





Some patients with idiopathic hypogonadotropic hypogonadism (IHH) undergo spontaneous activation of their hypothalamic-pituitary-gonadal axis resulting in normalization of steroidogenesis and/or gametogenesis, a phenomenon termed reversal.
To assess the responsiveness of the GnRH neuronal network to exogenous kisspeptin administration in IHH patients who have undergone reversal.
Six men with congenital IHH and evidence for reversal.
Subjects underwent q10 min blood sampling to measure GnRH-induced LH secretion at baseline and in response to iv boluses of kisspeptin (0.24–2.4 nmol/kg) and GnRH (75 ng/kg).
Individuals with sustained reversal of their hypogonadotropism (spontaneous LH pulses) responded to exogenous kisspeptin with a GnRH-induced LH pulse. Individuals who had reversal but then subsequently suffered relapse of their IHH (loss of spontaneous LH pulsatility) did not respond to kisspeptin.
The ability of kisspeptin to stimulate a GnRH-induced LH pulse correlates with the presence of endogenous LH pulses. These data suggest that reversal of hypogonadotropism, and by extension sexual maturation, may be due to the acquisition of kisspeptin responsiveness.
Kisspeptin responsiveness only correlates with the presence of endogenous LH pulses in patients with idiopathic hypogonadotropic hypogonadism who undergo spontaneous activation of their hypothalamic-pituitary-gonadal axis.
Characterized by low sex steroids and low/normal gonadotropins in adulthood, idiopathic hypogonadotropic hypogonadism (IHH) is traditionally thought to be a lifelong condition. However, a significant proportion of patients with IHH experience reversal and demonstrate improvement in their reproductive endocrine function, including normalization of circulating sex steroids, spontaneous increases in testicular volume (TV), and paternity/maternity without the use of medications (1). Although several factors could contribute to the mechanisms underlying reversal, clinical features that can predict reversal have not yet been identified with confidence. One factor that has been hypothesized to contribute to reversal is steroid hormone treatment, the most common therapy used to treat hypogonadotropic patients (1). However, because virtually all patients with IHH are treated with exogenous sex steroids or drugs that induce endogenous sex-steroid production, yet only up to 22% of IHH patients undergo reversal, it seems unlikely that restoration of a normal sex-steroid milieu is the sole trigger for reversal (2, 3). A second factor that has been hypothesized to contribute to reversal is the underlying severity of the hypogonadotropic hypogonadism. Although reversal might be considered a “mild” subphenotype of IHH, this phenomenon is not restricted to individuals with mild forms of this disease. Rather, reversal can occur in patients with severe hypogonadotropic hypogonadism as evidenced by cryptorchidism, micropenis, complete absence of pubertal development, and/or absence of pulsatile LH secretion (3, 4). Finally, given the growing complexity of the genetic landscape of IHH, it is reasonable to postulate that reversal is associated with specific genetic causes of IHH. Although reversal is not restricted to mutations in any one IHH gene, there is an association between reversal and heterozygous rare, deleterious variants affecting the neurokinin B signaling pathway (3, 5), a pathway that is closely linked to kisspeptin biology.
Despite these challenges in identifying the factors that trigger reversal, patients who have experienced the reversal phenomenon represent an important biologic opportunity to explore hypotheses related to activators and suppressors of the reproductive cascade, and by extension, to explore the physiologic triggers of puberty. Theoretically, reversal could represent an awakening of the hypothalamic pituitary gonadal axis of a magnitude on par with the awakening of the reproductive endocrine cascade at puberty, a process that requires kisspeptin, a potent stimulator of GnRH secretion (6, 7). In the rodent, kisspeptin administration accelerates and kisspeptin blockade delays sexual maturation (8, 9). In the rhesus monkey, both hypothalamic kisspeptin expression and the GnRH-induced LH response to kisspeptin administration increase with pubertal maturation (10, 11). In the human, loss-of-function mutations in the genes encoding kisspeptin (KISS1) and its receptor (KISS1R) cause IHH (6, 12, 13), and a gain-of-function mutation in KISS1R has been reported in a patient with central precocious puberty (14).
