





Abstract
Dopamine β-hydroxylase (DBH) deficiency is a rare genetic disorder characterized by failure to convert dopamine to norepinephrine. DBH-deficient patients lack sympathetic adrenergic function and are therefore predisposed to orthostatic hypotension. DBH-deficient mice exhibit hyperinsulinemia, lower plasma glucose levels, and insulin resistance due to loss of tonic sympathetic inhibition of insulin secretion. The impact of DBH deficiency on glucose homeostasis in humans is unknown.
We describe the metabolic profile of an adolescent female DBH-deficient patient. The patient underwent genetic testing, cardiovascular autonomic function testing, and evaluation of insulin secretion and sensitivity with hyperglycemic clamp under treatment-naive conditions. All procedures were repeated after 1 year of treatment with the norepinephrine prodrug droxidopa (300 mg, 3 times a day). Genetic testing showed a homozygous mutation in the DBH gene (rs74853476). Under treatment-naive conditions, she had undetectable plasma epinephrine and norepinephrine levels, resulting in sympathetic noradrenergic failure and orthostatic hypotension (−32 mm Hg supine to seated). She had high adiposity (41%) and fasting plasma insulin levels (25 μU/mL), with normal glucose (91 mg/dL). Hyperglycemic clamp revealed increased glucose-stimulated insulin secretion and insulin resistance. Droxidopa restored plasma norepinephrine and improved orthostatic tolerance, with modest effects on glucose homeostasis.
We provide evidence for impairment in cardiovascular autonomic regulation, hyperinsulinemia, enhanced glucose-stimulated insulin secretion, and insulin resistance in a DBH-deficient patient. These metabolic derangements were not corrected by chronic droxidopa treatment. These findings provide insight into the pathophysiology and treatment of DBH deficiency and into the importance of catecholaminergic mechanisms to resting metabolism.
Dopamine β-hydroxylase (DBH; OMIM 223360) deficiency is an autosomal recessive disorder described in approximately 20 patients worldwide (1). These patients have undetectable plasma norepinephrine and epinephrine, resulting in loss of sympathetic noradrenergic function and orthostatic hypotension (1). The presence of intact catecholamine release mechanisms results in elevated plasma dopamine. Management of orthostatic hypotension in DBH deficiency includes droxidopa (L-threo-3,4-dihydroxyphenylserine), which restores norepinephrine and epinephrine in noradrenergic nerve terminals and plasma while reducing dopamine concentrations (2). Norepinephrine also plays an important role in physiologic regulation of blood glucose levels by tonically inhibiting insulin secretion. Mice lacking a functional DBH gene (DBH−/−) exhibit hyperinsulinemia, reduced glucose levels, and insulin resistance (3). The impact of DBH deficiency on glucose homeostasis in humans is unknown. We hypothesized that DBH-deficient patients would have hyperinsulinemia and defects in insulin action, similar to findings in the mouse model.
Materials and Methods
Clinical history
We evaluated a 15-year-old female non-Hispanic white patient with DBH deficiency. She developed symptoms in early childhood, including syncopal episodes with standing and during bowel movements. In late childhood, she developed orthostatic hypotension that was managed with midodrine (10 mg, 3 times a day) and fludrocortisone (0.2 mg, 4 times a day).
General protocol
The Vanderbilt Institutional Review Board approved all procedures. Written informed consent was obtained (http://clinicaltrials.gov, NCT00748059). Midodrine and fludrocortisone were withheld >5 half-lives before admission. Routine safety screening tests were within normal limits (e.g., 12-lead electrocardiogram, complete blood count, comprehensive metabolic panel). DBH gene sequencing was performed using automated fluorescence dideoxy sequencing (MNG Laboratories, Atlanta, GA). She underwent standardized autonomic function testing (4). Plasma catecholamines were measured by high-performance liquid chromatography with electrochemical detection (5).
