





Abstract
Juvenile Paget’s disease (JPD), an ultra-rare, debilitating bone disease due to loss of functional osteoprotegerin (OPG), is caused by recessive mutations in TNFRFSF11B. A genotype–phenotype correlation spanning from mild to very severe forms is described.
This study aimed to describe the complexity of the human phenotype of OPG deficiency in more detail and to investigate heterozygous mutation carriers for clinical signs of JPD.
We investigated 3 children with JPD from families of Turkish, German, and Pakistani descent and 19 family members (14 heterozygous).
A new disease-causing 4 bp-duplication in exon 1 was detected in the German patient, and a microdeletion including TNFRFSF11B in the Pakistani patient. Skeletal abnormalities in all affected children included bowing deformities and fractures, contractures, short stature and skull involvement. Complex malformation of the inner ear and vestibular structures (2 patients) resulted in early deafness. Patients were found to be growth hormone deficient (2), displayed nephrocalcinosis (1), and gross motor (3) and mental (1) retardation. Heterozygous family members displayed low OPG levels (12), elevated bone turnover markers (7), and osteopenia (6). Short stature (1), visual impairment (2), and hearing impairment (1) were also present.
Diminished OPG levels cause complex changes affecting multiple organ systems, including pituitary function, in children with JPD and may cause osteopenia in heterozygous family members. Diagnostic and therapeutic measures should aim to address the complex phenotype.
Juvenile Paget’s disease (JPD; Online Mendelian Inheritance in Man entry #239000) is an extremely rare disease characterized by loss of function of osteprotegerin (OPG) (1–3). OPG, which is mainly secreted by osteoblasts, functions as a regulator of bone resorption by neutralizing the receptor activator of nuclear factor κB-ligand (RANKL) and thereby limiting the osteoclast activation due to the receptor activator of nuclear factor κB (RANK) pathway.
Unopposed RANKL action in JPD results in increased stimulation of osteoclast differentiation and action with pathologically elevated bone resorption (4, 5), which cannot be counterbalanced by the bone-forming osteoblasts. As a consequence, the reshaped long bones are widened, deformed, undermineralized, and often unable to sufficiently bear weight. Involvement of the skull is common and described as widening of the diploic space (3, 6, 7).
The skeletal phenotype in patients with JPD is primarily characterized by the bone disease. Serum levels of tissue-specific and bone-specific alkaline phosphatase are up to 10-fold elevated, demonstrating excessive osteoblast activity. In severely affected infants and young children, the painful inflammation of long bones results in gross motor developmental delay or inability to walk. In older children, deformities and fractures may lead to loss of ambulation.
In addition to the skeletal phenotype, most patients with JPD exhibit hearing impairment or complete hearing loss over time. This hearing loss is based on a conductive hearing loss due to misshaped and widened ossicles (8, 9) and on a sensorineural hearing loss, due to degeneration of the acoustic nerve, as observed in OPG deficient animals and cells (10).
Older patients with JPD also develop retinal changes: angioid streaks and calcification of retinal vessels have been described and can lead to vision loss over time (11). Although the pathogenesis of the vascular changes is currently not well understood, the observed calcifications may be the result of excess calcium availability due to the elevated bone turnover.
JPD is inherited in an autosomal recessive way, with most described cases being caused by homozygous mutations in the TNFRSF11B gene on chromosome 8. A genotype–phenotype correlation to some extent has been described (12, 13). Hereby, the variation in severity of the clinical phenotype seems to be determined by the amount of functional circulating OPG. Accordingly, heterozygous family members of patients with JPD, who may display diminished levels of circulating OPG, may be at risk for milder forms of the disease. With this study, we describe the complex clinical phenotype of 3 pediatric JPD patients as well as clinical signs of OPG deficiency in heterozygous family members.
Patients and Methods
Three children with JPD from families of Turkish, German, and Pakistani descent and 19 family members (14 heterozygous) were investigated for this study. Clinic visits were scheduled at the University Hospital Essen (Families 1 and 2) and Birmingham Children’s Hospital (Family 3). Informed consent was obtained from the participants. The ethical committee of the Medical Faculty, University Dusiburg-Essen, Germany, has approved the study (#16-7167-BO).
Childhood development and skeletal findings
Patient 1
Patient 1 initially presented at 3 months of age with a painful inflammation of the right femur, accompanied with elevated serum markers of inflammation (5). Gross motor development was delayed with delayed walking at age 21 months. Treatment with pamidronate commenced at 3 years of age and resulted in pain reduction and developmental progress to age-appropriate motor development. Intermittent treatment with denosumab was discontinued after 2 injections because of severe hypo- and hypercalcemia (5). At the age of 11 years, the patient is fully ambulatory and attends a school for hearing-impaired children. There are no cognitive deficits.
The long bones of the lower extremities showed marked bowing and hyperostosis from the time of first evaluation at the age of 4 months. Long bones appeared demineralized and broadened with a coarse trabecular pattern (5). She sustained two fractures at ages 3 and 7 years. Severe coxa vara developed bilaterally with Shepherd’s crook deformity of the proximal femur and with anterior bowing of both tibiae. The Shepherd’s crook deformity necessitated an osteotomy at age 9 years, which healed without complications.
Currently, she presents with coxa vara bilaterally with an 80° angle and a shortened right leg with 5-cm difference in length. She has a limping gait but is fully ambulatory and does not complain of bone pain. She has developed joint contractures with limitations of movement in both elbows (160°) and the left knee.
Patient 2
Patient 2 was born after an uneventful pregnancy at 34 weeks’ gestation. He was prenatally found to have widened femurs bilaterally. Postnatally widening and inflammation of long bones, especially of the lower extremities, were present, and muscular hypotonia was also noted. He was transferred to pediatric endocrinology at age 12 months. At this time, his gross motor development was delayed corresponding to a 4-month-old child (14, 15). Treatment with Pamidronate commenced and resulted in a swift improvement of his gross motor function. At the age of 3 years, he is able to walk with little support. He attends a regular preschool program with assistance for his hearing impairment and communicates with sign language.
At present, there is nonprogressive, mild bowing of both femurs, and coxa vara on the left side. He sustained two fractures at the ages of 15 months (femur) and 3 years (humerus) and has developed mild contractures of both elbows (170°). The boy was found to have a Chiari I malformation without accompanying hydrocephalus at 12 months of age.