When administered to healthy men and women, kisspeptin stimulates GnRH-induced LH secretion (15–19). Given that kisspeptin provides an opportunity to interrogate the functional integrity of the GnRH neuronal population in the hypothalamus, it has been employed as a physiologic probe in patients with disordered GnRH secretion and, in particular, those with IHH (2). Our initial experience utilizing exogenous kisspeptin administration to IHH patients with abiding disease, at the same dose and method of administration that yielded a robust response in healthy men, did not result in a GnRH-induced LH response (2). However, because reversal patients develop spontaneous GnRH-induced LH pulsations, and kisspeptin is a powerful stimulus for GnRH release, we hypothesized that the GnRH neuronal population of reversal patients would be kisspeptin “responsive.” In other words, we predicted that the GnRH neurons of patients who have experienced reversal of their hypogonadotropism, possibly a late onset form of delayed puberty, would respond to exogenous kisspeptin.
Materials and Methods
Subjects and eligibility criteria
This study was approved by the Institutional Review Board of Massachusetts General Hospital/Partners Healthcare, and all subjects gave written informed consent. IHH was defined as hypogonadal sex-steroid levels (testosterone <100 ng/dL in men; estradiol <20 pg/mL in women) in the setting of low or normal gonadotropin levels at age more than or equal to 18 years and the absence of any identifiable medical condition that could cause hypogonadotropic hypogonadism (20). As in our previous report (3), reversal of IHH in men was defined as: 1) fertility without use of exogenous GnRH or gonadotropin therapy; 2) serum testosterone greater than or equal to 250 ng/dL (after an appropriate washout of preexisting medical therapy); 3) TVs greater than or equal to 4 mL and at least 2 mL increase in TV in the absence of GnRH or gonadotropin therapy; or 4) LH pulse frequency and amplitude within the normal range for men (frequency, 5.1 ± 3.4 pulses/12 h; amplitude, 2.5 ± 2.1 IU/L [mean ± 2 SD]) (21). Of these 4 criteria used to define reversal, we previously observed that testicular growth in the absence of GnRH/gonadotropin therapy is the most frequently observed sign of attaining reproductive endocrine activity (3). Therefore, given the rarity of patients with reversal of IHH (up to 22% of 1 in 30 000) (3, 22), we focused recruitment on those individuals who demonstrated the requisite degree of TV growth on physical examination as this was interpreted as a surrogate marker of endogenous gonadotropin secretion.
Subjects also participated in a genetics study which included smell testing. Patient DNA was screened for rare sequence variants (RSVs) in CHD7 (MIM 608892), FGF8 (MIM 600483), FGFR1 (MIM 136350), GNRH1 (MIM 152760), GNRHR (MIM 138850), HS6ST1 (MIM 604846), KAL1 (MIM 300836), KISS1 (MIM 603286), KISS1R (MIM 604161), NSMF, previously called NELF (MIM 60813), PROK2 (MIM 607002), PROKR2 (MIM 607123), TAC3 (MIM 162330), and TACR3 (MIM 162332) by PCR amplification of exons followed by Sanger sequencing, as described previously (23). RSVs were defined as having a minor allele frequency of less than 1% in the 1000 Genomes Project and the National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project (24). RSVs were reported if they were predicted to be damaging by at least 2 out of 4 in silico prediction programs: PolyPhen-2 (25), SIFT (26), Mutation Taster (27), and Panther (28). Both self-report of olfaction as well as University of Pennsylvania Smell Identification Test scores, from a 40-item smell test, were used to classify olfactory capabilities (29).
