





Abstract
Lipodystrophy syndromes are characterized by generalized or partial absence of adipose tissue.
We conducted a systematic review to synthesize data on clinical and metabolic features of lipodystrophy (age at onset, < 18 years).
Sources included Medline, Embase, Cochrane Library, Scopus and Non-Indexed Citations from inception through January 2016.
Search terms included lipodystrophy, and age 0 to 18 years. Patients with unambiguous diagnosis of lipodystrophy were included. Lipodystrophy secondary to HIV treatment was excluded.
We identified 1141 patients from 351 studies. Generalized fat loss involving face, neck, abdomen, thorax, and upper and lower limbs was explicitly reported in 65% to 93% of patients with congenital generalized lipodystrophy (CGL) and acquired generalized lipodystrophy (AGL). In familial partial lipodystrophy (FPL), fat loss occurred from upper and lower limbs, with sparing of face and neck. In acquired partial lipodystrophy (APL), upper limbs were involved while lower limbs were spared. Other features were prominent musculature, acromegaloid, acanthosis nigricans and hepatosplenomegaly. Diabetes mellitus was diagnosed in 48% (n = 222) of patients with CGL (mean age at onset, 5.3 years). Hypertriglyceridemia was observed in CGL, AGL and FPL. Multiple interventions were used, with most patients receiving ≥ 3 interventions and being ≥ 18 years of age at the initiation of interventions.
To our knowledge, this is the largest reported pooled database describing lipodystrophy patients with age at onset < 18 years. We have suggested core and supportive clinical features and summarized data on available interventions, outcomes and mortality.
Lipodystrophy is a group of rare, congenital or acquired syndromes with an estimated prevalence of <1 per million (1). Lipodystrophy is characterized by the generalized or partial absence of adipose tissue, frequently with a muscular appearance (1–7). It is usually associated with evidence of insulin resistance (diabetes mellitus [DM], hypertriglyceridemia, and acanthosis nigricans) and can also be associated with transaminitis, pancreatitis, cardiomyopathy, nephropathy, and leptin deficiency (8–12).
Lipodystrophies are clinically classified as generalized or partial according to the distribution of fat loss and as congenital or acquired. This yields 4 major categories: congenital generalized lipodystrophy (CGL; or Berardinelli-Seip syndrome), acquired generalized lipodystrophy (AGL; or Lawrence syndrome), familial partial lipodystrophy (FPL; or Kobberling syndrome or Dunnigan syndrome), and acquired partial lipodystrophy (APL; or Barraquer-Simons syndrome) (13). Most of the inherited generalized lipodystrophies are autosomal recessive, and most of the inherited partial lipodystrophies are autosomal dominant (13). Various genetic mutations, including AGPAT2, BSCL2, CAV1, PTRF, PPARG, LMNA, AKT2, PLIN1, and ZMPSTE24, have been identified as causes of lipodystrophy (14–22). However, not all patients with lipodystrophy have a confirmed genetic diagnosis, leading to the potential for discovery of novel genes (14, 16, 23). Multiple other syndromes, including mandibuloacral dysplasia, Wiedemann-Rautenstrauch syndrome, Hutchinson-Gilford progeria syndrome, and Donohue syndrome (leprechaunism) have lipodystrophy as a feature of a multisystem disease, and some of these syndromes share common genetic etiologies with the major categories of lipodystrophy described above (19, 22, 24–26).
Management of lipodystrophy is aimed at correcting the associated metabolic abnormalities (27). High-dose insulin, a well-balanced diet, metformin, oral hypoglycemic agents, and lipid-lowering drugs have been used (14, 23, 28). Metreleptin, a recombinant human methionyl leptin, was approved in February 2014 by the US Food and Drug Administration for the treatment of generalized lipodystrophy (29), following evidence of substantial improvement in the metabolic complications of lipodystrophy with metreleptin replacement (7, 14, 30–34). Metreleptin continues to be an experimental drug for partial lipodystrophy.
Owing to the rarity of lipodystrophy syndromes, limited evidence is available regarding their diagnostic criteria and management, in particular, for patients <18 years of age (35). The Pediatric Endocrine Society formed a task force of experts to develop clinical practice guidelines to aid practicing clinicians in the diagnosis and treatment of patients with lipodystrophy in this age group. Consistent with the National Academy of Medicine standards (36), the National Guideline Clearinghouse criteria for inclusion of clinical practice guidelines (37), and Guidelines International Network (38), the Pediatric Endocrine Society commissioned the conduct of the present systematic review to support the development of key recommendations.
In the present systematic review, we synthesized the clinical, metabolic, and outcomes data of 1141 patients with lipodystrophy syndromes. These patients had onset of lipodystrophy at <18 years of age; however, one-third were reported during adulthood. To the best of our knowledge, this is the largest pooled analysis of pediatric-onset lipodystrophy, to date. In the present review, the core and supportive clinical features of various forms of lipodystrophy are presented. The association of metabolic outcomes with different interventions, including metreleptin, and the mortality data from these patients is discussed. Our systematic review, although subject to the reporting biases from each study from which the data were derived, nonetheless represents the most comprehensive review to date on lipodystrophy. It has the advantage of representing, not only experiences from large centers with expertise in lipodystrophy, but also individual cases from small centers, including under-resourced countries.
Materials and Methods
The search methods, eligibility criteria, and outcomes of interest were specified in advance in a protocol developed by the study investigators. The outcomes of interest were chosen by their importance to patients and their necessity for decision making. The clinical outcomes included the distribution of fat, menstrual irregularities, hirsutism, growth, and mortality (39). Metabolic outcomes such as blood glucose, serum insulin, hemoglobin A1c, lipid profile, liver function tests, and leptin were included.
Search methods
An expert reference librarian, following the protocol, conducted an electronic search strategy (Supplemental Table 1). A comprehensive search of databases (Medline, Embase, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, Scopus, and Medline In-Process and Other Non-Indexed Citations) was conducted in any language from the inception of each database through August 31, 2015. We identified additional candidate studies from the reference lists of the eligible primary studies and reviews, and we queried the content experts. We also obtained de-identified individual patient data from the National Institutes of Health (NIH) lipodystrophy database. Additional searches were performed to update the original electronic search up to January 2016.
We used the search terms “lipodystrophy,” “congenital generalized lipodystrophy,” and “familial partial lipodystrophy.” We limited our search to the pediatric age group (age, 0 to 18 years). However, because some of the reports were case series across all age groups, some patients aged >18 years at the time of being reported were captured in our database. Patients aged >18 years were included in our review if they had the onset of lipodystrophy at <18 years of age.
Eligibility criteria
We included original prospective and retrospective studies that reported data on lipodystrophy. Studies that contained a definite description of body fat distribution consistent with that seen in lipodystrophy were selected. Owing to the rarity of lipodystrophy, most data were extracted from case reports and case series. We have reported the clinical and metabolic features, interventions, and mortality of patients with CGL, AGL, FPL, and APL. Rare syndromes, including Donohue syndrome, mandibuloacral dysplasia (type A and type B), Wiedemann-Rautenstrauch syndrome, Hutchinson-Gilford progeria syndrome, SHORT syndrome (short stature, hyperextensibility, hernia, ocular depression, Rieger anomaly, and teething delay), atypical progeroid syndrome, Emery-Dreifuss muscular dystrophy, Brunzell syndrome, CANDLE syndrome (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature), cephalothoracobrachial lipodystrophy, Cockayne syndrome, faciotroncular lipodystrophy, Lafora disease, Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy, marfanoid-progeroid syndrome, neonatal-onset lipodystrophy syndrome, Nicolaides-Baraitser syndrome, Werner syndrome, and mandibular hypoplasia, deafness, and progeroid features, have also been reported.
Exclusion criteria
We excluded lipodystrophy related to HIV treatment, secondary to drug administration (insulin, growth hormone, steroids, antibiotics, and vaccinations), recurrent pressure, infantile centrifugal abdominal lipodystrophy, localized lipodystrophy, and juvenile-onset dermatomyositis. Lipodystrophy secondary to systemic diseases such as uncontrolled diabetes mellitus, thyrotoxicosis, anorexia nervosa, malnutrition, malignancy, and chronic infections were excluded. Cases that were described more than once in the published data were reported only once in the present review. Duplicate reporting of cases was detected by highly meticulous data extraction, including the year of the study, the first author’s surname, name and city of the institution from which the patient had been enrolled, and the coded identifier of each patient, if specified in the report. Adult patients captured in our search were excluded if the onset of lipodystrophy had occurred after 18 years of age.
