the distal common bile duct, consistent with a GIST. Endoscopic ultrasonography and magnetic resonance imaging did not demonstrate metastatic or nodal disease. Open transduodenal excision was performed to remove the tumor. Histologic evaluation confirmed the clinical diagnosis of a GIST, with positive staining for DOG1 and KIT. The mitotic count was low (1 per 50 high-power fields). Immunohistochemistry for SDHB was negative in the presence of an internal control. SDHA expression was retained. No somatic mutations were identified in KIT (exons 9, 11, 13, and 17) or PDGFRA (exons 12, 14, and 18). The germline SDHB mutation and loss of heterozygosity were confirmed on molecular testing of the tumor.
We describe an SDH-deficient GIST occurring outside of the stomach. This case indicates that SDH-deficient GISTs may also arise in the small intestine.
Gastrointestinal stromal tumors (GISTs) arise from the interstitial cells of Cajal and are the most common mesenchymal neoplasm of the gastrointestinal tract, with an estimated annual incidence of up to 14.5 per million (1). Approximately 80% to 85% of GISTs in adults are driven by somatic mutations in KIT or PDGFRA (2). In 2010, a new class of GIST, now known as succinate dehydrogenase (SDH)–deficient GIST, was recognized (3). SDH-deficient GISTs are not associated with KIT or PDGFRA mutation but rather are driven by dysfunction of the SDH subunits: either biallelic mutation/inactivation of SDHA, SDHB, SDHC, or SDHD or epimutation of SDHC (4–6). Loss of immunohistochemical staining for SDHB has proven a robust and reliable marker for SDH-deficient GIST (3). SDH-deficient GISTs demonstrate unique morphologic and clinical features, including a prognosis not predicted by size and mitotic rate, a propensity to metastasize to lymph nodes, and lack of response to imatinib (3, 7, 8). Approximately half of all patients with SDH-deficient GIST will be found to have a germline SDHx mutation, with attendant risk for paraganglioma/pheochromocytoma as the Carney-Stratakis syndrome (4–6); many (if not all) of the remainder are susceptible to the syndromic but nonhereditary Carney triad of paraganglioma, pulmonary chondroma, and GIST (6).
Although GISTs as a group can occur anywhere in the gastrointestinal tract, to date SDH-deficient GISTs have been reported to arise only in the stomach, where they account for 5% to 7.5% of GISTs (3, 4, 7, 8). We report a well-characterized case of an SDH-deficient GIST arising in the gastrointestinal tract outside the stomach.
Case
A 29-year-old man presented with a 3-day history of melena. He had no history of upper gastrointestinal symptoms. He carried a known germline SDHB mutation (c.268C>T, p.Arg90*) diagnosed at age 19 years on the basis of his family history. He had no history of pheochromocytoma/paraganglioma but had a poor record of attendance for follow-up. His magnetic resonance imaging (MRI) scans and plasma free metanephrines 4 years earlier were unremarkable. He had a family history of paraganglioma (maternal aunt and two cousins), whereas his mother, an SDHB carrier, was asymptomatic. There was no known family history of GIST.
Because of ongoing bleeding, urgent gastroscopy was performed. Gastroscopy demonstrated an actively bleeding 2.0 × 1.5–cm lesion in the second part of the duodenum proximal to the intramural portion of the distal common bile duct, compressing the distal common bile duct. Appearances were consistent with a GIST. No other lesions were identified. Endoscopic ultrasonography confirmed the oval, hypoechoic 20 × 15–mm intramural (subepithelial) lesion in the second part of the duodenum, which appeared to originate from within the muscularis propria. There was no evidence of pheochromocytoma/paraganglioma or metastatic or local nodal disease on MRI. Plasma free metanephrine and chromogranin A levels were normal. An open transduodenal excision of the lesion was performed with cholecystectomy and transpapillary stenting of the distal common bile duct. Surgery was uncomplicated.
Histologic evaluation revealed a GIST composed of a mixture of epithelioid and spindle-shaped cells growing in a somewhat lobulated pattern. The mitotic rate was low (<1 mitosis per 50 high-powered fields) [Fig. 1(A) and 1(C)]. Immunohistochemical staining for the GIST-specific markers KIT and DOG1 was diffusely strongly positive. SDHB immunohistochemistry demonstrated complete loss of expression in all neoplastic cells, with preserved staining in nonneoplastic endothelial, epithelial, and stromal cells [Fig. 1(B) and 1(D)], confirming the diagnosis of SDH-deficient GIST. Immunohistochemical staining for SDHA was positive. Mutation testing for KIT (exons 9, 11, 13, and 17) and PDGFRA (exons 12, 14, and 18) performed on formalin-fixed, paraffin-embedded tumor block by next-generation sequencing was negative. SDHB exon 3 Sanger sequencing of DNA extracted from formalin-fixed, paraffin-embedded tumor tissue and compared with sequencing from peripheral blood leukocyte DNA confirmed the germline SDHB mutation (c.268C>T, p.Arg90*) in the tumor, accompanied by loss of the wild-type allele consistent with loss of heterozygosity (Fig. 2).

(a) Hematoxylin and eosin–stained image showing a mixed spindled epithelioid tumor in the deep connective tissue underlying definite duodenal mucosa, characterized by a villous architecture and submucosal Brunner glands (original magnification, ×40). (b) SDHB immunohistochemical staining showing negative staining for the tumor area and positive staining for the epithelial cells (top of section) and surrounding nontumor tissue (original magnification, × 40). (c) Higher-magnification hematoxylin and eosin stain revealing mixed spindle and epithelioid cells characteristic of GIST (original magnification, ×200). (d) SDHB immunohistochemical staining showing negative tumor staining and densely positive SDHB staining of the muscularis propria (original magnification, ×400).