Source of peptides
Kisspeptin 112–121, the 10-amino acid isoform of kisspeptin (corresponding to amino acids 112–121 of the preprohormone), and GnRH were synthesized using good manufacturing practices by NeoMPS (PolyPeptide Laboratories). Kisspeptin was made available through the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Study design
Subjects were admitted to the Clinical Research Center at Massachusetts General Hospital for detailed neuroendocrine phenotyping in which blood sampling was performed every 10 minutes for 8–24 hours to evaluate endogenous GnRH-induced LH secretion. Subjects then received iv bolus(es) of kisspeptin-10 at doses ranging from 0.24 to 2.4 nmol/kg. All subjects received at least one dose of kisspeptin at 0.24 nmol/kg, as previous work by our group demonstrated that this dose consistently elicits GnRH-induced LH pulses of physiologic amplitude in healthy men and healthy luteal-phase women (17, 18). A bolus of 75 ng/kg GnRH was administered at the conclusion of the study as our group has previously shown that this dose results in robust GnRH-induced LH responses (2) in individuals with intact gonadotrope function.
All subjects had evidence of reversal before study enrollment and one subject, subject 6, had previous evidence of reversal with relapse. Subject 6 had undergone baseline pulse profiling at the NIH, which demonstrated relapse of his hypogonadotropism (no pulsatility and hypogonadal testosterone levels) (Table 1 and see Figure 4B below); at his request, he was not removed from his exogenous testosterone therapy. He was treated with exogenous pulsatile 25 ng/kg GnRH every 2 hours for 6 days to ensure pituitary responsiveness before admission to the Clinical Research Center (2).
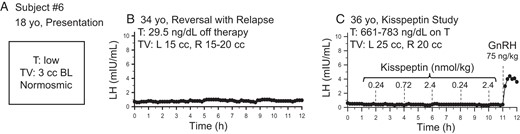
One male subject with reversal and relapse of hypogonadotropic hypogonadism.
Dashed lines indicate iv boluses. Note his initial clinical presentation (A), the flat baseline (B) and failure to respond to escalating kisspeptin doses (C). T, testosterone; TV, testicular volume.
Study Subject Characteristics
| ID | Initial Clinical Diagnosis | Evidence for Reversal | Evidence for Relapse | Kisspeptin Study | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at Dx | Smell Defect UPSIT | TV | LH | FSH | T | Age at Reversal | TV | LH | FSH | T | TV | LH | FSH | T | Age at Study | TV | LH | FSH | T | Kiss-10 Response | Category | |
| 1 | 19 y | Mild hyposmia | 12/12 | 16 | 21 | 15/15 | 265 | 26 | 15/20 | 2.3 | 3.6 | 276 | + | Reversal | ||||||||
| 2 | 17 y 11 mo | Mild hyposmia | 12/12 | 0.6 | 1.1 | 53 | 21 | 15/15 | 21 | 25/25 | 5 | 2.6 | 555 | + | Reversal | |||||||
| 3 | 18 y 8 mo | Normal | 12/12 | 1.15 | 0.32 | 28 | 19 y 9 mo | 20/20 | 520 | 21 | 25/25 | 379 | + | Reversal | ||||||||
| 4 | 18 y 2 mo | Mild hyposmia | Inguinal/4 | 2.3 | 2.3 | 63 | 19 | 10/12 | 24 | 10/15 | 2.3 | 3 | 374 | + | Reversal | |||||||
| 5 | 27 y | Reports no smell | 4/4 | 16 | 31 | 12/12 | 32 | 12/12 | 0.49 | 0.3 | 18 | − | Relapsed | |||||||||
| 6 | 17–18 y | Normal | 3/3 | Low | Low | Low | 34 | 15/20 | 15/20 | 0.8 | 0.9 | <20 | 36 | 25/20 | On exogenous T | − | Relapsed | |||||
| ID | Initial Clinical Diagnosis | Evidence for Reversal | Evidence for Relapse | Kisspeptin Study | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at Dx | Smell Defect UPSIT | TV | LH | FSH | T | Age at Reversal | TV | LH | FSH | T | TV | LH | FSH | T | Age at Study | TV | LH | FSH | T | Kiss-10 Response | Category | |
| 1 | 19 y | Mild hyposmia | 12/12 | 16 | 21 | 15/15 | 265 | 26 | 15/20 | 2.3 | 3.6 | 276 | + | Reversal | ||||||||
| 2 | 17 y 11 mo | Mild hyposmia | 12/12 | 0.6 | 1.1 | 53 | 21 | 15/15 | 21 | 25/25 | 5 | 2.6 | 555 | + | Reversal | |||||||
| 3 | 18 y 8 mo | Normal | 12/12 | 1.15 | 0.32 | 28 | 19 y 9 mo | 20/20 | 520 | 21 | 25/25 | 379 | + | Reversal | ||||||||
| 4 | 18 y 2 mo | Mild hyposmia | Inguinal/4 | 2.3 | 2.3 | 63 | 19 | 10/12 | 24 | 10/15 | 2.3 | 3 | 374 | + | Reversal | |||||||
| 5 | 27 y | Reports no smell | 4/4 | 16 | 31 | 12/12 | 32 | 12/12 | 0.49 | 0.3 | 18 | − | Relapsed | |||||||||
| 6 | 17–18 y | Normal | 3/3 | Low | Low | Low | 34 | 15/20 | 15/20 | 0.8 | 0.9 | <20 | 36 | 25/20 | On exogenous T | − | Relapsed | |||||
Dx, diagnosis; TV, testicular volume in cc; LH, luteinizing hormone in mIU/mL; FSH, follicle stimulating hormone in mIU/mL; T, testosterone in ng/dL; UPSIT, University of Pennsylvania Smell Identification Test.