Patient 3
Patient 3 was born at term with a birth weight of 2.92 kg. She presented with heart failure age 3 weeks due to a patent ductus, which was subsequently ligated. Bilateral sensory neural hearing loss was identified at the age of 2 months. She had failure to thrive and developmental delay in her first year. At the age of 11 months, she was noted to have scoliosis and osteopenic bones. She was unable to sit until the age of 18 months, and at the age of 3 years, she could roll but was unable to stand or crawl. She only spoke a few single words at this age. At the age of 4 years, after the removal of many carious teeth, she commenced treatment with intravenous Zoledronate given every 4 months, which was adjusted to every 6 months over the age of 5 years. The parents reported an immediate improvement in that she was no longer sweating and was sleeping better. There was a progressive improvement in her gross motor skills, and at the age of 6 years, she was able to walk with her hands held. She has evidence of microcephaly, and at the age of 4 years, she was identified as having a Chiari malformation with significant cerebellar tonsillar descent. She has evidence of widening of long bones of her upper and lower limbs with fixed flexion deformity of both knees. She has evidence of coxa vara bilaterally.
None of the patients with JPD developed retinal abnormalities at the time of last examination (Patient 1, 11 years; Patient 2, 2 years; and Patient 3, 6 years of age).
Hearing impairment and shape/structure of the inner ear
Patient 1
At 5 years of age, the girl received hearing aids. A detailed analysis of her hearing abilities at 8 years of age showed an air conduction threshold at 75-80-105-90-90 dB (0.25-0.5-1-2-4 kHz) on the right and 100-96-110-90-100 on the left. The bone-conduction threshold was 25-30-40-70-60 dB on the right and 30-30-40-50-70 dB on the left, which is consistent with the diagnosis of a severe to profound combined middle-ear/inner ear hearing loss.
Treatment with denosumab resulted in 18% improvement in audiograms (5). However, after treatment with denosumab was discontinued, a sudden deterioration in her hearing abilities resulted in the necessity of cochlear implants.
Computed tomography (CT) scans revealed an incomplete partition of the cochlea type III on the right and a common cavity on the left (Fig. 1A). The middle and inner ear are clearly separated, with increased sclerosis of the petrous bone in general.
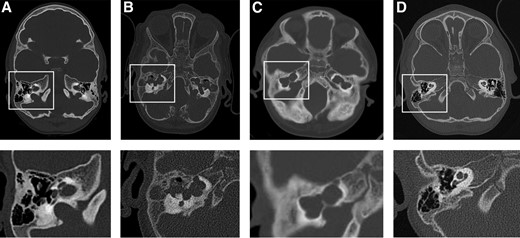
Malformation of the inner ear and vestibular structures in the patients with JPD show similar changes in all three patients: CT scans of head (upper panel), including inner ear and vestibular structures (lower panel) of (A) Patient 1 at 5 years of age, (B) Patient 2 at 15 months of age, (C) Patient 3 at age 4 years, and (D) a healthy child (boy, 34 months of age).
Patient 2
Newborn hearing screening hinted at a hearing impairment. Brainstem evoked response audiometry analysis showed a click threshold of 70 dB (right) and 60 dB (left). At 2 months of age, the thresholds increased to 100 dB (right) and 70 dB (left), and hearing aids were provided.
Whole brain magnetic resonance imaging (MRI; 1.5 T) revealed a distinct common cavity formation on both sides. CT scans at the age of 4 months and 2 years showed a complex cystic malformation of the inner ear and vestibular structures, without separation of inner and middle ear. Ossicles were not identifiable (Fig. 1B).
Further progression of hearing loss was diagnosed at 15 months of age (brainstem evoked response audiometry click threshold was 95 dB bilateral) and at 20 months of age (a threshold was unverifiable, consistent with deafness). A decision against cochlear implantation was made by the parents and physicians based on the increased perioperative risk and unclear prognosis due to the complex malformation. Communication successfully occurs by sign language.
Patient 3
She was noted to have bilateral sensory neural hearing loss at the age of 2 months and initially wore bilateral hearing aids. At age 4 years, a CT scan in the patient showed marked dysplasia of cochlea and vestibular structures bilaterally, consistent with severe cochlear dysplasia, with an 8-shaped common cavity as described in Patient 2 (Fig. 1C). Her hearing was retested, and it was found that she had no response even when using hearing aids. These have since been discontinued, and she communicates by sign language.
So far, none of the patients has developed calcification of the auricles.
Growth
The two patients with mutations in the TNFRSF11B gene (Patients 1 and 2) displayed height below the 3rd percentile starting at 12 months of age. Biochemical assessment repeatedly revealed low-to-normal or decreased insulin-like growth factor (IGF)-1 and IGF binding protein (BP)-3 levels and growth hormone (GH) stimulation tests were initiated. Other pituitary hormones (thyroid-stimulating hormone, prolactin, luteinizing hormone, and follicle-stimulating hormone) were within age-appropriate norms in both children.
Patient 1
In Patient 1, GH stimulation tests were initiated at 5 years of age and revealed insufficient GH levels after stimulation, consistent with a diagnosis of GH deficiency (maximum level of GH 6.27 ng/mL). No structural changes of her pituitary or compromise by surrounding skeletal structures were detected on MRI.
Treatment with GH at 0.035 mg/kg BW/d was initiated at age 5 years. At that time, her actual height was 3 cm below the 3rd percentile, and the bone age was 2 years advanced. Her target height is on the 50th percentile, and she initially showed sufficient catch-up growth. However, at age 9, low IGF-1 and IGFBP-3 serum levels developed [–2.1 and –0.99 standard deviation score (SDS), respectively]. With increasing doses of GH to 0.04 mg/kg BW/d serum levels of IGF-1 and IGFBP-3 remained low (–1.55 and –1.28, respectively), and growth velocity was unsatisfactory with 3.3 cm in the last year. Antibodies against GH were not detectable. The patient’s current height plots 12 cm below the 3rd percentile, whereas her sitting height plots on the 50th percentile. There is marked rhizomelia (arm: upper segment 14 cm, lower segment 19 cm). The GH therapy has been discontinued due to suspected compliance problems.
Patient 2
In Patient 2, GH stimulation tests were initiated at 2 years of age with a maximum GH level of 6.08 ng/mL (norm >7.25 ng/mL), consistent with GH deficiency. Growth is below the 3rd percentile, but with satisfactory growth velocity. He has thus not commenced treatment with GH. No structural changes of his pituitary or compromise by surrounding skeletal structures were detected on MRI.
Patient 3
Patient 3 is extremely short, displaying a height SDS of –5.2 with a growth velocity of 4.2 cm/y (height velocity SDS –2.05). She has a normal IGF-1 level of 17.2 nmol/l (normal range: 10.4 to 31.7) and, thus, did not undergo a GH stimulation test (Fig. 2).