Selection of studies
Two reviewers working independently considered the potential eligibility of each abstract and title that resulted from the initial search. The full-text versions of the eligible studies were reviewed. Two independent reviewers extracted data from each full-text report using a standardized form. Disagreements were harmonized by consensus and, if not possible, through arbitration by a third reviewer.
Data extraction and management
Using a standardized web-based data extraction form, we extracted the following descriptive data from each study: demographic data (age, sex, ethnicity, and form of lipodystrophy), clinical features, interventions the patients had received, if any (type and duration), measures of outcome (specifically defined as an event or measure and period for the ascertainment of this outcome), and mortality data. We extracted the outcomes of interest at the longest point of complete follow-up. Owing to the heterogeneity in patient description, all clinical and metabolic features were not explicitly reported for each patient. We extracted and reported only unambiguously stated data. Data on clinical features were also extracted from the patient photographs included in a few studies. Evaluation of publication bias was not feasible because of heterogeneity and because most of the included studies were case reports and case series.
Statistical analysis
Descriptive analyses were used to present the patient characteristics. We conducted random-effects meta-analyses of the individual patient data to pool the metabolic outcomes before and after the interventions. Because of the limited number of patients included in each intervention and the lack of standardization of assays across the numerous laboratories included in our review, we were unable to statistically compare the changes in metabolic outcomes. Statistical analysis was performed using Stata, version 14.1 (StataCorp LLP, College Station, TX).
Results
Search results and study description
The initial search identified 1303 studies, of which 351 were eligible (Fig. 1; Supplemental Table 2). The original study design in 219 studies was case report (single patient), 111 were cohort studies (≥2 patients), and 21 studies reported data from the NIH database. The data from the NIH studies were extracted from the individual patient data we received from the NIH and not from the published studies. The systematic review included 1141 patients (219 from case reports, 811 from cohort studies, and 111 from the NIH database). Within each cohort, only those patients who met the inclusion criteria were included. The mean age at reporting of all patients was 15.4 ± 14.0 years (range, 0.01 to 76.5 years). Of the total 1141 patients in our database, 744 were reported at <18 years of age and 397 at ≥18 years of age. The mean age at the onset of lipodystrophy for all patients was 3.1 ± 4.3 years (range, 0.01 to 17.5 years).

The process of study selection. 1We extracted data from studies in Russian, French, Polish, Spanish, Italian, and Portuguese and excluded studies in German, Japanese, Dutch, Hebrew, Hungarian, Korean, Norwegian, and Slovakian. 2Lipodystrophy related to HIV treatment, secondary to drug administration (insulin, growth hormone, steroids, antibiotics, and vaccinations), secondary to recurrent pressure, infantile centrifugal abdominal lipodystrophy, localized lipodystrophy, juvenile-onset dermatomyositis, and secondary to systemic diseases such as uncontrolled DM, thyrotoxicosis, anorexia nervosa, malnutrition, malignancy, and chronic infections were excluded. 3Age at onset of lipodystrophy, <18 y vs ≥18 y.
Congenital generalized lipodystrophy
We identified 519 patients with CGL (Table 1). Most patients were white (32%), followed by Asian (24%), and black (17%). Of 286 patients for whom data were available, 174 (61%) had a history of parental consanguinity. The mean age at onset of fat loss was 0.3 year (range, 0.0 to 12 years). Fat loss from the face and neck, abdomen, thorax, and upper and lower limbs was explicitly reported for 89% to 93% of patients with CGL [Table 1; Fig. 2(A)]. Sixteen patients were specifically reported to have sparing of the palms and soles. DM (commonly without ketosis) was diagnosed in 222 patients (48%) with CGL, with a mean age at onset of 5.3 years (range, 0.1 to 34 years).
Clinical Features of 4 Major Lipodystrophies (CGL, AGL, FPL, APL)
| Clinical Feature | CGL | Patients (n)a | AGL | Patients (n)a | FPL | Patients (n)a | APL | Patients (n)a |
|---|---|---|---|---|---|---|---|---|
| Sex (male/female), %b | 45/55 | 457 | 33/77 | 86 | 17/83 | 124 | 17/83 | 124 |
| Ethnicity (W/H/B/A/O), % | 32/12/17/24/15 | 231 | 72/4/10/10/4 | 69 | 79/3/5/3/10 | 67 | 73/7/0/10/10 | 41 |
| History of consanguinity, % | 61 | 286 | 47 | 15 | 22 | 9 | 31 | 16 |
| Age at onset of fat loss, yc | 0.3 ± 1.5 (0.0–12) | 340 | 5.0 ± 3.5 (0.0–15.0) | 53 | 9.9 ± 5.6 (0.0–16.0) | 16 | 8.2 ± 3.9 (0.5–16) | 106 |
| Fat loss, % (n)d | ||||||||
| Face and neck | 93 (481) | 519 | 74 (64) | 86 | 6 (7) | 124 | 90 (111) | 124 |
| Abdomen | 90 (469) | 519 | 65 (56) | 86 | 19 (23) | 124 | 57 (70) | 124 |
| Thorax | 90 (469) | 519 | 65 (56) | 86 | 9 (11) | 124 | 74 (92) | 124 |
| Upper limbs | 92 (476) | 519 | 73 (63) | 86 | 66 (82) | 124 | 82 (102) | 124 |
| Lower limbs | 89 (467) | 519 | 73 (63) | 86 | 73 (90) | 124 | 14 (17) | 124 |
| Fat sparing, ne | ||||||||
| Face and neck | 0 | 3 | 30 | 2 | ||||
| Abdomen | 0 | 3 | 6 | 11 | ||||
| Upper limb | 0 | 2 | 0 | 1 | ||||
| Lower limb | 0 | 2 | 0 | 28 | ||||
| Palms | 16 | 3 | 1 | 1 | ||||
| Soles | 16 | 2 | 1 | 7 | ||||
| Other features | ||||||||
| Prominent musculature, % (n) | 97 (284) | 294 | 94 (44) | 47 | 67 (26) | 39 | 65 (15) | 23 |
| Infectious illness preceding fat loss onset, % (n) | 31 (15) | 49 | 85 (17) | 20 | 25 (1) | 4 | 67 (16) | 24 |
| Presence of DM, % (n) | 48 (222) | 467 | 70 (46) | 66 | 53 (50) | 95 | 35 (27) | 78 |
| Age at onset of DM, yc | 5.3 ± 5.8 (0.1–34.0) | 146 | 16.2 ± 12.9 (1.3–62.0) | 33 | 24.2 ± 12.7 (8–57) | 35 | 14.8 ± 5.5 (3–22) | 8 |