Study Subject Characteristics
| ID | Initial Clinical Diagnosis | Evidence for Reversal | Evidence for Relapse | Kisspeptin Study | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at Dx | Smell Defect UPSIT | TV | LH | FSH | T | Age at Reversal | TV | LH | FSH | T | TV | LH | FSH | T | Age at Study | TV | LH | FSH | T | Kiss-10 Response | Category | |
| 1 | 19 y | Mild hyposmia | 12/12 | 16 | 21 | 15/15 | 265 | 26 | 15/20 | 2.3 | 3.6 | 276 | + | Reversal | ||||||||
| 2 | 17 y 11 mo | Mild hyposmia | 12/12 | 0.6 | 1.1 | 53 | 21 | 15/15 | 21 | 25/25 | 5 | 2.6 | 555 | + | Reversal | |||||||
| 3 | 18 y 8 mo | Normal | 12/12 | 1.15 | 0.32 | 28 | 19 y 9 mo | 20/20 | 520 | 21 | 25/25 | 379 | + | Reversal | ||||||||
| 4 | 18 y 2 mo | Mild hyposmia | Inguinal/4 | 2.3 | 2.3 | 63 | 19 | 10/12 | 24 | 10/15 | 2.3 | 3 | 374 | + | Reversal | |||||||
| 5 | 27 y | Reports no smell | 4/4 | 16 | 31 | 12/12 | 32 | 12/12 | 0.49 | 0.3 | 18 | − | Relapsed | |||||||||
| 6 | 17–18 y | Normal | 3/3 | Low | Low | Low | 34 | 15/20 | 15/20 | 0.8 | 0.9 | <20 | 36 | 25/20 | On exogenous T | − | Relapsed | |||||
| ID | Initial Clinical Diagnosis | Evidence for Reversal | Evidence for Relapse | Kisspeptin Study | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at Dx | Smell Defect UPSIT | TV | LH | FSH | T | Age at Reversal | TV | LH | FSH | T | TV | LH | FSH | T | Age at Study | TV | LH | FSH | T | Kiss-10 Response | Category | |
| 1 | 19 y | Mild hyposmia | 12/12 | 16 | 21 | 15/15 | 265 | 26 | 15/20 | 2.3 | 3.6 | 276 | + | Reversal | ||||||||
| 2 | 17 y 11 mo | Mild hyposmia | 12/12 | 0.6 | 1.1 | 53 | 21 | 15/15 | 21 | 25/25 | 5 | 2.6 | 555 | + | Reversal | |||||||
| 3 | 18 y 8 mo | Normal | 12/12 | 1.15 | 0.32 | 28 | 19 y 9 mo | 20/20 | 520 | 21 | 25/25 | 379 | + | Reversal | ||||||||
| 4 | 18 y 2 mo | Mild hyposmia | Inguinal/4 | 2.3 | 2.3 | 63 | 19 | 10/12 | 24 | 10/15 | 2.3 | 3 | 374 | + | Reversal | |||||||
| 5 | 27 y | Reports no smell | 4/4 | 16 | 31 | 12/12 | 32 | 12/12 | 0.49 | 0.3 | 18 | − | Relapsed | |||||||||
| 6 | 17–18 y | Normal | 3/3 | Low | Low | Low | 34 | 15/20 | 15/20 | 0.8 | 0.9 | <20 | 36 | 25/20 | On exogenous T | − | Relapsed | |||||
Dx, diagnosis; TV, testicular volume in cc; LH, luteinizing hormone in mIU/mL; FSH, follicle stimulating hormone in mIU/mL; T, testosterone in ng/dL; UPSIT, University of Pennsylvania Smell Identification Test.