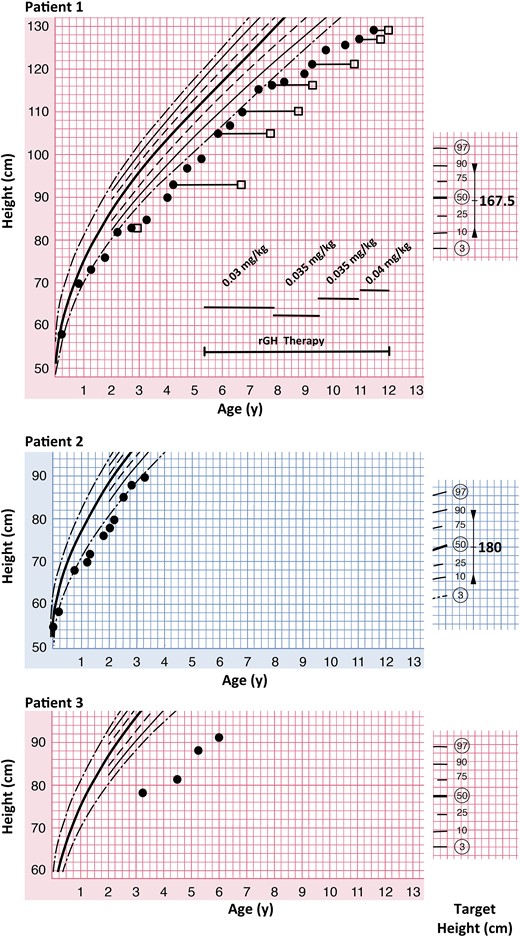
Growth is impaired in patients with JPD. Patient 1 is displaying growth (closed circles) below the 3rd percentile and was found to be GH deficient. Treatment with recombinant GH (rGH) was commenced at 5 years 4 months of age and initially resulted in growth along the 3rd percentile. At age 9 years, growth velocity deteriorated despite adjustment of the GH doses. The bone age (open squares) was initially advanced, but is almost age appropriate at age 11 years 6 months. Patient 2 is growing just below the 3rd percentile. He has a confirmed diagnosis of GH deficiency but is not treated with GH so far. Patient 3 is growing well below the 3rd percentile, despite normal IGF 1 serum levels. She has not been tested for GH deficiency.
Genetic evaluation
Family 1
The female patient (Patient 1: Family I: IV-9) is the second child of consanguineous parents of Turkish descent. As reported previously (5), a homozygous mutation in TNFRSF11B:c. [2T>G]; [2T>G] (Genbank NM_002546.3, ATG = 1 to 3) results in a methionine to arginine exchange at position 1. Nine family members are heterozygous carriers of this mutation; 3 are unaffected (Fig. 3).
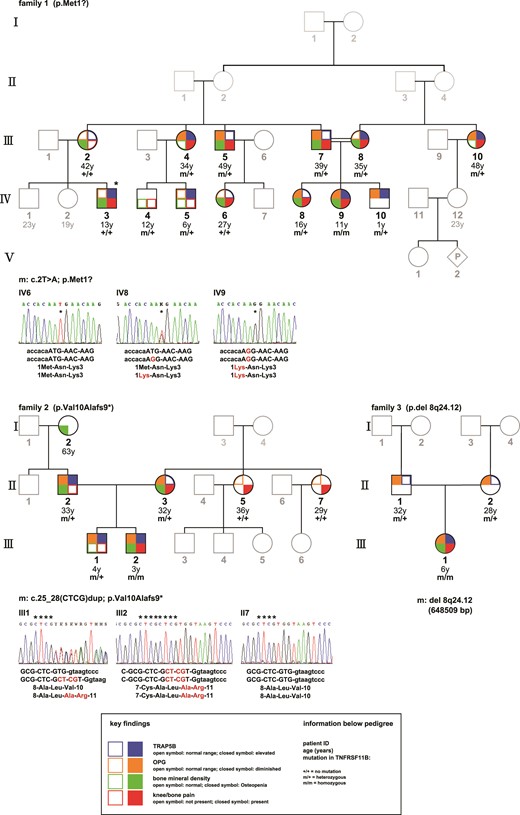
Pedigree of Families 1 to 3. Investigated family members are highlighted; the mutational status is indicated as m = mutation or + = regular sequencing results. The respective mutations in TNFRSF-11B are displayed for each family. The main findings are shown as following: left upper corner (brown color): diminished (closed symbol) or normal (open symbol) serum levels of OPG; left lower corner (green color): lower than normal (closed symbol) or within normal range (open symbol) bone mineral density as assessed by a dual-energy X-ray absorptiometry measurement (Lunar Prodigy); right lower corner (red color): presence (closed symbol) or absence (open symbol) of bone pain, described as frequent knee and bone pain after physical exercise and/or pain at lumbar and thoracic spine; right upper corner (blue color): elevated (closed symbol) or normal (open symbol) serum levels of TRAP 5b. *Individual IV-3 is suffering from rheumatoid arthritis.
Family 2
The patient (Patient 2: Family 2: III-2) is the second child of a German couple. To the knowledge of the family, the parents are nonconsanguineous. A novel homozygous 4 bp insertion in the TNFRFS11B gene was identified c.[25-28dup];[25-28dup] in exon 1. A CTCG insertion at position 25, resulting in a frameshift with a valine to alanine exchange at position 10 (Val10Alafs*9) and a premature stop codon. The parents of the patient and the 2-year-older brother are carriers; 2 maternal aunts are unaffected.
Family 3
The patient (Patient 3: Family 3: III-1) is the only child of a British couple of Pakistani descent. The parents are distantly related. A homozygous microdeletion at 8q24.12 including the entire TNFRSF11B gene was detected. The deletion also contains 3 other HGNC genes: COLEC10, MAL2, and NOV. Both parents are heterozygous carriers of the microdeletion.
Bone turnover markers
In all 3 patients, circulating levels of OPG were below the detection limit of the assay (<0.4 pmol/L). Adult heterozygous family members displayed diminished OPG levels, ranging from 1 pmol/L to 3.6 pmol/L, when compared with unaffected family members and healthy controls. In heterozygous children, circulating OPG levels were higher [4.12 (infant) and 3.1 pmol/L] than in adults.
Circulating levels of RANKL were elevated in the patients (9.1 to 23.2 pmol/L) and, to a lesser extent, in heterozygous carriers (0.2 to 1.9 pmol/L), compared with healthy controls and unaffected family members (0.05 to 0.4 pmol/L), with the exception of a 13-year-old boy (Family 1, IV-3), with juvenile arthritis who was found to have elevated levels of RANKL. The RANKL/OPG ratio was grossly elevated in patients (13 to 33.3) and about fivefold elevated in mutation carriers (0.06 to 0.46) compared with healthy subjects (0.01 to 0.1) (Fig. 4).