| Presence of ketosis, % (n) | 5 (3) | 56 | 13 (4) | 30 | 4 (1) | 29 | 0 (0) | 15 |
| Acromegaloid features, % (n) | 76 (174) | 230 | 88 (14) | 16 | 33 (1) | 3 | 23 (3) | 13 |
| Protuberant abdomen, % (n) | 80 (93) | 116 | 88 (14) | 16 | 17 (1) | 6 | 40 (2) | 5 |
| Polycystic ovaries, % (n) | 64 (9) | 14 | 100 (1) | 1 | 100 (8) | 8 | 30 (3) | 10 |
| Menstrual irregularities, % (n) | 61 (14) | 23 | 100 (9) | 9 | 86 (6) | 7 | 38 (5) | 13 |
| Prominent veins, % (n) | 86 (80) | 93 | 88 (22) | 25 | 100 (3) | 3 | 100 (3) | 3 |
| Acanthosis nigricans, % (n) | 71 (141) | 198 | 74 (34) | 46 | 70 (23) | 33 | 13 (6) | 46 |
| Hirsutism, % (n) | 73 (121) | 165 | 57 (9) | 16 | 75 (9) | 12 | 42 (8) | 19 |
| Family history of LD, % (n) | 66 (149) | 224 | 8 (4) | 53 | 62 (51) | 82 | 4 (1) | 28 |
| Hepatomegaly, % (n) | 84 (251) | 299 | 80 (42) | 53 | 54 (7) | 13 | 29 (17) | 58 |
| Splenomegaly, % (n) | 64 (78) | 121 | 51 (18) | 35 | 67 (2) | 3 | 7 (1) | 14 |
| Cardiovascular disease, % (n) | 46 (144) | 313 | 19 (6) | 31 | 6 (3) | 50 | 15 (3) | 20 |
| Pancreatitis, % (n) | 31 (20) | 64 | 21 (6) | 28 | 49 (21) | 43 | 7 (1) | 14 |
| Percentage of body fatf | 7.2 ± 3.2 (1.9–17.7) | 68 | 9.2 ± 4.5 (0.3–27.0) | 28 | 23.1 ± 6.2 (10.0–44.8) | 52 | 21.6 ± 5.9 (12.5–29.0) | 5 |
| Appetite (I/D/NA), % | 54/15/31 | 26 | 29/42/29 | 7 | 0/0/100 | 2 | 0/14/86 | 7 |
| Height velocity (I/D/NA), % | 32/9/59 | 202 | 47/17/36 | 30 | 0/50/50 | 12 | 7/0/93 | 15 |
| Weight (I/D/NA), % | 18/20/62 | 188 | 18/43/39 | 28 | 12/44/44 | 9 | 0/29/71 | 17 |
| Bone age (Ad/De/NA), % | 76/10/14 | 103 | 40/13/47 | 15 | 20/40/40 | 5 | 0/0/100 | 3 |
| Intellectual disability, % (n) | 47 (108) | 229 | 50 (7) | 14 | 43 (3) | 7 | 8 (1) | 12 |
| Autoimmune diseases, % (n) | 6 (4) | 61 | 67 (18) | 27 | 9 (4) | 43 | 31 (19) | 62 |
| Clinical Feature | CGL | Patients (n)a | AGL | Patients (n)a | FPL | Patients (n)a | APL | Patients (n)a |
|---|---|---|---|---|---|---|---|---|
| Sex (male/female), %b | 45/55 | 457 | 33/77 | 86 | 17/83 | 124 | 17/83 | 124 |
| Ethnicity (W/H/B/A/O), % | 32/12/17/24/15 | 231 | 72/4/10/10/4 | 69 | 79/3/5/3/10 | 67 | 73/7/0/10/10 | 41 |
| History of consanguinity, % | 61 | 286 | 47 | 15 | 22 | 9 | 31 | 16 |
| Age at onset of fat loss, yc | 0.3 ± 1.5 (0.0–12) | 340 | 5.0 ± 3.5 (0.0–15.0) | 53 | 9.9 ± 5.6 (0.0–16.0) | 16 | 8.2 ± 3.9 (0.5–16) | 106 |
| Fat loss, % (n)d | ||||||||
| Face and neck | 93 (481) | 519 | 74 (64) | 86 | 6 (7) | 124 | 90 (111) | 124 |
| Abdomen | 90 (469) | 519 | 65 (56) | 86 | 19 (23) | 124 | 57 (70) | 124 |
| Thorax | 90 (469) | 519 | 65 (56) | 86 | 9 (11) | 124 | 74 (92) | 124 |
| Upper limbs | 92 (476) | 519 | 73 (63) | 86 | 66 (82) | 124 | 82 (102) | 124 |
| Lower limbs | 89 (467) | 519 | 73 (63) | 86 | 73 (90) | 124 | 14 (17) | 124 |
| Fat sparing, ne | ||||||||
| Face and neck | 0 | 3 | 30 | 2 | ||||
| Abdomen | 0 | 3 | 6 | 11 | ||||
| Upper limb | 0 | 2 | 0 | 1 | ||||
| Lower limb | 0 | 2 | 0 | 28 | ||||
| Palms | 16 | 3 | 1 | 1 | ||||
| Soles | 16 | 2 | 1 | 7 | ||||
| Other features | ||||||||
| Prominent musculature, % (n) | 97 (284) | 294 | 94 (44) | 47 | 67 (26) | 39 | 65 (15) | 23 |
| Infectious illness preceding fat loss onset, % (n) | 31 (15) | 49 | 85 (17) | 20 | 25 (1) | 4 | 67 (16) | 24 |
| Presence of DM, % (n) | 48 (222) | 467 | 70 (46) | 66 | 53 (50) | 95 | 35 (27) | 78 |
| Age at onset of DM, yc | 5.3 ± 5.8 (0.1–34.0) | 146 | 16.2 ± 12.9 (1.3–62.0) | 33 | 24.2 ± 12.7 (8–57) | 35 | 14.8 ± 5.5 (3–22) | 8 |
| Presence of ketosis, % (n) | 5 (3) | 56 | 13 (4) | 30 | 4 (1) | 29 | 0 (0) | 15 |
| Acromegaloid features, % (n) | 76 (174) | 230 | 88 (14) | 16 | 33 (1) | 3 | 23 (3) | 13 |
| Protuberant abdomen, % (n) | 80 (93) | 116 | 88 (14) | 16 | 17 (1) | 6 | 40 (2) | 5 |
| Polycystic ovaries, % (n) | 64 (9) | 14 | 100 (1) | 1 | 100 (8) | 8 | 30 (3) | 10 |
| Menstrual irregularities, % (n) | 61 (14) | 23 | 100 (9) | 9 | 86 (6) | 7 | 38 (5) | 13 |
| Prominent veins, % (n) | 86 (80) | 93 | 88 (22) | 25 | 100 (3) | 3 | 100 (3) | 3 |
| Acanthosis nigricans, % (n) | 71 (141) | 198 | 74 (34) | 46 | 70 (23) | 33 | 13 (6) | 46 |
| Hirsutism, % (n) | 73 (121) | 165 | 57 (9) | 16 | 75 (9) | 12 | 42 (8) | 19 |
| Family history of LD, % (n) | 66 (149) | 224 | 8 (4) | 53 | 62 (51) | 82 | 4 (1) | 28 |
| Hepatomegaly, % (n) | 84 (251) | 299 | 80 (42) | 53 | 54 (7) | 13 | 29 (17) | 58 |
| Splenomegaly, % (n) | 64 (78) | 121 | 51 (18) | 35 | 67 (2) | 3 | 7 (1) | 14 |
| Cardiovascular disease, % (n) | 46 (144) | 313 | 19 (6) | 31 | 6 (3) | 50 | 15 (3) | 20 |
| Pancreatitis, % (n) | 31 (20) | 64 | 21 (6) | 28 | 49 (21) | 43 | 7 (1) | 14 |
| Percentage of body fatf | 7.2 ± 3.2 (1.9–17.7) | 68 | 9.2 ± 4.5 (0.3–27.0) | 28 | 23.1 ± 6.2 (10.0–44.8) | 52 | 21.6 ± 5.9 (12.5–29.0) | 5 |
| Appetite (I/D/NA), % | 54/15/31 | 26 | 29/42/29 | 7 | 0/0/100 | 2 | 0/14/86 | 7 |
| Height velocity (I/D/NA), % | 32/9/59 | 202 | 47/17/36 | 30 | 0/50/50 | 12 | 7/0/93 | 15 |
| Weight (I/D/NA), % | 18/20/62 | 188 | 18/43/39 | 28 | 12/44/44 | 9 | 0/29/71 | 17 |
| Bone age (Ad/De/NA), % | 76/10/14 | 103 | 40/13/47 | 15 | 20/40/40 | 5 | 0/0/100 | 3 |
| Intellectual disability, % (n) | 47 (108) | 229 | 50 (7) | 14 | 43 (3) | 7 | 8 (1) | 12 |
| Autoimmune diseases, % (n) | 6 (4) | 61 | 67 (18) | 27 | 9 (4) | 43 | 31 (19) | 62 |
Abbreviations: A, Asian; Ad, advanced; B, black; D, decreased; De, delayed; H, Hispanic; I, increased; LD, lipodystrophy; NA, not affected; O, other; W, white.
Total number of patients for whom data were reported in published studies; data not explicitly reported were not included in the analysis.
Percentage of patients for whom data were reported.
Reported as mean ± standard deviation (range).
Reported as the percentage and number of patients for whom this specific site was reported as being involved; loss of fat from palms and soles was explicitly reported in 2% to 10% patients.
Reported as the number of patients for whom this site was explicitly reported as being spared; this does not imply that the site was not spared in the remaining patients. Reported as mean ± standard deviation (range); measurements and reference ranges may vary depending on the method used.