Laboratory assays and pulse analysis
For 5 of the subjects, LH for each sample and testosterone on 2-hour pools were measured by direct immunoassay using the automated Abbott ARCHITECT system (Abbott Laboratories, Inc) as previously described (2). For one subject (number 3), LH and FSH were measured using Siemens IMMULITE 2000 (Siemens Healthcare Diagnostics, Inc), which has previously been shown to have good concordance with ARCHITECT (30). We further tested the concordance between IMMULITE and ARCHITECT LH assays in 132 samples representing a range of physiologic and pathophysiologic states and found tight correlation (R2 = 0.98), although IMMULITE values were about 20% higher than ARCHITECT. To allow data analysis across all 6 subjects, the data from subject 3 were converted to Architect units using the following formula: ARCHITECT = (IMMULITE + 0.35)/1.2. For subject 3, testosterone was measured by Elecsys (Roche Diagnostics) (31).
LH pulses were identified using a validated modification of the Santen and Bardin method (32, 33) augmented by a deconvolution algorithm as previously described (17). Pulse amplitude was calculated as the difference between time 0 of kisspeptin administration and peak of the pulse. An amplitude of 0 was assigned if LH decreased after kisspeptin. Area under the curve (AUC) for each pulse was calculated by trapezoid integration as previously described (34). For these calculations, the time of kisspeptin administration was always considered the start of a pulse, even if no pulse was present (in which case AUC was close to 0). For subjects who underwent reversal of their IHH and who therefore produced endogenous LH pulses, 2 analyses were performed. 1) The likelihood that kisspeptin administration coincided with endogenous pulses by chance was calculated based on the assumption that these constitute Bernoulli trials, where “success” consists of the coincidence of a pulse within a 10-minute window of the exogenous administration of kisspeptin (18). The probability of an observed number of successes occurring strictly by chance is given by the binomial probability, where the underlying probability of a pulse is computed from the frequency of endogenous pulses during baseline frequent sampling. Equivalently, the null hypothesis is that all increases in LH occurred independently of kisspeptin administration and were due to endogenous, and temporally independent, reproductive endocrine activity (35). Note that temporal independence ignores the refractory period both after endogenous pulses as well as kisspeptin-induced pulses. The extent to which this is a confounding effect remains an open question and therefore a second method of analysis was undertaken to examine kisspeptin as a pulse generator. 2) The second method of analysis was through examination of the interpulse intervals (IPIs). In particular, the IPI would be expected to decrease if exogenous kisspeptin stimulated an LH pulse independent of those occurring spontaneously. The IPIs were therefore compared between baseline and over the course of kisspeptin administration. P < .05 was considered significant. Unless otherwise noted, values are reported as mean ± SD. Clinicaltrials.gov registration number, NCT00914823.
Results
Subject characteristics
Six men with documented evidence of IHH and later evidence for reversal participated in this study. After age 18, subjects demonstrated spontaneous increases in TV in the absence of exogenous gonadotropins or GnRH therapy (Table 1). Around the time that reversal occurred, all men were treated with testosterone therapy. One man was anosmic, and 3 men had mild hyposmia by formal smell testing (36).