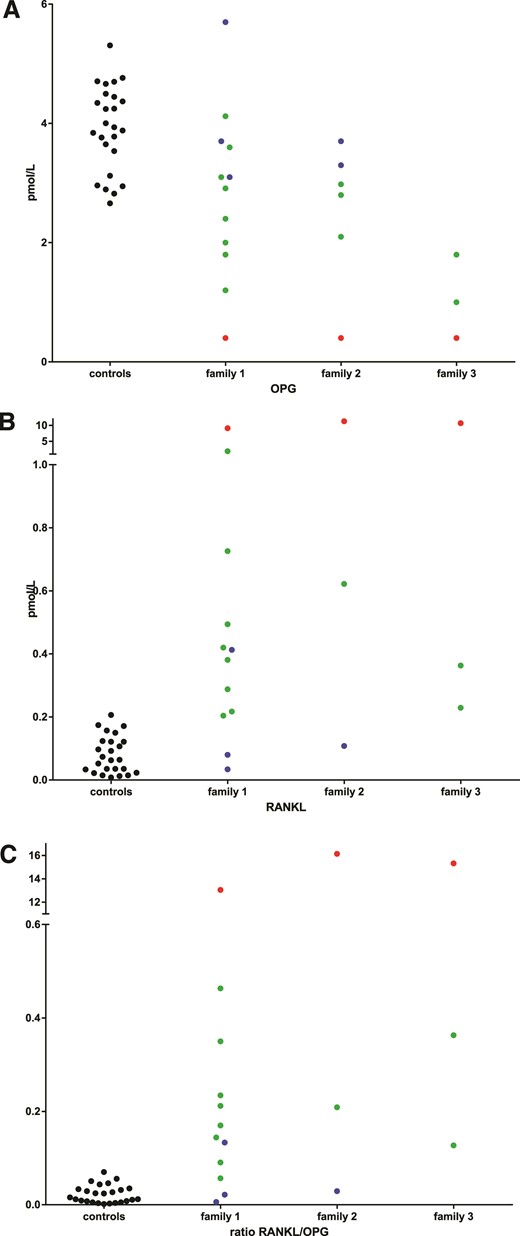
Plasma levels of OPG are diminished in patients with JPD and heterozygous family members. Plasma levels of (A) OPG (in pmol/L), (B) RANKL (in pmol/L), and (C) RANKL/OPG ratio in healthy adults (black circles), unaffected family members (blue circles), mutation carriers (green circles), and patients with JPD (red circles).
Accordingly, tartrate-resistant acid phosphatase (TRAP) 5b serum levels as a marker of osteoclast activity were elevated in the patients, osteocalcin levels were in the upper normal range, and deoxypyridinoline excretion in the urine was elevated in Patients 1 and 2 (not done in Patient 3). Heterozygous family members displayed elevated TRAP 5b levels and normal levels of deoxypyridinoline and osteocalcin (Table 1).
Clinical, Biochemical, and Radiological Characteristics of the 3 Index Patients With JPD, Heterozygous Family Members, and Unaffected Family Members
| ID | Sex | Genotype TNRFSF11B | Age (y) | HT-SDS | CrP (mg/dL) | TSAP (U/L) | OPG (pmol/L) | RANKL (pmol/L) | RANKL/OPG | TRAP 5b (U/L) | OC (ng/mL) | DPD (µg/g creatinine) | IGF1-SDS | DEXAZ/T Score | BP (mm Hg) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JPD | |||||||||||||||
| IV.9 | Female | c. [2T:G]; [2T:G] | 11.17 | –2.65 | 0.6 | 375 | 0.4 | 9.140 | 22.85 | 14.9 | 299.5 | 391 | –1.05 | −0.9 | 128/63 |
| III.2 | Male | c. [25_28dupCTCG]; [25_28dupCTCG] | 2.61 | −1.50 | 1.2 | 301 | 0.4 | 14.271 | 35.68 | 17.2 | 133.7 | 518 | |||
| III.1 | Female | del 8q24.12; del 8q24.1 | 6.0 | –5.2 | 0.4 | 23.32 | 58.30 | 12.7 | 167.2 | ||||||
| Heterozygous family members | |||||||||||||||
| IV.8 | Female | c. [2T:G]; [=] | 16.05 | 1.02 | <0.5 | 126 | 1.8 | 0.381 | 0.21 | 87.9 | 97 | −1 | –1.1 | ||
| IV.10 | Male | c. [2T:G]; [=] | 1.00 | <0.5 | 398 | 4.1 | 1.909 | 0.46 | 21.2 | 81.2 | |||||
| III.7 | Male | c. [2T:G]; [=] | 39.70 | 0.59 | <0.5 | 96 | 2 | 0.288 | 0.14 | 4.5 | 24.6 | 20 | –1.4 | 122/70 | |
| III.8 | Female | c. [2T:G]; [=] | 35.20 | −0.20 | 1.6 | 122 | 3.6 | 0.204 | 0.06 | 7 | 66.0 | 89 | –2.5 | 122/73 | |
| III.4 | Female | c. [2T:G]; [=] | 34.51 | 0.67 | <0.5 | 66 | 1.2 | 0.420 | 0.35 | 5,1 | 31.3 | −0.4 | 133/76 | ||
| IV.5 | Male | c. [2T:G]; [=] | 6.37 | −0.22 | <0.5 | 318 | 3.1 | 0.726 | 0.23 | 29 | 94.6 | 0.16 | −0.3 | 118/58 | |
| III.10 | Female | c. [2T:G]; [=] | 47.72 | −0.92 | <0.5 | 87 | 2.4 | 0.217 | 0.09 | 5.2 | 28.5 | −0.4 | |||
| III.5 | Male | c. [2T:G]; [=] | 49.80 | −0.30 | 1 | 150 | 2.9 | 0.494 | 0.17 | 4,4 | 27 | –1.8 | 131/90 | ||
| IV.4 | Male | c. [2T:G]; [=] | 11.24 | <0.5 | 242 | 130 | 284 | −0.14 | 0.5 | ||||||
| II.3 | Female | c. [25_28dupCTCG]; [=] | 30.80 | −0.78 | <0.5 | 99 | 2.8 | 0.094 | 0.03 | 2.3 | 19.1 | 31 | |||
| II.2 | Male | c. [25_28dupCTCG]; [=] | 32.11 | 0.64 | <0.5 | 83 | 2.1 | 0.346 | 0.16 | 5.1 | 22 | 40 | |||
| III.1 | Male | c. [25_28dupCTCG]; [=] | 3.16 | 0.22 | <0.5 | 257 | 2.9 | 0.622 | 0.21 | 10.8 | 76 | 273 | −0.59 | ||
| II.2 | Female | del 8q24.1; [=] | 1 | 0.363 | 0.36 | 2.6 | 26.1 | ||||||||
| II.1 | Male | del 8q24.1; [=] | 1.8 | 0.229 | 0.13 | 2.9 | 34.4 | ||||||||
| Unaffected family members | |||||||||||||||
| III.2 | Female | c. [=]; [=] | 42.38 | 0.02 | 1.2 | 107 | 5.7 | 0.034 | 0.01 | 4.6 | 22.2 | −1 | |||
| IV.3a | Male | c. [=]; [=] | 13.60 | −0.02 | <0.5 | 368 | 3.1 | 0.413 | 0.13 | 33 | 241.7 | 283 | 1.11 | –1.3 | 134/63 |
| IV.6 | Female | c. [=]; [=] | 27.10 | 0.78 | <0.5 | 56 | 3.7 | 0.080 | 0.02 | 3.6 | 28.2 | −1 | |||