Metabolic measurements
Body composition was measured by air displacement plethysmography (BOD POD; COSMED, Concord, CA). A hyperglycemic clamp was performed as previously described (6). A primed 20% dextrose (Hospira, Lake Forest, IL) infusion was administered at a fixed rate (200 mg/kg) for 10 minutes, with infusion rates adjusted thereafter to maintain plasma glucose at 200 mg/dL. Acute glucose-stimulated insulin response was calculated as maximum insulin level in the first 10 minutes of infusion above mean baseline value. Late glucose-stimulated insulin response was calculated as mean insulin level between 90 and 120 minutes of infusion above mean baseline value. The insulin sensitivity index (ISI) was calculated by dividing the average glucose infusion rate (mg/kg/min) by the average insulin concentration (μU/mL) between 90 and 120 minutes. Reference data were obtained from 5 healthy female non-Hispanic white volunteers studied using identical methods.
Chronic droxidopa treatment
The patient was readmitted after 1 year of droxidopa treatment (300 mg by mouth, 3 times a day) (Sumitomo Pharmaceuticals, Osaka, Tokyo, Japan). Droxidopa was held constant during admission, with procedures performed >1 hour after administration.
Results
Genetic testing
The patient was homozygous for single nucleotide variant IVS1+2T→C (rs74853476; GrCh37 9:136501834), reflecting replacement of cytosine for thymine in the splice donor site in intron 1. Heterozygosity for this variant has been described in two other DBH-deficient patients (7).
Cardiovascular autonomic profile
The respiratory sinus arrhythmia ratio was 1.28 (>1.2 healthy), suggesting intact parasympathetic function. Evidence for sympathetic noradrenergic failure included the absence of systolic blood pressure (SBP) increase during phase IV of the Valsalva maneuver (baseline: 109 mm Hg; post: 91 mm Hg; Δ: −10 mm Hg; +20 mm Hg healthy) and the absence of cold pressor response (baseline: 102 mm Hg; post: 98 mm Hg; Δ: −4 mm Hg; +20 mm Hg healthy) (4). Her SBP decreased by 32 mm Hg from supine to sitting (101 mm Hg versus 69 mm Hg), and she was unable to tolerate standing. Appropriate reflex tachycardia was observed [supine: 69 beats per minute (bpm); sitting: 91 bpm]. Plasma norepinephrine and epinephrine were below detection limits, with elevated dopamine (Table 1).
Metabolic and Neurohumoral Profile of a DBH-Deficient Patient
| Parameter | DBH-Deficient Patient (Naive) | DBH-Deficient Patient (Droxidopa) | Healthya |
|---|---|---|---|
| Age, y | 15 | 16 | 26 (22–30) |
| Weight, kg | 79 | 83 | 74 (62–91) |
| BMI, kg/m2 | 25 | 27 | 25 (22–28) |
| Fat mass, % | 41 | 39 | 22 (11–33) |
| Fat-free mass, % | 59 | 61 | 78 (67–88) |
| Supine catecholamines | |||
| Plasma norepinephrine, pg/mL | <4 | 190 | 80–520 |
| Plasma epinephrine, pg/mL | <4 | <4 | 0-40 |
| Plasma dopamine, pg/mL | 169 | 23 | <20 |
| Upright catecholamines | |||
| Plasma norepinephrine, pg/mL | <4 | 553 | 80–520 |
| Plasma epinephrine, pg/mL | <4 | 5 | 4–66 |
| Plasma dopamine, pg/mL | 223 | 147 | <20 |
| Fasting insulin, μU/mL | 25 | 36 | 7 (4–11) |
| Fasting glucose, mg/dL | 91 | 91 | 94 (83–104) |
| HOMA2-IR | 3.2 | 4.4 | 1.0 (0.7–1.4) |
| Insulin secretion | |||
| InsulinΔ0–10 min, μU/mL | 170 | 191 | 42 (24–60) |