Clinical Features of 4 Major Lipodystrophies (CGL, AGL, FPL, APL)
| Clinical Feature | CGL | Patients (n)a | AGL | Patients (n)a | FPL | Patients (n)a | APL | Patients (n)a |
|---|---|---|---|---|---|---|---|---|
| Sex (male/female), %b | 45/55 | 457 | 33/77 | 86 | 17/83 | 124 | 17/83 | 124 |
| Ethnicity (W/H/B/A/O), % | 32/12/17/24/15 | 231 | 72/4/10/10/4 | 69 | 79/3/5/3/10 | 67 | 73/7/0/10/10 | 41 |
| History of consanguinity, % | 61 | 286 | 47 | 15 | 22 | 9 | 31 | 16 |
| Age at onset of fat loss, yc | 0.3 ± 1.5 (0.0–12) | 340 | 5.0 ± 3.5 (0.0–15.0) | 53 | 9.9 ± 5.6 (0.0–16.0) | 16 | 8.2 ± 3.9 (0.5–16) | 106 |
| Fat loss, % (n)d | ||||||||
| Face and neck | 93 (481) | 519 | 74 (64) | 86 | 6 (7) | 124 | 90 (111) | 124 |
| Abdomen | 90 (469) | 519 | 65 (56) | 86 | 19 (23) | 124 | 57 (70) | 124 |
| Thorax | 90 (469) | 519 | 65 (56) | 86 | 9 (11) | 124 | 74 (92) | 124 |
| Upper limbs | 92 (476) | 519 | 73 (63) | 86 | 66 (82) | 124 | 82 (102) | 124 |
| Lower limbs | 89 (467) | 519 | 73 (63) | 86 | 73 (90) | 124 | 14 (17) | 124 |
| Fat sparing, ne | ||||||||
| Face and neck | 0 | 3 | 30 | 2 | ||||
| Abdomen | 0 | 3 | 6 | 11 | ||||
| Upper limb | 0 | 2 | 0 | 1 | ||||
| Lower limb | 0 | 2 | 0 | 28 | ||||
| Palms | 16 | 3 | 1 | 1 | ||||
| Soles | 16 | 2 | 1 | 7 | ||||
| Other features | ||||||||
| Prominent musculature, % (n) | 97 (284) | 294 | 94 (44) | 47 | 67 (26) | 39 | 65 (15) | 23 |
| Infectious illness preceding fat loss onset, % (n) | 31 (15) | 49 | 85 (17) | 20 | 25 (1) | 4 | 67 (16) | 24 |
| Presence of DM, % (n) | 48 (222) | 467 | 70 (46) | 66 | 53 (50) | 95 | 35 (27) | 78 |
| Age at onset of DM, yc | 5.3 ± 5.8 (0.1–34.0) | 146 | 16.2 ± 12.9 (1.3–62.0) | 33 | 24.2 ± 12.7 (8–57) | 35 | 14.8 ± 5.5 (3–22) | 8 |
| Presence of ketosis, % (n) | 5 (3) | 56 | 13 (4) | 30 | 4 (1) | 29 | 0 (0) | 15 |
| Acromegaloid features, % (n) | 76 (174) | 230 | 88 (14) | 16 | 33 (1) | 3 | 23 (3) | 13 |
| Protuberant abdomen, % (n) | 80 (93) | 116 | 88 (14) | 16 | 17 (1) | 6 | 40 (2) | 5 |
| Polycystic ovaries, % (n) | 64 (9) | 14 | 100 (1) | 1 | 100 (8) | 8 | 30 (3) | 10 |
| Menstrual irregularities, % (n) | 61 (14) | 23 | 100 (9) | 9 | 86 (6) | 7 | 38 (5) | 13 |
| Prominent veins, % (n) | 86 (80) | 93 | 88 (22) | 25 | 100 (3) | 3 | 100 (3) | 3 |
| Acanthosis nigricans, % (n) | 71 (141) | 198 | 74 (34) | 46 | 70 (23) | 33 | 13 (6) | 46 |
| Hirsutism, % (n) | 73 (121) | 165 | 57 (9) | 16 | 75 (9) | 12 | 42 (8) | 19 |
| Family history of LD, % (n) | 66 (149) | 224 | 8 (4) | 53 | 62 (51) | 82 | 4 (1) | 28 |
| Hepatomegaly, % (n) | 84 (251) | 299 | 80 (42) | 53 | 54 (7) | 13 | 29 (17) | 58 |
| Splenomegaly, % (n) | 64 (78) | 121 | 51 (18) | 35 | 67 (2) | 3 | 7 (1) | 14 |
| Cardiovascular disease, % (n) | 46 (144) | 313 | 19 (6) | 31 | 6 (3) | 50 | 15 (3) | 20 |
| Pancreatitis, % (n) | 31 (20) | 64 | 21 (6) | 28 | 49 (21) | 43 | 7 (1) | 14 |
| Percentage of body fatf | 7.2 ± 3.2 (1.9–17.7) | 68 | 9.2 ± 4.5 (0.3–27.0) | 28 | 23.1 ± 6.2 (10.0–44.8) | 52 | 21.6 ± 5.9 (12.5–29.0) | 5 |
| Appetite (I/D/NA), % | 54/15/31 | 26 | 29/42/29 | 7 | 0/0/100 | 2 | 0/14/86 | 7 |
| Height velocity (I/D/NA), % | 32/9/59 | 202 | 47/17/36 | 30 | 0/50/50 | 12 | 7/0/93 | 15 |
| Weight (I/D/NA), % | 18/20/62 | 188 | 18/43/39 | 28 | 12/44/44 | 9 | 0/29/71 | 17 |
| Bone age (Ad/De/NA), % | 76/10/14 | 103 | 40/13/47 | 15 | 20/40/40 | 5 | 0/0/100 | 3 |
| Intellectual disability, % (n) | 47 (108) | 229 | 50 (7) | 14 | 43 (3) | 7 | 8 (1) | 12 |
| Autoimmune diseases, % (n) | 6 (4) | 61 | 67 (18) | 27 | 9 (4) | 43 | 31 (19) | 62 |
| Clinical Feature | CGL | Patients (n)a | AGL | Patients (n)a | FPL | Patients (n)a | APL | Patients (n)a |
|---|---|---|---|---|---|---|---|---|
| Sex (male/female), %b | 45/55 | 457 | 33/77 | 86 | 17/83 | 124 | 17/83 | 124 |
| Ethnicity (W/H/B/A/O), % | 32/12/17/24/15 | 231 | 72/4/10/10/4 | 69 | 79/3/5/3/10 | 67 | 73/7/0/10/10 | 41 |
| History of consanguinity, % | 61 | 286 | 47 | 15 | 22 | 9 | 31 | 16 |
| Age at onset of fat loss, yc | 0.3 ± 1.5 (0.0–12) | 340 | 5.0 ± 3.5 (0.0–15.0) | 53 | 9.9 ± 5.6 (0.0–16.0) | 16 | 8.2 ± 3.9 (0.5–16) | 106 |
| Fat loss, % (n)d | ||||||||
| Face and neck | 93 (481) | 519 | 74 (64) | 86 | 6 (7) | 124 | 90 (111) | 124 |
| Abdomen | 90 (469) | 519 | 65 (56) | 86 | 19 (23) | 124 | 57 (70) | 124 |
| Thorax | 90 (469) | 519 | 65 (56) | 86 | 9 (11) | 124 | 74 (92) | 124 |
| Upper limbs | 92 (476) | 519 | 73 (63) | 86 | 66 (82) | 124 | 82 (102) | 124 |
| Lower limbs | 89 (467) | 519 | 73 (63) | 86 | 73 (90) | 124 | 14 (17) | 124 |
| Fat sparing, ne | ||||||||
| Face and neck | 0 | 3 | 30 | 2 | ||||
| Abdomen | 0 | 3 | 6 | 11 | ||||
| Upper limb | 0 | 2 | 0 | 1 | ||||
| Lower limb | 0 | 2 | 0 | 28 | ||||
| Palms | 16 | 3 | 1 | 1 | ||||
| Soles | 16 | 2 | 1 | 7 | ||||
| Other features | ||||||||
| Prominent musculature, % (n) | 97 (284) | 294 | 94 (44) | 47 | 67 (26) | 39 | 65 (15) | 23 |
| Infectious illness preceding fat loss onset, % (n) | 31 (15) | 49 | 85 (17) | 20 | 25 (1) | 4 | 67 (16) | 24 |
| Presence of DM, % (n) | 48 (222) | 467 | 70 (46) | 66 | 53 (50) | 95 | 35 (27) | 78 |
| Age at onset of DM, yc | 5.3 ± 5.8 (0.1–34.0) | 146 | 16.2 ± 12.9 (1.3–62.0) | 33 | 24.2 ± 12.7 (8–57) | 35 | 14.8 ± 5.5 (3–22) | 8 |
| Presence of ketosis, % (n) | 5 (3) | 56 | 13 (4) | 30 | 4 (1) | 29 | 0 (0) | 15 |
| Acromegaloid features, % (n) | 76 (174) | 230 | 88 (14) | 16 | 33 (1) | 3 | 23 (3) | 13 |
| Protuberant abdomen, % (n) | 80 (93) | 116 | 88 (14) | 16 | 17 (1) | 6 | 40 (2) | 5 |
| Polycystic ovaries, % (n) | 64 (9) | 14 | 100 (1) | 1 | 100 (8) | 8 | 30 (3) | 10 |
| Menstrual irregularities, % (n) | 61 (14) | 23 | 100 (9) | 9 | 86 (6) | 7 | 38 (5) | 13 |
| Prominent veins, % (n) | 86 (80) | 93 | 88 (22) | 25 | 100 (3) | 3 | 100 (3) | 3 |
| Acanthosis nigricans, % (n) | 71 (141) | 198 | 74 (34) | 46 | 70 (23) | 33 | 13 (6) | 46 |
| Hirsutism, % (n) | 73 (121) | 165 | 57 (9) | 16 | 75 (9) | 12 | 42 (8) | 19 |
| Family history of LD, % (n) | 66 (149) | 224 | 8 (4) | 53 | 62 (51) | 82 | 4 (1) | 28 |