Although men with IHH are traditionally thought to have prepubertal size testes (TV <4 cc), some have evidence of partial pubertal development (37). Indeed, 3 of the 6 men in this study had robust TVs at the time of diagnosis; these volumes had been achieved in the absence of exogenous gonadotropin or GnRH therapy (Table 1). All men, even those who initially presented with robust TV, had further increases in TV, a testimony to their reversal. In addition to increases in TV, 2 men (subjects 1 and 3) had normalization of endogenous testosterone levels.
All subjects were screened for mutations in IHH genes. In 5 of the 6 subjects, we identified no RSVs in IHH genes that were predicted to be deleterious. In subject 2, we identified the variant FGFR1 p.V427L, which is predicted to be pathogenic by 2 prediction programs (PolyPhen-2 and Mutation Taster). In this European family, all affected individuals (the study subject and 2 aunts with IHH) were found to carry this variant. The study subject inherited this variant from his father, who did not have IHH, but who reported delayed puberty. The variant was not found in 33 000 non-Finnish European individuals in the Exome Aggregation Consortium (38). Although no in vitro studies on the functional consequences of this variant are published, the available data suggests that this variant may contribute to the reproductive endocrine phenotypes in this family. No subjects carried variants in KISS1R, GNRH1, or GNRHR, suggesting that all subjects possessed the receptors and ligands needed to respond to exogenously administered kisspeptin and GnRH.
Response to kisspeptin administration in subjects with sustained reversal
Four subjects demonstrated spontaneous pulsatile LH secretion at baseline, with a mean LH pulse amplitude of 1.5 ± 0.3 mIU/mL and a pulse frequency of 1.0 ± 0.3 pulse every 2 hours, testifying to the robust nature of their reversal. All 4 of these subjects responded to exogenous kisspeptin with an LH pulse within 30 minutes of kisspeptin administration. To demonstrate that the kisspeptin iv bolus did not correspond to the appearance of a LH pulse by chance, 3 subjects were given multiple boluses of kisspeptin (Figure 1, A, C, and D). The likelihood that a resulting pulse occurred within 10 minutes of kisspeptin administration due to an endogenous pulse was found to be statistically unlikely, P < .01 in all 3 study subjects. In addition, IPIs between endogenous, and kisspeptin-induced pulses, were examined: in all 3 subjects, the mean IPI from the nadir of the pulse preceding kisspeptin administration to the nadir of pulse induced by kisspeptin administration was shorter by 17–28 minutes per individual compared to the IPI between endogenous pulses and were similar to the same kisspeptin induced intervals seen in healthy men as previously published (Supplemental Figure 1) (17). In addition, the decrease between median IPI of a pulse “with” as compared with “without” kisspeptin ranged from 10–35 minutes for all 3 individuals.
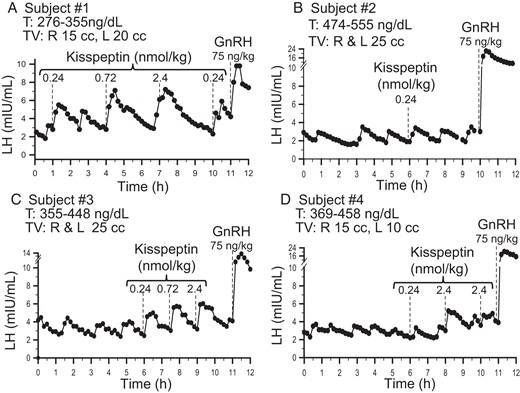
Four male subjects (A–D) with reversal of hypogonadotropic hypogonadism.
Dashed lines indicate iv boluses. T, testosterone; TV: testicular volume.
A range of doses of kisspeptin was administered (0.24–2.4 nmol/kg) to examine the dose-response relationship between kisspeptin and LH AUC. In general, increasing the kisspeptin dose produced pulses with increasing LH AUC. As was seen in previous published data on kisspeptin responsiveness in healthy men, subject 1 had initiated an endogenous LH pulse when he received exogenous kisspeptin (2.4 nmol/kg) (Figure 1A), thereby altering pulse morphology (17). In subject 4, only the kisspeptin 2.4 nmol/kg dose induced a LH pulse larger than his endogenous pulses (Figure 2).