| InsulinΔ90–120 min, μU/mL | 224 | 159 | 42 (22–62) |
| ISI, μU/mL × 100 | 3.5 | 8.9 | 30 (14–45) |
| Parameter | DBH-Deficient Patient (Naive) | DBH-Deficient Patient (Droxidopa) | Healthya |
|---|---|---|---|
| Age, y | 15 | 16 | 26 (22–30) |
| Weight, kg | 79 | 83 | 74 (62–91) |
| BMI, kg/m2 | 25 | 27 | 25 (22–28) |
| Fat mass, % | 41 | 39 | 22 (11–33) |
| Fat-free mass, % | 59 | 61 | 78 (67–88) |
| Supine catecholamines | |||
| Plasma norepinephrine, pg/mL | <4 | 190 | 80–520 |
| Plasma epinephrine, pg/mL | <4 | <4 | 0-40 |
| Plasma dopamine, pg/mL | 169 | 23 | <20 |
| Upright catecholamines | |||
| Plasma norepinephrine, pg/mL | <4 | 553 | 80–520 |
| Plasma epinephrine, pg/mL | <4 | 5 | 4–66 |
| Plasma dopamine, pg/mL | 223 | 147 | <20 |
| Fasting insulin, μU/mL | 25 | 36 | 7 (4–11) |
| Fasting glucose, mg/dL | 91 | 91 | 94 (83–104) |
| HOMA2-IR | 3.2 | 4.4 | 1.0 (0.7–1.4) |
| Insulin secretion | |||
| InsulinΔ0–10 min, μU/mL | 170 | 191 | 42 (24–60) |
| InsulinΔ90–120 min, μU/mL | 224 | 159 | 42 (22–62) |
| ISI, μU/mL × 100 | 3.5 | 8.9 | 30 (14–45) |
Values represent mean (95% confidence interval).
Healthy represents reference data obtained, using identical methods, from 5 previously studied healthy female volunteers.
Metabolic and Neurohumoral Profile of a DBH-Deficient Patient
| Parameter | DBH-Deficient Patient (Naive) | DBH-Deficient Patient (Droxidopa) | Healthya |
|---|---|---|---|
| Age, y | 15 | 16 | 26 (22–30) |
| Weight, kg | 79 | 83 | 74 (62–91) |
| BMI, kg/m2 | 25 | 27 | 25 (22–28) |
| Fat mass, % | 41 | 39 | 22 (11–33) |
| Fat-free mass, % | 59 | 61 | 78 (67–88) |
| Supine catecholamines | |||
| Plasma norepinephrine, pg/mL | <4 | 190 | 80–520 |
| Plasma epinephrine, pg/mL | <4 | <4 | 0-40 |
| Plasma dopamine, pg/mL | 169 | 23 | <20 |
| Upright catecholamines | |||
| Plasma norepinephrine, pg/mL | <4 | 553 | 80–520 |
| Plasma epinephrine, pg/mL | <4 | 5 | 4–66 |
| Plasma dopamine, pg/mL | 223 | 147 | <20 |
| Fasting insulin, μU/mL | 25 | 36 | 7 (4–11) |
| Fasting glucose, mg/dL | 91 | 91 | 94 (83–104) |
| HOMA2-IR | 3.2 | 4.4 | 1.0 (0.7–1.4) |
| Insulin secretion | |||
| InsulinΔ0–10 min, μU/mL | 170 | 191 | 42 (24–60) |
| InsulinΔ90–120 min, μU/mL | 224 | 159 | 42 (22–62) |
| ISI, μU/mL × 100 | 3.5 | 8.9 | 30 (14–45) |
| Parameter | DBH-Deficient Patient (Naive) | DBH-Deficient Patient (Droxidopa) | Healthya |
|---|---|---|---|
| Age, y | 15 | 16 | 26 (22–30) |
| Weight, kg | 79 | 83 | 74 (62–91) |
| BMI, kg/m2 | 25 | 27 | 25 (22–28) |
| Fat mass, % | 41 | 39 | 22 (11–33) |
| Fat-free mass, % | 59 | 61 | 78 (67–88) |
| Supine catecholamines | |||
| Plasma norepinephrine, pg/mL | <4 | 190 | 80–520 |
| Plasma epinephrine, pg/mL | <4 | <4 | 0-40 |
| Plasma dopamine, pg/mL | 169 | 23 | <20 |
| Upright catecholamines | |||
| Plasma norepinephrine, pg/mL | <4 | 553 | 80–520 |
| Plasma epinephrine, pg/mL | <4 | 5 | 4–66 |
| Plasma dopamine, pg/mL | 223 | 147 | <20 |
| Fasting insulin, μU/mL | 25 | 36 | 7 (4–11) |
| Fasting glucose, mg/dL | 91 | 91 | 94 (83–104) |
| HOMA2-IR | 3.2 | 4.4 | 1.0 (0.7–1.4) |
| Insulin secretion | |||
| InsulinΔ0–10 min, μU/mL | 170 | 191 | 42 (24–60) |
| InsulinΔ90–120 min, μU/mL | 224 | 159 | 42 (22–62) |
| ISI, μU/mL × 100 | 3.5 | 8.9 | 30 (14–45) |
Values represent mean (95% confidence interval).