| II.5 | Female | c. [=]; [=] | 35.92 | 0.59 | 3.7 | 0.108 | 0.03 | ||||||||
| II.7 | Female | c. [=]; [=] | 28.33 | −0.24 | 3.3 | 0.159 | 0.05 | ||||||||
| ID | Sex | Genotype TNRFSF11B | Age (y) | HT-SDS | CrP (mg/dL) | TSAP (U/L) | OPG (pmol/L) | RANKL (pmol/L) | RANKL/OPG | TRAP 5b (U/L) | OC (ng/mL) | DPD (µg/g creatinine) | IGF1-SDS | DEXAZ/T Score | BP (mm Hg) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JPD | |||||||||||||||
| IV.9 | Female | c. [2T:G]; [2T:G] | 11.17 | –2.65 | 0.6 | 375 | 0.4 | 9.140 | 22.85 | 14.9 | 299.5 | 391 | –1.05 | −0.9 | 128/63 |
| III.2 | Male | c. [25_28dupCTCG]; [25_28dupCTCG] | 2.61 | −1.50 | 1.2 | 301 | 0.4 | 14.271 | 35.68 | 17.2 | 133.7 | 518 | |||
| III.1 | Female | del 8q24.12; del 8q24.1 | 6.0 | –5.2 | 0.4 | 23.32 | 58.30 | 12.7 | 167.2 | ||||||
| Heterozygous family members | |||||||||||||||
| IV.8 | Female | c. [2T:G]; [=] | 16.05 | 1.02 | <0.5 | 126 | 1.8 | 0.381 | 0.21 | 87.9 | 97 | −1 | –1.1 | ||
| IV.10 | Male | c. [2T:G]; [=] | 1.00 | <0.5 | 398 | 4.1 | 1.909 | 0.46 | 21.2 | 81.2 | |||||
| III.7 | Male | c. [2T:G]; [=] | 39.70 | 0.59 | <0.5 | 96 | 2 | 0.288 | 0.14 | 4.5 | 24.6 | 20 | –1.4 | 122/70 | |
| III.8 | Female | c. [2T:G]; [=] | 35.20 | −0.20 | 1.6 | 122 | 3.6 | 0.204 | 0.06 | 7 | 66.0 | 89 | –2.5 | 122/73 | |
| III.4 | Female | c. [2T:G]; [=] | 34.51 | 0.67 | <0.5 | 66 | 1.2 | 0.420 | 0.35 | 5,1 | 31.3 | −0.4 | 133/76 | ||
| IV.5 | Male | c. [2T:G]; [=] | 6.37 | −0.22 | <0.5 | 318 | 3.1 | 0.726 | 0.23 | 29 | 94.6 | 0.16 | −0.3 | 118/58 | |
| III.10 | Female | c. [2T:G]; [=] | 47.72 | −0.92 | <0.5 | 87 | 2.4 | 0.217 | 0.09 | 5.2 | 28.5 | −0.4 | |||
| III.5 | Male | c. [2T:G]; [=] | 49.80 | −0.30 | 1 | 150 | 2.9 | 0.494 | 0.17 | 4,4 | 27 | –1.8 | 131/90 | ||
| IV.4 | Male | c. [2T:G]; [=] | 11.24 | <0.5 | 242 | 130 | 284 | −0.14 | 0.5 | ||||||
| II.3 | Female | c. [25_28dupCTCG]; [=] | 30.80 | −0.78 | <0.5 | 99 | 2.8 | 0.094 | 0.03 | 2.3 | 19.1 | 31 | |||
| II.2 | Male | c. [25_28dupCTCG]; [=] | 32.11 | 0.64 | <0.5 | 83 | 2.1 | 0.346 | 0.16 | 5.1 | 22 | 40 | |||
| III.1 | Male | c. [25_28dupCTCG]; [=] | 3.16 | 0.22 | <0.5 | 257 | 2.9 | 0.622 | 0.21 | 10.8 | 76 | 273 | −0.59 | ||
| II.2 | Female | del 8q24.1; [=] | 1 | 0.363 | 0.36 | 2.6 | 26.1 | ||||||||
| II.1 | Male | del 8q24.1; [=] | 1.8 | 0.229 | 0.13 | 2.9 | 34.4 | ||||||||
| Unaffected family members | |||||||||||||||
| III.2 | Female | c. [=]; [=] | 42.38 | 0.02 | 1.2 | 107 | 5.7 | 0.034 | 0.01 | 4.6 | 22.2 | −1 | |||
| IV.3a | Male | c. [=]; [=] | 13.60 | −0.02 | <0.5 | 368 | 3.1 | 0.413 | 0.13 | 33 | 241.7 | 283 | 1.11 | –1.3 | 134/63 |
| IV.6 | Female | c. [=]; [=] | 27.10 | 0.78 | <0.5 | 56 | 3.7 | 0.080 | 0.02 | 3.6 | 28.2 | −1 | |||
| II.5 | Female | c. [=]; [=] | 35.92 | 0.59 | 3.7 | 0.108 | 0.03 | ||||||||
| II.7 | Female | c. [=]; [=] | 28.33 | −0.24 | 3.3 | 0.159 | 0.05 | ||||||||
Given are the genotype at the TNFRSF11B locus; the age at investigation in years; the height (HT) SDS; serum levels of CrP (in mg/dL), TSAP (in U/L), OPG (in pmol/L), RANKL (in pmol/L), the RANKL/OPG ratio, TRAP 5b (in U/L), osteocalcin (OC, in ng/mL), DPD in urine (in µg/g creatinine), and IGF-1 (in ng/mL); IGF1-SDS; DEXA Z score or T score (depending on the age at time of investigation); and BP (in mm Hg). Pathological readings (aberrant from the applicable age appropriate norm) are highlighted by bold letters. Age-dependent reference values are provided as supplemental material.