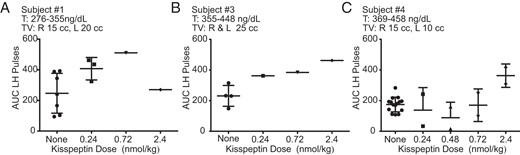
Area under the curve (AUC) for all LH pulses for 3 male subjects (A–C) who received multiple boluses of kisspeptin.
Bars are mean ± SD. T, testosterone; TV, testicular volume.
Responses to kisspeptin in subjects with reversal and subsequent relapse of hypogonadotropism
In contrast to the 4 subjects who had robust endogenous pulsatile LH secretion, 2 subjects did not have LH pulses at baseline, consistent with “relapse” back to the hypogonadotropic state. Subject 5 reported hypogonadal symptoms upon discontinuing his testosterone therapy for study participation, suggesting that he had undergone relapse. Consistent with this, subject 5 demonstrated no spontaneous LH pulses during the study (Figure 3B). In addition, he failed to respond to all kisspeptin boluses (Figure 3B). Despite his failure to exhibit robust responses to even high doses of exogenous kisspeptin, he clearly responded to exogenous GnRH (LH pulse amplitude, 3.5 mIU/mL) (Figure 3B), demonstrating the functional integrity of his pituitary gonadotropes.
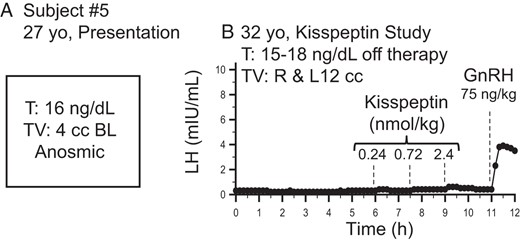
One male subject with reversal and relapse of hypogonadotropic hypogonadism at initial clinical presentation (A) and during the kisspeptin study (B).
Dashed lines indicate iv boluses. T, testosterone; TV, testicular volume; BL, bilateral.
Subject 6 was already known to have relapsed at the time of study participation. Because he had already been placed on sex-steroid treatment and was reluctant to discontinue this therapy, he participated in the study protocol while remaining on testosterone. To ensure that his pituitary was capable of responding to exogenous kisspeptin administration, he was treated with exogenous pulsatile GnRH for 1 week before study admission. Similar to subject 5 above, subject 6 failed to respond to exogenous kisspeptin but did respond to exogenous GnRH (LH pulse amplitude, 4.0 mIU/mL) (Figure 4C).
Discussion
Our study subjects divided themselves into 2 groups, those with sustained reversal (as evidenced by the presence of spontaneous LH pulses) and those with relapse of their reversal (as evidenced by an absence of LH pulses). Initially, we hypothesized that all subjects, whether their reversal was sustained or halted, would respond to kisspeptin, but our data did not support this hypothesis. Assumptions of this hypothesis are that 1) part of the mechanism underlying reversal involves the acquisition of kisspeptin responsiveness; and 2) the ability to respond to kisspeptin is retained over time. At the conclusion of this study, we observed that only those individuals with sustained reversal responded to kisspeptin with a GnRH-induced LH response; those with relapse did not. Thus, the ability to respond to kisspeptin with a GnRH-induced LH pulse correlated with the activity of the endogenous GnRH pulse generator.
Variability in the response to kisspeptin is already known to occur in several in vivo models. In the human adult, we and others have demonstrated that healthy women are sensitive to exogenous kisspeptin administration in the luteal and periovulatory phases of the menstrual cycle but relatively resistant in the follicular phase (18, 39), an observation that is poorly understood but might relate to differences in the sex-steroid milieu (40). Although no data are yet available in children, in prepubertal and juvenile animals, the LH response to exogenous kisspeptin administration increases across sexual maturation (41, 42). Finally, women with hypothalamic amenorrhea (an acquired, as opposed to a congenital, form of hypogonadotropism) respond robustly to kisspeptin (43). Taken together, these observations, from several different animal and human models, lend credence to the possibility that reversal of IHH is due to the acquisition of kisspeptin responsiveness, a hypothesis supported by the data generated in those reversal individuals with spontaneous LH pulses. Therefore, the sensitivity to kisspeptin may be conceptualized as a spectrum of dose responsiveness, with the extremes of the curve represented on one side by healthy males (sensitive), and on the other side, abiding IHH males (resistant). Patients with sustained reversal of their IHH represent a shift of the healthy male dose response curve (Figure 5).