| Hepatomegaly, % (n) | 84 (251) | 299 | 80 (42) | 53 | 54 (7) | 13 | 29 (17) | 58 |
| Splenomegaly, % (n) | 64 (78) | 121 | 51 (18) | 35 | 67 (2) | 3 | 7 (1) | 14 |
| Cardiovascular disease, % (n) | 46 (144) | 313 | 19 (6) | 31 | 6 (3) | 50 | 15 (3) | 20 |
| Pancreatitis, % (n) | 31 (20) | 64 | 21 (6) | 28 | 49 (21) | 43 | 7 (1) | 14 |
| Percentage of body fatf | 7.2 ± 3.2 (1.9–17.7) | 68 | 9.2 ± 4.5 (0.3–27.0) | 28 | 23.1 ± 6.2 (10.0–44.8) | 52 | 21.6 ± 5.9 (12.5–29.0) | 5 |
| Appetite (I/D/NA), % | 54/15/31 | 26 | 29/42/29 | 7 | 0/0/100 | 2 | 0/14/86 | 7 |
| Height velocity (I/D/NA), % | 32/9/59 | 202 | 47/17/36 | 30 | 0/50/50 | 12 | 7/0/93 | 15 |
| Weight (I/D/NA), % | 18/20/62 | 188 | 18/43/39 | 28 | 12/44/44 | 9 | 0/29/71 | 17 |
| Bone age (Ad/De/NA), % | 76/10/14 | 103 | 40/13/47 | 15 | 20/40/40 | 5 | 0/0/100 | 3 |
| Intellectual disability, % (n) | 47 (108) | 229 | 50 (7) | 14 | 43 (3) | 7 | 8 (1) | 12 |
| Autoimmune diseases, % (n) | 6 (4) | 61 | 67 (18) | 27 | 9 (4) | 43 | 31 (19) | 62 |
Abbreviations: A, Asian; Ad, advanced; B, black; D, decreased; De, delayed; H, Hispanic; I, increased; LD, lipodystrophy; NA, not affected; O, other; W, white.
Total number of patients for whom data were reported in published studies; data not explicitly reported were not included in the analysis.
Percentage of patients for whom data were reported.
Reported as mean ± standard deviation (range).
Reported as the percentage and number of patients for whom this specific site was reported as being involved; loss of fat from palms and soles was explicitly reported in 2% to 10% patients.
Reported as the number of patients for whom this site was explicitly reported as being spared; this does not imply that the site was not spared in the remaining patients. Reported as mean ± standard deviation (range); measurements and reference ranges may vary depending on the method used.
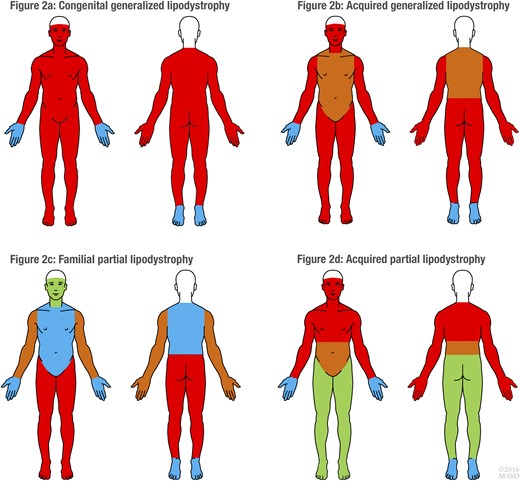
Distribution of adipose tissue in 4 major lipodystrophies (CGL, AGL, FPL, APL). Red, fat loss in >72% patients; brown, fat loss in 57% to 72% patients; blue, fat loss in <57% patients; and green, fat sparing. Please refer to Table 1 for the exact reported frequencies of the sites of fat loss in each of the 4 major lipodystrophies. For the purposes of uniformity in the pictorial representation, frequency cutoffs of >72%, 57% to 72%, and <57% were used.
The other clinical features were prominent musculature (97%), acromegaloid appearance (76%), protuberant abdomen (80%), prominent veins (86%), acanthosis nigricans (71%), hirsutism (73%), hepatomegaly (84%), and advanced bone age (76%). Most patients had a normal height velocity (59%; n = 119) and normal weight (62%; n = 117). The mean percentage of body fat was 7.2% (range, 1.9% to 17.7%), although the measurements and reference ranges for the percentage of body fat could have varied, depending on the method used. Two-thirds of patients (n = 149) reported a family history of a similar physical appearance and/or history of fat loss. Polycystic ovaries and menstrual irregularities were present in 9 and 14 of 14 and 23 female patients, respectively, for whom data on menstruation were reported.
The metabolic features of CGL patients were important for elevated mean serum insulin, hemoglobin A1c, triglycerides, and transaminases (Supplemental Table 3). Leptin deficiency was also noted.
Acquired generalized lipodystrophy
We identified 86 patients with AGL (Table 1). A female predominance (female/male ratio, 2.3) was noted. Most patients were white (72%). The mean age at the onset of fat loss was 5 years (range, 0.0 to 15 years). Progressive loss of fat occurred from the face and neck, abdomen, thorax, and upper and lower limbs in 65% to 74% patients [Table 1; Fig. 2(B)]. Infectious illness preceding the onset of fat loss had occurred in 17 of 20 patients for whom these data were reported. The prevalence of DM was 70% (n = 46), and the mean age at the onset of DM was 16.2 years (range, 1.3 to 62 years).
Similar to CGL, patients with AGL had prominent musculature (94%), acanthosis nigricans (74%), hepatomegaly (80%), and splenomegaly (51%). Data on acromegaloid features, protuberant abdomen, menstrual irregularities, hirsutism, appetite, physical growth, and bone age were available for too few patients to draw conclusions. Also, 18 of 27 patients (67%) had associated autoimmune diseases.
Hypertriglyceridemia was observed in patients with AGL (Supplemental Table 3), and elevated serum insulin, hemoglobin A1c, total cholesterol, and transaminase levels were also reported.
Familial partial lipodystrophy
We identified 124 patients with FPL (Table 1). White race (79%) and a female predominance (83%) were noted. The mean age at the onset of fat loss was 9.9 years (range, 0.0 to 16 years). Fat loss was prominent from the upper and lower limbs (66% and 73% patients, respectively), with sparing of the face and neck explicitly reported for 30 of 124 patients [Fig. 2(C)]. The prevalence of DM was 53%, with a mean age at onset of 24.2 years (range, 8 to 57 years).
The other clinical features were prominent musculature (67%), acanthosis nigricans (70%), and a positive family history of a similar physical appearance and/or history of fat loss (62%). A high prevalence of pancreatitis (49%; n = 21) was reported, likely secondary to hypertriglyceridemia (Supplemental Table 3).
Acquired partial lipodystrophy
We identified 124 patients with APL (Table 1). White race (73%) and a female predominance (83%) were noted. The mean age at the onset of fat loss was 8.2 years (range, 0.5 to 16 years). The most frequent sites of fat loss were face and neck (90%) and upper limbs (82%), followed by the thorax (74%) and abdomen [57%; Fig. 2(D)]. The lower limbs were reportedly spared in 28 of 124 patients. A lower prevalence of DM (35%) was noted compared with the prevalence in AGL (70%). A smaller proportion of patients with APL had acanthosis nigricans (13%), hepatomegaly (29%), and associated autoimmune diseases (31%).