Abbreviations: BP, blood pressure; CrP, C-reactive protein; DPD, deoxypyridinoline; DEXA, dual-emission X-ray absorptiometry; TSAP, tissue-specific alkaline phosphatase.
a Individual IV.3 is suffering from rheumatoid arthritis.
Clinical, Biochemical, and Radiological Characteristics of the 3 Index Patients With JPD, Heterozygous Family Members, and Unaffected Family Members
| ID | Sex | Genotype TNRFSF11B | Age (y) | HT-SDS | CrP (mg/dL) | TSAP (U/L) | OPG (pmol/L) | RANKL (pmol/L) | RANKL/OPG | TRAP 5b (U/L) | OC (ng/mL) | DPD (µg/g creatinine) | IGF1-SDS | DEXAZ/T Score | BP (mm Hg) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JPD | |||||||||||||||
| IV.9 | Female | c. [2T:G]; [2T:G] | 11.17 | –2.65 | 0.6 | 375 | 0.4 | 9.140 | 22.85 | 14.9 | 299.5 | 391 | –1.05 | −0.9 | 128/63 |
| III.2 | Male | c. [25_28dupCTCG]; [25_28dupCTCG] | 2.61 | −1.50 | 1.2 | 301 | 0.4 | 14.271 | 35.68 | 17.2 | 133.7 | 518 | |||
| III.1 | Female | del 8q24.12; del 8q24.1 | 6.0 | –5.2 | 0.4 | 23.32 | 58.30 | 12.7 | 167.2 | ||||||
| Heterozygous family members | |||||||||||||||
| IV.8 | Female | c. [2T:G]; [=] | 16.05 | 1.02 | <0.5 | 126 | 1.8 | 0.381 | 0.21 | 87.9 | 97 | −1 | –1.1 | ||
| IV.10 | Male | c. [2T:G]; [=] | 1.00 | <0.5 | 398 | 4.1 | 1.909 | 0.46 | 21.2 | 81.2 | |||||
| III.7 | Male | c. [2T:G]; [=] | 39.70 | 0.59 | <0.5 | 96 | 2 | 0.288 | 0.14 | 4.5 | 24.6 | 20 | –1.4 | 122/70 | |
| III.8 | Female | c. [2T:G]; [=] | 35.20 | −0.20 | 1.6 | 122 | 3.6 | 0.204 | 0.06 | 7 | 66.0 | 89 | –2.5 | 122/73 | |
| III.4 | Female | c. [2T:G]; [=] | 34.51 | 0.67 | <0.5 | 66 | 1.2 | 0.420 | 0.35 | 5,1 | 31.3 | −0.4 | 133/76 | ||
| IV.5 | Male | c. [2T:G]; [=] | 6.37 | −0.22 | <0.5 | 318 | 3.1 | 0.726 | 0.23 | 29 | 94.6 | 0.16 | −0.3 | 118/58 | |
| III.10 | Female | c. [2T:G]; [=] | 47.72 | −0.92 | <0.5 | 87 | 2.4 | 0.217 | 0.09 | 5.2 | 28.5 | −0.4 | |||
| III.5 | Male | c. [2T:G]; [=] | 49.80 | −0.30 | 1 | 150 | 2.9 | 0.494 | 0.17 | 4,4 | 27 | –1.8 | 131/90 | ||
| IV.4 | Male | c. [2T:G]; [=] | 11.24 | <0.5 | 242 | 130 | 284 | −0.14 | 0.5 | ||||||
| II.3 | Female | c. [25_28dupCTCG]; [=] | 30.80 | −0.78 | <0.5 | 99 | 2.8 | 0.094 | 0.03 | 2.3 | 19.1 | 31 | |||
| II.2 | Male | c. [25_28dupCTCG]; [=] | 32.11 | 0.64 | <0.5 | 83 | 2.1 | 0.346 | 0.16 | 5.1 | 22 | 40 | |||
| III.1 | Male | c. [25_28dupCTCG]; [=] | 3.16 | 0.22 | <0.5 | 257 | 2.9 | 0.622 | 0.21 | 10.8 | 76 | 273 | −0.59 | ||
| II.2 | Female | del 8q24.1; [=] | 1 | 0.363 | 0.36 | 2.6 | 26.1 | ||||||||
| II.1 | Male | del 8q24.1; [=] | 1.8 | 0.229 | 0.13 | 2.9 | 34.4 | ||||||||
| Unaffected family members | |||||||||||||||
| III.2 | Female | c. [=]; [=] | 42.38 | 0.02 | 1.2 | 107 | 5.7 | 0.034 | 0.01 | 4.6 | 22.2 | −1 | |||
| IV.3a | Male | c. [=]; [=] | 13.60 | −0.02 | <0.5 | 368 | 3.1 | 0.413 | 0.13 | 33 | 241.7 | 283 | 1.11 | –1.3 | 134/63 |
| IV.6 | Female | c. [=]; [=] | 27.10 | 0.78 | <0.5 | 56 | 3.7 | 0.080 | 0.02 | 3.6 | 28.2 | −1 | |||
| II.5 | Female | c. [=]; [=] | 35.92 | 0.59 | 3.7 | 0.108 | 0.03 | ||||||||
| II.7 | Female | c. [=]; [=] | 28.33 | −0.24 | 3.3 | 0.159 | 0.05 | ||||||||
| ID | Sex | Genotype TNRFSF11B | Age (y) | HT-SDS | CrP (mg/dL) | TSAP (U/L) | OPG (pmol/L) | RANKL (pmol/L) | RANKL/OPG | TRAP 5b (U/L) | OC (ng/mL) | DPD (µg/g creatinine) | IGF1-SDS | DEXAZ/T Score | BP (mm Hg) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JPD | |||||||||||||||
| IV.9 | Female | c. [2T:G]; [2T:G] | 11.17 | –2.65 | 0.6 | 375 | 0.4 | 9.140 | 22.85 | 14.9 | 299.5 | 391 | –1.05 | −0.9 | 128/63 |
| III.2 | Male | c. [25_28dupCTCG]; [25_28dupCTCG] | 2.61 | −1.50 | 1.2 | 301 | 0.4 | 14.271 | 35.68 | 17.2 | 133.7 | 518 | |||
| III.1 | Female | del 8q24.12; del 8q24.1 | 6.0 | –5.2 | 0.4 | 23.32 | 58.30 | 12.7 | 167.2 | ||||||
| Heterozygous family members | |||||||||||||||
| IV.8 | Female | c. [2T:G]; [=] | 16.05 | 1.02 | <0.5 | 126 | 1.8 | 0.381 | 0.21 | 87.9 | 97 | −1 | –1.1 | ||
| IV.10 | Male | c. [2T:G]; [=] | 1.00 | <0.5 | 398 | 4.1 | 1.909 | 0.46 | 21.2 | 81.2 | |||||
| III.7 | Male | c. [2T:G]; [=] | 39.70 | 0.59 | <0.5 | 96 | 2 | 0.288 | 0.14 | 4.5 | 24.6 | 20 | –1.4 | 122/70 | |
| III.8 | Female | c. [2T:G]; [=] | 35.20 | −0.20 | 1.6 | 122 | 3.6 | 0.204 | 0.06 | 7 | 66.0 | 89 | –2.5 | 122/73 | |
| III.4 | Female | c. [2T:G]; [=] | 34.51 | 0.67 | <0.5 | 66 | 1.2 | 0.420 | 0.35 | 5,1 | 31.3 | −0.4 | 133/76 | ||
| IV.5 | Male | c. [2T:G]; [=] | 6.37 | −0.22 | <0.5 | 318 | 3.1 | 0.726 | 0.23 | 29 | 94.6 | 0.16 | −0.3 | 118/58 | |
| III.10 | Female | c. [2T:G]; [=] | 47.72 | −0.92 | <0.5 | 87 | 2.4 | 0.217 | 0.09 | 5.2 | 28.5 | −0.4 | |||