Healthy represents reference data obtained, using identical methods, from 5 previously studied healthy female volunteers.
Metabolic profile
The patient reported fatigue and weakness in her lower extremities with orthostasis that limited her ability to engage in physical activities. She had normal body mass index (BMI) with high adiposity (Table 1). Fasting plasma insulin was elevated, with normal glucose. Her homeostatic model assessment 2 index of insulin resistance (HOMA2-IR) was higher than defined cutoff levels for insulin resistance (8). Her acute and late glucose-stimulated insulin secretion was higher than the 95th percentile of reference data (Fig. 1) (6). Her ISI was lower than the 95th percentile of reference data (Table 1), consistent with insulin resistance.
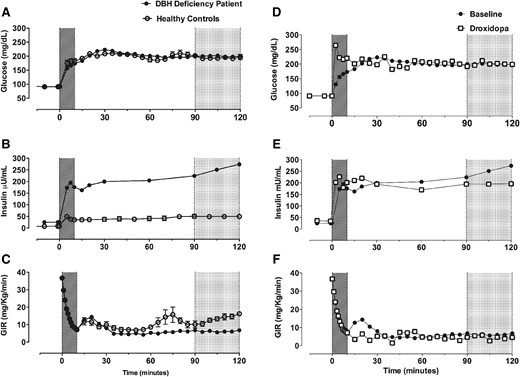
β-Cell function and insulin sensitivity in a DBH-deficient patient under treatment-naive conditions (baseline) and after norepinephrine replacement (droxidopa). (A) Plasma glucose was similarly clamped at ∼200 mg/dL during hyperglycemic clamps in the DBH-deficient patient (at baseline) and in healthy female control subjects (n = 5). (B) Plasma glucose was similarly clamped at ∼200 mg/dL during hyperglycemic clamps on baseline and droxidopa study days. (C) There was a greater increase in plasma insulin in response to hyperglycemia in the DBH-deficient patient at baseline. (D) Droxidopa did not alter the exaggerated increase in plasma insulin in response to hyperglycemia. (E) The glucose infusion rate (GIR) required to maintain hyperglycemia, a measure of insulin sensitivity, was lower in the DBH-deficient patient compared with healthy control subjects at baseline. (F) Droxidopa did not alter the GIR in the patient with DBH deficiency. Acute glucose-stimulated insulin response (times 0–10 min, hatched area); late glucose-stimulated insulin response (90–120 minutes, gray area)
Effect of droxidopa treatment
Droxidopa restored plasma norepinephrine and epinephrine and reduced dopamine (Table 1). The patient had ongoing evidence of sympathetic noradrenergic failure during autonomic function testing. She had improved orthostatic tolerance and was able to stand for 5 minutes despite decreased SBP (supine: 104 mm Hg; seated: 100 mm Hg; standing: 65 mm Hg). Appropriate reflex tachycardia was observed (supine: 62 bpm; seated: 81 bpm; standing: 100 bpm). Droxidopa did not improve BMI, adiposity, fasting insulin and glucose, or HOMA2-IR (Table 1). Acute glucose-stimulated insulin secretion was higher, and late glucose-stimulated insulin secretion was lower, after droxidopa (Fig. 1). The ISI was improved but was still below normal levels (Table 1).