The metabolic abnormalities associated with APL were less severe than those associated with other forms of lipodystrophy, except for the serum blood urea nitrogen, which was elevated to 26 mg/dL (range, 0.2 to 72 mg/dL; Supplemental Table 3).
Other lipodystrophy syndromes
The clinical and metabolic features of the other lipodystrophy syndromes are summarized in Supplemental Tables 4 and 5, respectively. Owing to the small number of reported patients with even rarer and novel lipodystrophy syndromes, these were combined under “Other syndromes” in Supplemental Tables 4 and 5.
Interventions and outcomes
The interventions reported in the included studies were metreleptin, diet, omega-3 polyunsaturated fatty acids and/or fish oil, insulin, metformin, oral hypoglycemic agents, statins, fibrates, nicotinic acid, and plasmapheresis. The association of various interventions (single, double, and ≥3 interventions) with the metabolic outcomes is described in Supplemental Table 6. Most patients were receiving ≥3 interventions and were ≥18 years of age at the initiation of intervention. Owing to the heterogeneity in the dose, route, and duration of treatment and the small sample size, it was difficult to draw statistically significant conclusions regarding the management of lipodystrophy in the included patients. A diet consisting of ~30% fat, ~20% protein, and ~50% carbohydrates, with limited simple carbohydrates, was recommended by most centers. However, the degree to which patients followed the recommendation was not captured systematically in the reported studies. Broadly, metreleptin and diet for a mean duration of 29 months lowered the serum insulin and triglyceride levels in 7 patients with CGL.
Mortality data
Patient age and the cause of mortality for lipodystrophy syndromes are reported in Supplemental Table 7. Of the 502 patients with CGL whose mortality status was known at the time of being reported (mean age at reporting, 12.6 years), 33 were dead. The mean patient age at death in the CGL group was 12.5 years (range, 0.4 to 46.0 years), with respiratory infection the most frequently reported cause of death, followed by cardiac failure. Donohue syndrome resulted in a high mortality rate of 50% (21 of 42 patients dead at reporting) and a relatively early mean age at death (1.2 years; range, 0.03 to 8.3 years), with respiratory infection the most common cause.
Discussion
We conducted a systematic review to summarize the existing data on clinical and metabolic features of non-HIV–related lipodystrophy in children. This in-depth review of the data from 1141 patients with lipodystrophy is the largest pooled database, to the best of our knowledge, reported to date. Because some of the included reports were case series across all age groups, some patients aged >18 years at inclusion into the specific study (although with an age at onset of lipodystrophy of <18 years) were captured in our database. We have suggested the core and supportive clinical features of 4 major lipodystrophies (CGL, AGL, FPL, and APL) to help clinicians in diagnosis and management decisions (Table 2). The features explicitly reported as present or absent in ≥30% to 50% patients for each type of lipodystrophy were examined and categorized as core (if present in ≥50% patients) or supportive (if present in ≥25% patients).
Diagnostic Features of 4 Major Lipodystrophiesa
| Lipodystrophy Type |
|---|
| Congenital generalized lipodystrophy |
| Core features |
| 1. Generalized lack of subcutaneous fat from entire body (with or without sparing of mechanical fat), resulting in prominence of muscles and veins |
| 2. Onset of fat loss usually occurs during infancy (mean age, 0.3 ± 1.5 years; range, 0–12 years) |
| 3. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| Supportive features |
| 1. Acromegaloid features |
| 2. Family history of similar physical appearance and/or history of fat loss in autosomal recessive patternb |
| 3. Hepatomegaly/NAFLD |
| 4. Cardiomyopathy |
| Common associated genes: AGPAT2, BSCL2, PTRF, CAV1 |
| Acquired generalized lipodystrophy |
| Core features |
| 1. Generalized lack of subcutaneous fat from entire body, often without sparing of mechanical fat, resulting in prominence of muscles and veins |
| 2. Onset of fat loss usually occurs during childhood (mean age, 5.0 ± 3.5 y; range, 0–15 y) |
| 3. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| Supportive features |
| 1. Panniculitis or autoimmune disease preceding onset of lipodystrophy |
| 2. Hepatomegaly/NAFLD |
| Familial partial lipodystrophy |
| Core features |
| 1. Regional lack of subcutaneous fat (legs more than arms), resulting in a peripheral muscular appearance, often with fat sparing or fat accumulation in the face and neck |
| 2. Onset of fat loss usually occurs during late childhood through early adolescence (mean age, 9.9 ± 5.6 y; range, 0–16 y) |
| Supportive features |
| 1. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| 2. Family history of similar physical appearance and/or history of fat loss in autosomal dominant patternb |
| 3. Pancreatitis |
| Common associated genes: LMNA, PPARG |
| Acquired partial lipodystrophy |
| Core features |
| 1. Cephalocaudal pattern of subcutaneous fat loss starting in face and variably including the neck, arms, and thorax, with sparing of lower extremities |
| 2. Onset of fat loss usually occurs during late childhood through early adolescence (mean age, 8.2 ± 3.9 y; range, 0.5–16 y) |
| Supportive features |
| 1. Infectious or autoimmune disease preceding onset of lipodystrophy |
| 2. Evidence of membranoproliferative glomerulonephritis |
| Lipodystrophy Type |
|---|
| Congenital generalized lipodystrophy |
| Core features |
| 1. Generalized lack of subcutaneous fat from entire body (with or without sparing of mechanical fat), resulting in prominence of muscles and veins |
| 2. Onset of fat loss usually occurs during infancy (mean age, 0.3 ± 1.5 years; range, 0–12 years) |
| 3. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| Supportive features |
| 1. Acromegaloid features |
| 2. Family history of similar physical appearance and/or history of fat loss in autosomal recessive patternb |
| 3. Hepatomegaly/NAFLD |
| 4. Cardiomyopathy |
| Common associated genes: AGPAT2, BSCL2, PTRF, CAV1 |
| Acquired generalized lipodystrophy |
| Core features |
| 1. Generalized lack of subcutaneous fat from entire body, often without sparing of mechanical fat, resulting in prominence of muscles and veins |
| 2. Onset of fat loss usually occurs during childhood (mean age, 5.0 ± 3.5 y; range, 0–15 y) |
| 3. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| Supportive features |
| 1. Panniculitis or autoimmune disease preceding onset of lipodystrophy |
| 2. Hepatomegaly/NAFLD |
| Familial partial lipodystrophy |
| Core features |
| 1. Regional lack of subcutaneous fat (legs more than arms), resulting in a peripheral muscular appearance, often with fat sparing or fat accumulation in the face and neck |
| 2. Onset of fat loss usually occurs during late childhood through early adolescence (mean age, 9.9 ± 5.6 y; range, 0–16 y) |
| Supportive features |
| 1. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| 2. Family history of similar physical appearance and/or history of fat loss in autosomal dominant patternb |
| 3. Pancreatitis |
| Common associated genes: LMNA, PPARG |
| Acquired partial lipodystrophy |
| Core features |
| 1. Cephalocaudal pattern of subcutaneous fat loss starting in face and variably including the neck, arms, and thorax, with sparing of lower extremities |
| 2. Onset of fat loss usually occurs during late childhood through early adolescence (mean age, 8.2 ± 3.9 y; range, 0.5–16 y) |
| Supportive features |
| 1. Infectious or autoimmune disease preceding onset of lipodystrophy |
| 2. Evidence of membranoproliferative glomerulonephritis |
Abbreviations: AGPAT2, 1-acylglycerol-3-phosphate O-acyltransferase 2; BSCL2, Berardinelli-Seip congenital lipodystrophy type 2; CAV1, gene encoding caveolin 1; LMNA, gene encoding lamin A; NAFLD, nonalcoholic fatty liver disease; PPARG, peroxisome proliferator-activated receptor-γ; PTRF, polymerase I and transcript release factor.
These features should serve as a useful general framework for the clinical diagnosis of lipodystrophy; they do not account for the vast heterogeneity of clinical features seen in various lipodystrophy syndromes. The features explicitly reported as present or absent in ≥30% to 50% of patients for each type of lipodystrophy were examined and categorized as core (if present in ≥50% patients) or supportive (if present in ≥25% patients).