| III.5 | Male | c. [2T:G]; [=] | 49.80 | −0.30 | 1 | 150 | 2.9 | 0.494 | 0.17 | 4,4 | 27 | –1.8 | 131/90 | ||
| IV.4 | Male | c. [2T:G]; [=] | 11.24 | <0.5 | 242 | 130 | 284 | −0.14 | 0.5 | ||||||
| II.3 | Female | c. [25_28dupCTCG]; [=] | 30.80 | −0.78 | <0.5 | 99 | 2.8 | 0.094 | 0.03 | 2.3 | 19.1 | 31 | |||
| II.2 | Male | c. [25_28dupCTCG]; [=] | 32.11 | 0.64 | <0.5 | 83 | 2.1 | 0.346 | 0.16 | 5.1 | 22 | 40 | |||
| III.1 | Male | c. [25_28dupCTCG]; [=] | 3.16 | 0.22 | <0.5 | 257 | 2.9 | 0.622 | 0.21 | 10.8 | 76 | 273 | −0.59 | ||
| II.2 | Female | del 8q24.1; [=] | 1 | 0.363 | 0.36 | 2.6 | 26.1 | ||||||||
| II.1 | Male | del 8q24.1; [=] | 1.8 | 0.229 | 0.13 | 2.9 | 34.4 | ||||||||
| Unaffected family members | |||||||||||||||
| III.2 | Female | c. [=]; [=] | 42.38 | 0.02 | 1.2 | 107 | 5.7 | 0.034 | 0.01 | 4.6 | 22.2 | −1 | |||
| IV.3a | Male | c. [=]; [=] | 13.60 | −0.02 | <0.5 | 368 | 3.1 | 0.413 | 0.13 | 33 | 241.7 | 283 | 1.11 | –1.3 | 134/63 |
| IV.6 | Female | c. [=]; [=] | 27.10 | 0.78 | <0.5 | 56 | 3.7 | 0.080 | 0.02 | 3.6 | 28.2 | −1 | |||
| II.5 | Female | c. [=]; [=] | 35.92 | 0.59 | 3.7 | 0.108 | 0.03 | ||||||||
| II.7 | Female | c. [=]; [=] | 28.33 | −0.24 | 3.3 | 0.159 | 0.05 | ||||||||
Given are the genotype at the TNFRSF11B locus; the age at investigation in years; the height (HT) SDS; serum levels of CrP (in mg/dL), TSAP (in U/L), OPG (in pmol/L), RANKL (in pmol/L), the RANKL/OPG ratio, TRAP 5b (in U/L), osteocalcin (OC, in ng/mL), DPD in urine (in µg/g creatinine), and IGF-1 (in ng/mL); IGF1-SDS; DEXA Z score or T score (depending on the age at time of investigation); and BP (in mm Hg). Pathological readings (aberrant from the applicable age appropriate norm) are highlighted by bold letters. Age-dependent reference values are provided as supplemental material.
Abbreviations: BP, blood pressure; CrP, C-reactive protein; DPD, deoxypyridinoline; DEXA, dual-emission X-ray absorptiometry; TSAP, tissue-specific alkaline phosphatase.
a Individual IV.3 is suffering from rheumatoid arthritis.
Plasma levels of OPG, RANKL, and TRAP 5b were measured using commercially available enzyme-linked immunosorbent assays, according to the manufacturers’ instructions (OPG: Quidel; soluble RANKL: Biomedica; and TRAP 5b: Quidel, all by TECOmedical, Sissach, Switzerland).
Heterozygous carriers
Skeletal findings
In 60% of the investigated adult/adolescent carriers osteopenia [t < –1, at the lumbar spine (L1 to L4)] as detected by a dual-energy X-ray absorptiometry scan (Lunar Prodigy, GE, Boston, MA; precision 0.010 g/cm2 at lumbar spine) was present; one individual showed osteoporosis (t < –2.5). Bone pain in the form of back pain or recurrent knee pain after physical exercise (climbing stairs) was described by 85% of adult heterozygous carriers, but there was no evidence of increased fracture rates.
Hearing impairment
The father of Patient 2 suffers from a progressive severe-to-profound sensorineural hearing loss affecting the middle and high frequencies on the right side and a moderate-to-severe sensorineural hearing loss of the same frequencies on the contralateral side. He is provided with hearing aids. A CT scan of his petrous bone showed a normal configuration of the cochlear and vestibular structures. The mother and brother of Patient 2 have normal hearing abilities (normal audiograms). Heterozygous members of Families 1 and 3 had no evidence of hearing impairment and were not further investigated.
Ophthalmological findings
Mutation carriers of Family 1 and 3 did not present with obvious impairments of visual function and were not investigated for retinal changes. However, in Family 2, both parents (heterozygous carriers) have irregular astigmatism but were found to have normal retinal appearances.
Growth
Adult mutation carriers have mostly reached a final height within their respective target height, with the exception of the mother of Patient 2 (Family 2: II-3). Heterozygous siblings and cousins of Patients 1and 2 show normal growth so far.
Discussion
JPD is an extremely rare skeletal disease with grossly elevated bone turnover due to loss of function of OPG. The known function of OPG as an antagonist to RANKL action implies that other functions of the RANK pathway might be altered in patients with JPD as well. RANK, a transmembrane glycoprotein, is expressed not only on monocytes (16), the osteoclast precursors, and mature osteoclasts (17), but also on dendritic cells (18) and epithelial cells of the mammary gland (19).