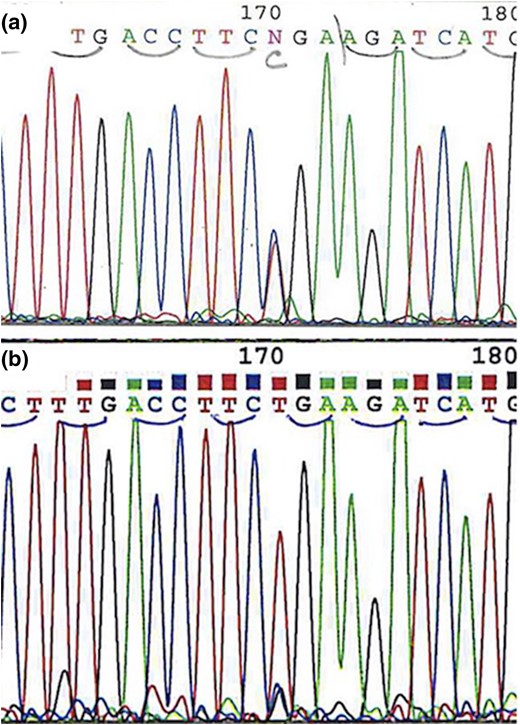
(a) Chromatogram demonstrating germline heterozygous SDHB c.268C>T, p.Arg90* mutation. (b) Chromatogram from the tumor demonstrating the SDHB c.268C>T, p.Arg90* mutation accompanied by loss of the wild type allele consistent with loss of heterozygosity.
The patient consented to undergo investigations, which were performed as part of clinical care and in accordance with the New Zealand National Health Advisory Committee’s Ethical Guidelines for Observational Studies.
Discussion
We report an SDH-deficient GIST occurring in the duodenum. Previously, these have been reported to exclusively occur in the stomach. For example, in a series of 378 nongastric GISTs, none were SDH deficient (7). The clinical presentation with upper gastrointestinal bleeding was typical, with tumors often manifesting with gastrointestinal bleeding or epigastric discomfort (3, 6, 7). Because the duodenum is part of the small bowel, it suggests that, although probably rare, SDH-deficient GISTs may also occur elsewhere in the small intestine. As such, the possibility of nongastric GIST should be considered in an SDHx patient with unexplained iron deficiency or vague gastrointestinal symptoms. Similarly, although we previously considered that a nongastric location almost excluded SDH-deficient GIST, we would now at least consider the diagnosis of SDH deficiency in GISTs arising in other locations if there was suggestive morphologic features, molecular findings (i.e.,KIT and PDGFRA wild type), or clinical features (i.e., multifocality or personal/family history of GIST or paraganglioma) and would recommend SDHB immunohistochemistry.
Although the tumor in this case had low mitotic activity, tumor size and mitotic count do not predict tumor behavior in SDH-deficient GISTs (4, 8). Metastases have been reported up to 42 years after primary tumor removal, and therefore it has been suggested that lifelong follow-up of these patients is needed (7). However, the optimal follow-up is uncertain. For pediatric patients with GIST, it has been suggested that postoperative surveillance be performed by imaging (chest radiography and abdominal/pelvic computed tomography or MRI) every 3 months for the first 2 years, then every 6 months for 2 years, and annually thereafter (1). In the event of recurrent disease, apart from surgical excision, there are limited treatment options because SDH-deficient GISTs typically respond poorly to tyrosine kinase inhibitors and traditional chemotherapy is generally ineffective (4). However, even in the presence of metastatic disease, patients may survive many years because disease is often indolent (3, 4, 6–8). The overall tumor mortality rate in this group of SDH-deficient GISTs is thought to be at least 25% (8).
In the case reported here, the patient had a known SDHB germline mutation. Approximately 20% to 30% of SDH-deficient GISTs have been reported to harbor a germline mutation in SDHB, SDHC, or SDHD, with an additional 30% being associated with germline SDHA mutation (6, 9). Of note, SDH-deficient GISTs not associated with a germline SDHx mutation are still often syndromic, with at least most, and perhaps all, being associated with postzygotic SDHC promoter hypermethylation (i.e.,SDHC epimutation)—the hallmark of the Carney triad of pulmonary chondroma, gastric GIST, and paraganglioma (4, 10).
Therefore, in addition to awareness of the distinct natural history, the diagnosis of SDH-deficient GIST can be a sentinel event for the presence of underlying syndromic disease—either germline SDHx mutation (Carney–Stratakis syndrome) or epimutation (Carney triad). We therefore recommend that such patients be screened for other components of these syndromes (4). Occurrence of other SDH-deficient tumors can be highly asynchronous, with paragangliomas being reported up to 25 years after the diagnosis of GIST, emphasizing the need for lifelong follow-up of patients with germline SDHx mutations (4).
Conclusion
We encountered a case of an SDH-deficient GIST arising in the duodenum, confirming that these tumors do not exclusively arise from the stomach, as previously thought. Pathologists and clinicians should be aware of the possibility that SDH-deficient GISTs may arise outside the stomach because of the distinct natural history and strong syndromic associations of this unique class of GIST.
Abbreviations:
Acknowledgments
We acknowledge Professor John Windsor, pancreatobiliary surgeon, for his surgical expertise in this case.
Disclosure Summary: The authors have nothing to disclose.
References
Author notes
Address all correspondence and requests for reprints to: Marianne S. Elston, PhD, Waikato Clinical Campus, University of Auckland, Waikato Hospital, Private Bag 3200, Hamilton 3240, New Zealand. E-mail: [email protected].

{kind=link}
{kind=link}