Although CGL and FPL are genetic conditions, the family history can be negative.
Diagnostic Features of 4 Major Lipodystrophiesa
| Lipodystrophy Type |
|---|
| Congenital generalized lipodystrophy |
| Core features |
| 1. Generalized lack of subcutaneous fat from entire body (with or without sparing of mechanical fat), resulting in prominence of muscles and veins |
| 2. Onset of fat loss usually occurs during infancy (mean age, 0.3 ± 1.5 years; range, 0–12 years) |
| 3. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| Supportive features |
| 1. Acromegaloid features |
| 2. Family history of similar physical appearance and/or history of fat loss in autosomal recessive patternb |
| 3. Hepatomegaly/NAFLD |
| 4. Cardiomyopathy |
| Common associated genes: AGPAT2, BSCL2, PTRF, CAV1 |
| Acquired generalized lipodystrophy |
| Core features |
| 1. Generalized lack of subcutaneous fat from entire body, often without sparing of mechanical fat, resulting in prominence of muscles and veins |
| 2. Onset of fat loss usually occurs during childhood (mean age, 5.0 ± 3.5 y; range, 0–15 y) |
| 3. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| Supportive features |
| 1. Panniculitis or autoimmune disease preceding onset of lipodystrophy |
| 2. Hepatomegaly/NAFLD |
| Familial partial lipodystrophy |
| Core features |
| 1. Regional lack of subcutaneous fat (legs more than arms), resulting in a peripheral muscular appearance, often with fat sparing or fat accumulation in the face and neck |
| 2. Onset of fat loss usually occurs during late childhood through early adolescence (mean age, 9.9 ± 5.6 y; range, 0–16 y) |
| Supportive features |
| 1. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| 2. Family history of similar physical appearance and/or history of fat loss in autosomal dominant patternb |
| 3. Pancreatitis |
| Common associated genes: LMNA, PPARG |
| Acquired partial lipodystrophy |
| Core features |
| 1. Cephalocaudal pattern of subcutaneous fat loss starting in face and variably including the neck, arms, and thorax, with sparing of lower extremities |
| 2. Onset of fat loss usually occurs during late childhood through early adolescence (mean age, 8.2 ± 3.9 y; range, 0.5–16 y) |
| Supportive features |
| 1. Infectious or autoimmune disease preceding onset of lipodystrophy |
| 2. Evidence of membranoproliferative glomerulonephritis |
| Lipodystrophy Type |
|---|
| Congenital generalized lipodystrophy |
| Core features |
| 1. Generalized lack of subcutaneous fat from entire body (with or without sparing of mechanical fat), resulting in prominence of muscles and veins |
| 2. Onset of fat loss usually occurs during infancy (mean age, 0.3 ± 1.5 years; range, 0–12 years) |
| 3. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| Supportive features |
| 1. Acromegaloid features |
| 2. Family history of similar physical appearance and/or history of fat loss in autosomal recessive patternb |
| 3. Hepatomegaly/NAFLD |
| 4. Cardiomyopathy |
| Common associated genes: AGPAT2, BSCL2, PTRF, CAV1 |
| Acquired generalized lipodystrophy |
| Core features |
| 1. Generalized lack of subcutaneous fat from entire body, often without sparing of mechanical fat, resulting in prominence of muscles and veins |
| 2. Onset of fat loss usually occurs during childhood (mean age, 5.0 ± 3.5 y; range, 0–15 y) |
| 3. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| Supportive features |
| 1. Panniculitis or autoimmune disease preceding onset of lipodystrophy |
| 2. Hepatomegaly/NAFLD |
| Familial partial lipodystrophy |
| Core features |
| 1. Regional lack of subcutaneous fat (legs more than arms), resulting in a peripheral muscular appearance, often with fat sparing or fat accumulation in the face and neck |
| 2. Onset of fat loss usually occurs during late childhood through early adolescence (mean age, 9.9 ± 5.6 y; range, 0–16 y) |
| Supportive features |
| 1. Evidence of insulin resistance (hyperinsulinemia, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia) |
| 2. Family history of similar physical appearance and/or history of fat loss in autosomal dominant patternb |
| 3. Pancreatitis |
| Common associated genes: LMNA, PPARG |
| Acquired partial lipodystrophy |
| Core features |
| 1. Cephalocaudal pattern of subcutaneous fat loss starting in face and variably including the neck, arms, and thorax, with sparing of lower extremities |
| 2. Onset of fat loss usually occurs during late childhood through early adolescence (mean age, 8.2 ± 3.9 y; range, 0.5–16 y) |
| Supportive features |
| 1. Infectious or autoimmune disease preceding onset of lipodystrophy |
| 2. Evidence of membranoproliferative glomerulonephritis |
Abbreviations: AGPAT2, 1-acylglycerol-3-phosphate O-acyltransferase 2; BSCL2, Berardinelli-Seip congenital lipodystrophy type 2; CAV1, gene encoding caveolin 1; LMNA, gene encoding lamin A; NAFLD, nonalcoholic fatty liver disease; PPARG, peroxisome proliferator-activated receptor-γ; PTRF, polymerase I and transcript release factor.
These features should serve as a useful general framework for the clinical diagnosis of lipodystrophy; they do not account for the vast heterogeneity of clinical features seen in various lipodystrophy syndromes. The features explicitly reported as present or absent in ≥30% to 50% of patients for each type of lipodystrophy were examined and categorized as core (if present in ≥50% patients) or supportive (if present in ≥25% patients).
Although CGL and FPL are genetic conditions, the family history can be negative.
CGL is a rare autosomal recessive disorder (17). Several CGL mutations have been previously identified in genes, including AGPAT2 (CGL type 1), BSCL2 (CGL type 2), CAV1, and PTRF (CGL type 4), with the first 2 accounting for about 95% of reported patients (18, 40–43). These mutations alter gene activation and signaling pathways in differentiation of mesenchymal stem cells into adipocytes (44, 45), resulting in loss of subcutaneous adipose tissue. Although the term “congenital” in CGL implies onset in the neonatal period, and we also noted generalized fat loss in CGL usually during the first 3 years of life, some patients were not identified as experiencing fat loss until early adolescence (15). It is not clear whether this relates to the use of the term “CGL” to describe genetic causes of generalized loss of fat after the first few years of life, such as commonly occurs in FPL, or simply represents a lack of recognition of fat loss until adolescence. The supportive features for CGL included acromegaloid appearance, hepatomegaly (because of hepatic steatosis) (46), protuberant abdomen (due to organomegaly), and cardiomyopathy. The onset of DM was during early childhood through adolescence, except in a group of 10 Brazilian CGL patients homozygous for a 1036 bp deletion in the AGPAT2 gene (BSCL1 locus), as described by Gomes et al. (47) and a single patient in the NIH lipodystrophy database (age at the onset of DM, 34 years). Gomes et al. (47) reported a mean age at onset of DM of 25.67 ± 11.25 years in the AGPAT2 group compared with 11.93 ± 8.29 years in the BSCL2 (669insA Seipin) group (P = 0.007) (47). DM in patients with CGL was ketosis resistant, with greater insulin levels and greater insulin resistance reported in BSCL2 patients than in AGPAT2 patients (47).
AGL was characterized by female predominance. Patients with AGL had a normal fat distribution at birth; however, these patients developed a generalized loss of subcutaneous adipose tissue during childhood or puberty. Preceding infectious illnesses, including Neisseria meningitidis, measles, varicella, and mumps (3, 48, 49), were associated with the onset of AGL. Histologic analysis of subcutaneous adipose tissue in AGL patients has previously revealed panniculitis (50). From a review of 79 patients with AGL, Misra and Garg (3) proposed subclassification of AGL into 3 varieties: type 1, panniculitis variety (25%); type 2, the autoimmune variety (25%); and type 3, the idiopathic variety (50%). In our review, the autoimmune conditions associated with AGL were vitiligo, autoimmune hepatitis, juvenile rheumatoid arthritis, Sjögren syndrome, Hashimoto’s thyroiditis, Graves’ disease, autoimmune hemolytic disease, nonthrombocytopenic purpura, and celiac disease (3, 51, 52). AGL can also be associated with juvenile dermatomyositis, which was not included in our review (53).
Consistent with previous reviews (3, 35), we found that the key distinguishing features between CGL and AGL were a later age at the onset of fat loss, panniculitis, and an onset of DM during adolescence, with somewhat worse dyslipidemia in those with AGL compared with those with CGL. Insulin-resistant DM and a low percentage of body fat were reported in CGL and AGL, which help to distinguish these conditions from type 1 and type 2 DM, respectively (35, 54, 55).