Conclusions
DBH deficiency offers a unique model to evaluate the impact of chronic catecholamine depletion on glucose homeostasis. As expected, our patient had impaired sympathetic noradrenergic control of cardiovascular function. Similar to the mouse model (3), she exhibited hyperinsulinemia, increased glucose-stimulated insulin secretion, and insulin resistance. Restoration of plasma norepinephrine and epinephrine with droxidopa improved orthostatic tolerance but produced modest changes in glucose homeostasis. These findings provide insight into the pathophysiology and treatment of DBH deficiency and into the catecholaminergic mechanisms contributing to metabolic regulation.
DBH−/− mice are hyperphagic but 20% smaller than wild-type mice (3). Our DBH-deficient patient had normal BMI but increased adiposity and a metabolic phenotype more consistent with obese patients or patients with type 2 diabetes. This could reflect deconditioning, but this was not tested. DBH−/− mice exhibit hyperinsulinemia, which appears milder than the human phenotype and effectively reduces blood glucose concentrations by 25% (3). Hyperinsulinemia in DBH−/− mice is associated with loss of inhibitory α2-adrenergic receptor tone, with normal pancreatic morphology and insulin content (3). In our patient, hyperinsulinemia did not alter glucose levels. This disparate finding could reflect the lean phenotype in DBH−/− mice. Episodic hypoglycemia has been reported in 16% of DBH-deficient patients studied, but it is unclear if this is related to hyperinsulinemia (1).
Droxidopa improved orthostatic tolerance but had modest effects on glucose homeostasis. It increased acute and late glucose-stimulated insulin secretion and improved the ISI, a measure of insulin sensitivity. Changes in ISI were dependent on increased late-phase insulin secretion and did not translate to improved HOMA2-IR. These findings indicate that plasma norepinephrine-independent mechanisms regulate insulin action in DBH deficiency. The continued elevation of plasma dopamine does not likely contribute to hyperinsulinemia because dopamine inhibits insulin release (9). It is possible, however, that DBH deficiency results in changes in brain catecholamine turnover, which was not corrected by droxidopa. There could also be sustained developmental changes in neural circuitry regulating insulin release or defects in α2-adrenergic receptor responsiveness. The latter could be tested by determining if α2-receptor agonists lower insulin in DBH-deficient patients, similar to findings with clonidine in the mouse model (3). DBH−/− mice also had increased parasympathetic tone, which could enhance insulin release. Finally, nonautonomic factors could contribute, including alterations in β-cell structure and function or in hormones regulating insulin secretion including estrogen, leptin, fatty acids, and incretins (10). Potential limitations include that observations were limited to 1 DBH-deficient patient, and maturation of this patient over the course of the study could have a confounding effect on insulin secretion and action.
Abbreviations:
- BMI
body mass index
- bpm
beats per minute
- DBH
dopamine β-hydroxylase
- HOMA2-IR
homeostatic model assessment 2 insulin resistance
- ISI
insulin sensitivity index
- SBP
systolic blood pressure
Acknowledgments
This work was supported by National Institutes of Health Grants K23HL103976, P01HL056693, R00HL122507, UL1TR000445 and by a Clinical Scientist Career Development Award to C.A.S. by the Doris Duke Foundation.
Clinical trial registry: ClinicalTrials.gov no. NCT00748059 (registered 5 September 2008).
Disclosure Summary: S.R.R. is a consultant for Lundbeck, NA Ltd. and GE Healthcare and receives research support from Medtronic and the Canadian Institutes of Health Research. I.B. is a consultant for Lundbeck, NA Ltd. and Astra Zeneca and receives research support from Astra Zeneca and Forest Laboratories. D.R. is a consultant for Lundbeck, NA Ltd. C.A.S. is a consultant for Lundbeck, NA Ltd. and Theravance Biopharma. The remaining authors have nothing to disclose.
References
Author notes
Address all correspondence and requests for reprints to: Cyndya A. Shibao, MD, MSCI, Division of Clinical Pharmacology, Vanderbilt University Medical Center, 2222 Pierce Avenue, Room 506 RRB, Nashville, Tennessee 37232-6602. E-mail: [email protected].

{kind=link}