With this report, we aimed to characterize the complex phenotype of JPD1 in patients and heterozygous carriers more fully. We identified several additional signs of severe forms of JPD.
Gross motor and mental development
Gross motor development was delayed in the described children of this report before the diagnosis and treatment were established. However, gross motor development progressed to almost age appropriate norms after treatment with bisphosphonates was initiated.
Gross motor developmental delay is common in children with severe forms of JPD (3), and likely associated with the pain from the inflammatory bone disease. Maintenance of ambulation seems to depend on effective treatment of the skeletal phenotype. Whereas most patients with JPD are described with normal cognitive function, there are reports describing mental retardation, as seen in Patient 3, in few patients with JPD (20, 21).
Contractures of joints are not specifically previously reported in patients with JPD, except for a restricted range of motion of the head, which was reported (3) in a 9-month-old patient. However, over time, all 3 patients of this report developed contractures of joints (knee, elbows), indicating that this might be a feature of JPD.
Growth hormone deficiency
Most patients with severe forms of JPD due to mutations in TNFRSF11B were reported to be of short stature (1–4, 7, 22, 23), whereas patients with milder forms might be of normal stature (12). A patient with an activating mutation of TNFRSF11A was reported to be of good height for age (height Z score +1.5) (24).
Whereas the short stature in JPD patients is at least partly attributed to the skeletal dysplasia with bowing deformities of femur and tibia and, by rhizomelia, a feature reported in another case by Naot et al. (3), GH deficiency might be another sign of this disease. Indeed, both children of this report with TNFRSF11B mutations were found to be GH deficient, and treatment with recombinant GH resulted initially in good catch-up growth in Patient 1. The accelerated bone age in this patient, which was present even before treatment with GH was initiated, is currently not well understood. There were no clinical or biochemical signs of untimely pubertal or adrenal hormone production at any time. We thus speculate that the bone age acceleration is most likely due to the elevated bone turnover in this patient.
Malformation of the skull and inner ear
Although almost all patients with JPD are described to have expansion of the skull base and the diploic space (3), Patients 2 and 3 described in this report were found to have a Chiari I malformation without hydrocephalus. Presumably, the Chiari I is a consecutive disorder due to the narrowing of the posterior fossa caused by the expansive bone formation. A similar finding has been described in a 5-year-old girl with a severe form of JPD (25), adding a Chiari I malformation to the signs of severe forms of JPD.
The malformations of the inner ear are unrelated to the described widening of the skull base. Whereas in Patient 1, the vestibular organ was normally developed, Patients 2 and 3 presented with an 8-shaped “common cavity” without identifiable cochlear or vestibular structures. This malformation indicates an effect as early as 4 to 5 weeks’ gestation in these patients. A similar malformation of the inner ear was described in 1 other patient with JPD (24, 26), as well as in a patient with an activating mutation in TNRFRSF11A (24), pointing toward a role for the RANKL pathway in the early development of the inner ear structures.
The distinct sensorineural hearing loss in patients with JPD has been recognized for a long time. It has been described in man and mice lacking OPG (10, 12) and might be responsive to treatment with bisphosphonates or denosumab (5, 9).
Vision
Despite the severity of the phenotype, the patients described in this report were not found to have visual impairment or retinal abnormalities, possibly due to the young age of the children. Kerr et al. (11) described retinal changes in a small cohort of 7 patients with JPD due to different underlying mutations. Three of these patients were adolescents, being 13, 14, and 15 years of age. Although the 13-year-old had not developed retinal abnormalities, these were identifiable in the older adolescents.
Whyte et al. (27) reported a patient with JPD who developed blindness despite normal bone turnover markers during treatment with bisphosphonates, implying a possible role for the lack of OPG, rather than elevated bone turnover or RANKL, for progression of the ophthalmological disease. It is unclear whether treatment with a RANKL antibody might be beneficial to prevent this progression.
Affected heterozygote carriers
Heterozygous carriers in this report showed diminished OPG levels and mildly increased bone turnover markers. In many of the mutation carriers, osteopenia or even osteoporosis was present. Bone pain was reported frequently; however, skeletal disease does not seem to be severe.
Other possibly JPD-associated signs included vision impairment and irregular astigmatism in Family 2 and sensorineural hearing loss in the father of Patient 2. However, it is unknown whether these changes are caused by reduced OPG levels, because astigmatism has not been described in JPD patients so far.
Conclusion
There is a substantial burden of disease on patients with severe forms of JPD. With this report, we suggest that malformation of the skull base and Chiari 1 malformation, GH deficiency, malformation of the inner ear, and contractures of joints are additional signs of severe forms of JPD due to mutations in TNFRSF11B. With the availability of new drugs interacting with the OPG/RANKL signaling, it is crucial to determine the effects of loss of OPG vs elevated RANKL levels on the progression of extraskeletal effects to tailor therapy in patients and carriers.
Heterozygous carriers of the TNFRSF11B mutations have lower circulating OPG levels and elevated RANKL levels compared with unaffected individuals. Carriers display elevated bone turnover, which can result in osteoporosis and elevated inflammatory laboratory work. Some tested individuals displayed hearing impairment and vision impairment. Monitoring of heterozygous family members—including a follow-up of growth, bone turnover, vision, and hearing screening—is therefore advisable.
Abbreviations:
- BP
binding protein
- CT
computed tomography
- GH
growth hormone
- IGF
insulin-like growth factor
- JPD
juvenile Paget’s disease
- MRI
magnetic resonance imaging
- OPG
osteoprotegerin
- RANK
receptor activator of nuclear factor κB
- RANKL
receptor activator of nuclear factor κB-ligand
- SDS
standard deviation score
- TRAP
tartrate-resistant acid phosphatase
Acknowledgments
We thank Jens Bauer from the University of Duisburg-Essen for excellent technical help with data management and figure conception. We also thank Marc Schlamann, University of Giessen, and Jan-Peter Thomas, University of Bochum, for help with the interpretation of CT scans from Patients 1 and 2.
M.M.S. has been funded by an Ifores stipend from the Medical Faculty of the University of Duisburg-Essen.
Disclosure Summary: The authors have nothing to disclose.
References
Köhler G, Egelkraut H. Münchener Funktionelle Entwicklungsdiagnostik Für Das Zweite Und Dritte Lebensjahr. Handanweis. München: Institut für Soziale Pädiatrie und Jugendmedizin, Universität München; 1984.
Author notes
Address all correspondence and requests for reprints to: Corinna Grasemann, MD, Pediatric Endocrinology and Diabetology, Klinik für Kinderheilkunde II, Universitätsklinikum Essen, Hufelandstr. 55, 45122 Essen, Germany. E-mail: [email protected].

{kind=link}
{kind=link}
{kind=link}
{kind=link}