FPL is an autosomal dominant disorder predominantly affecting females and whites (56). At least 5 subtypes of FPL have been described, including FPL type 1 (Kobberling syndrome; no causative genes identified), FPL type 2 (Dunnigan syndrome; from mutations in nuclear lamin A/C encoded by the LMNA gene), FPL type 3 (from mutations in the PPARG gene), FPL type 4 (from mutations in the AKT2 gene), and FPL type 5 (from mutations in the PLIN1 gene) (20, 21, 56, 57). Fat loss in FPL has traditionally been reported to occur around the onset of puberty (35); consistent with that, the mean age at the onset of fat loss in our study was 9.9 years. However, we found that some patients were reported to have experienced fat loss in early childhood. Fat loss commonly involved the upper and lower limbs. The face and neck were spared, leading to a Cushingoid appearance, primarily in FPL resulting from LMNA mutation (58). The physical appearance and metabolic abnormalities were more pronounced in women and developed at an earlier age compared with men in a Spanish family (59). However, other reports described males in German (60) and French (61) families with the metabolic phenotype of FPL and marked hypertriglyceridemia and insulin resistance. The onset of DM in FPL occurred during adulthood and was accompanied by pancreatitis secondary to severe hypertriglyceridemia. The underlying mechanism for dyslipidemia in FPL has not been clearly elucidated.
APL was characterized by a cephalocaudal pattern of subcutaneous fat loss, starting in the face and variably including the neck, arms, and thorax, with sparing of the lower extremities. The onset of fat loss occurred during childhood and rarely during infancy (4). Similar to AGL, preceding infectious illnesses (including measles, varicella, and scarlet fever) and associated autoimmune conditions (including autoimmune hepatitis, scleroderma, idiopathic thrombocytopenic purpura, primary hypothyroidism, rheumatoid arthritis, systemic lupus erythematosus, dermatomyositis, and vitiligo) were reported (4, 62, 63). In both FPL and APL, the mean percentage of body fat was normal, which was reflected by the relatively higher leptin levels and less severe phenotypic and metabolic abnormalities compared with the generalized variety of lipodystrophy.
Using the GRADE (grades of recommendation, assessment, development, and evaluation) approach (64), the current direct evidence supporting the management of lipodystrophy in children warrants lower certainty owing to the methodologic limitations of the available studies (heterogeneity of the case reports and case series) and imprecision (i.e., small number of cases). Indirect evidence (i.e., evidence derived from other relevant conditions, such as evidence on the management of DM or dyslipidemia in children without lipodystrophy) warrants greater certainty and can be extrapolated for decision making, when appropriate. Metreleptin has been approved by the Food and Drug Administration for the treatment of children with CGL and AGL based on improvement in surrogate endpoint data (e.g., triglycerides and hemoglobin A1c) in small numbers of patients; ongoing data collection from pediatric patients treated with metreleptin is needed to determine metreleptin’s effects on morbidity and mortality and its long-term safety and efficacy in this population.
Limitations and strengths
Our findings regarding the clinical and metabolic features of 4 major lipodystrophies are broadly consistent with those of the American Association of Clinical Endocrinologists consensus statement published in 2013 (35). However, in the present comprehensive review, we have provided objective data from patients reported in published studies. We have also described the features of other rarer and novel forms of lipodystrophy.
The major weakness of the present review resulted from biased reporting in the published studies, with likely underreporting of patients with milder manifestations of lipodystrophy. Also, the overall reporting in the included studies was likely biased toward more severe clinical and metabolic features. The features that were more likely to be reported, if present, included history of consanguinity, the sites of fat loss, the presence of prominent musculature, preceding infectious illness, reproductive data, and intellectual disability. Features less likely to be reported, if absent, were the sites of fat sparing, effects on appetite and growth, and associated autoimmune conditions. Additionally, the inclusion of pediatric age range in our search strategy might have excluded some cases of lipodystrophy that were reported as adults but with an onset of symptoms at <18 years of age. Furthermore, because several patients with an onset at <18 years old were reported after they had reached adulthood, some complications of lipodystrophy, and the management of those complications, occurred in adulthood. However, because we restricted our review to patients with an age of onset of <18 years, the ages at which various features of lipodystrophy occurred in our data set might have been systematically biased toward younger ages.
The strengths of the present review relate to the comprehensive nature of literature search and the measures undertaken to reduce the effect of bias and random error, including predefined protocol-driven work, duplicate data extraction and review, and expert contact. The present review represents not only the experience from large centers with expertise in lipodystrophy, but also individual cases from smaller centers and cases reported in 6 non-English languages. Additionally, limiting the present study to cases with an age at onset of <18 years allowed a focused description of pediatric lipodystrophy.
Implications for practice and research
The diagnosis of lipodystrophy in nonobese patients with metabolic aberrations requires a careful history and physical examination, with genetic testing guided by the patient’s specific phenotype. Several genetic mutations have been identified in association with lipodystrophy, each with unique clinical features (Supplemental Table 8). However, some genetic mutations remain to be identified; thus, genetic testing cannot be 100% sensitive, even in patients with inherited forms of lipodystrophy (1, 14, 16, 23). Patients with generalized lipodystrophy have relative leptin deficiency; however, serum leptin cannot be used as a diagnostic criterion because the serum leptin concentrations vary with age, body mass index, sex, nutritional status, and, even, emotional state (65–67). Moreover, the reference ranges for leptin have not yet been standardized (68, 69). Finally, considering the rarity of lipodystrophy syndromes, registry studies might facilitate comprehensive reporting of these patients and help to avoid publication bias and bias in phenotype description.
Abbreviations:
- AGL
acquired generalized lipodystrophy
- APL
acquired partial lipodystrophy
- CGL
congenital generalized lipodystrophy
- DM
diabetes mellitus
- FPL
familial partial lipodystrophy
- NIH
National Institutes of Health
Acknowledgments
The present review was commissioned by the Pediatric Endocrine Society. This work is a publication of the US Department of Agriculture, Agricultural Research Service, Children’s Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine (Houston, TX). The contents of this publication do not necessarily reflect the views or policies of the US Department of Agriculture, nor does the mention of trade names, commercial products, or organizations imply endorsement from the US Government. We thank Larry J. Prokop for expert reference search and Ahmed T. Ahmed for help with data extraction. We also thank Bruno Gonzalez Nolasco and Meinas Elmusharaf for translation of the French reports, Jerzy Bednarski for translation of the Polish reports, and Tahir Suleahria for translation of the Russian reports.
This study was funded by the Pediatric Endocrine Society through an educational grant from Astra Zeneca; the investigators had no contact with Astra Zeneca. This publication was also supported by CTSA Grant UL1 TR000135 from the National Center for Advancing Translational Sciences, a component of the NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Disclosure Summary: The authors have nothing to disclose.
References
U.S. Food and Drug Administration. FDA approves Myalept to treat rare metabolic disease. [New release]. 2014. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm387060.htm. Accessed 15 April 2016.
Institute of Medicine of the National Academies. Clinical Practice Guidelines We Can Trust: Standards for Developing Trustworthy Clinical Practice Guidelines (CPGs). 2011. Available at: http://www.nationalacademies.org/hmd/~/media/Files/Report%20Files/2011/Clinical-Practice-Guidelines-We-Can-Trust/Clinical%20Practice%20Guidelines%202011%20Insert.pdf. Accessed 29 June 2016.
National Guideline Clearinghouse. Criteria for Inclusion of Clinical Practice Guidelines in NGC. 2016. Available at: http://www.guideline.gov/about/inclusion-criteria.aspx. Accessed 29 June 2016.
Laudes M, Oberhauser F, Walgenbach K, Schubert M, Schulte DM, Faust M, Krone W. Comparison of phenotypes in male and female individuals of a new family with Dunnigan type of familial partial lipodystrophy due to a lamin A/C R482W mutation. Horm Metab Res 2009; 41:414–417.
Araujo-Vilar D, Loidi L, Dominguez F, Cabezas-Cerrato J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Horm Metab Res 2003;35:29–35.
Author notes
Address all correspondence and requests for reprints to: Mohammad Hassan Murad, MD, MPH, Evidence-Based Practice Center, Mayo Clinic College of Medicine, 200 First Street Southwest, Rochester, Minnesota 55905. E-mail: [email protected].

{kind=link}
{kind=link}