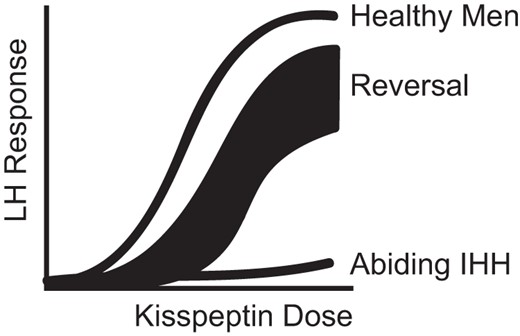
Model of kisspeptin responsiveness.
It appears that patients who have suffered relapse of their reversal lose their ability to respond to kisspeptin; the reasons for this loss are unclear. It is possible that these relapsed individuals, even if tested during a period of spontaneous hypothalamic-pituitary activity, might not be responsive to kisspeptin. Thus, it is possible that not all GnRH-induced LH pulsations are kisspeptin dependent in all physiological situations, a concept supported by our physiologic investigations in kisspeptin mutant mice, which revealed kisspeptin independent reproductive activity that was insufficient to restore the reproductive axis (44).
Although IHH is typically conceptualized as a “GnRH-deficient” state, some patients secrete enough GnRH to sustain nonpulsatile LH secretion. These individuals can evince robust responses to single boluses of exogenous GnRH even in the absence of formalized pituitary priming with pulsatile hormone administration (2). Subject 5 demonstrates this concept. Although he did not have LH pulses at baseline, and was not primed with exogenous pulsatile GnRH, he demonstrated a LH response to a single bolus of GnRH hormone. Extending this concept beyond the pituitary gonadotrophs, it is reasonable to consider whether priming is required at the hypothalamic level. Animal studies suggest that GnRH neurons do not require priming. The Kiss1−/− mouse responds to exogenous kisspeptin the first time it is injected; so do prepubertal monkeys (11, 45). Whether human GnRH neurons require long term priming with kisspeptin in order to respond to kisspeptin is less clear. We have administered multiple boluses of kisspeptin to IHH patients over 8–12 hours without effect, but it is possible that days/week of exposure are required (2). Conversely, for IHH individuals with reversal and relapse, GnRH neurons could have lost their ability to respond to exogenous kisspeptin in the time interval between relapse and study participation.
Clinically, testicular volume, typically considered a bedside surrogate for the ongoing activation of the hypothalamic-pituitary-gonadal axis, did not predict GnRH responsiveness to kisspeptin. For example, subject 5 had an increase in TV from 4 cc (27 y) to 12 cc (32 y). This testicular growth would typically be considered robust evidence of reversal. However, during his inpatient admission (32 y), subject 5 had no demonstrable spontaneous LH pulses and no response to kisspeptin administration. Subject 6 also had a marked increase in testicular size, no spontaneous LH pulses, and failed to mount a response to kisspeptin. Marked testicular growth in the absence of LH pulses and in the context of low testosterone levels is counter to the conventional wisdom that gonadotropin secretion is required for testicular growth. It is likely that sustained intervals of gonadotropin activation of the testes were present at some point in each of these subjects, but frequent, intensive, neuroendocrine monitoring would have been required to capture such activity. Regardless, the absence of LH pulses along with an absent response to kisspeptin suggests a fundamental absence of reproductive endocrine activity at least at the level of the GnRH neuron.
In addition to testicular volume, another factor which was not associated with kisspeptin responsiveness was the sex-steroid milieu. All 4 men with sustained reversal had robust LH pulses, normal testosterone levels, and unequivocal responses to kisspeptin and GnRH (testifying to the integrity of the GnRH neurons and the pituitary gonadotropes, respectively). However, subject 5, with relapse, was hypogonadal with a testosterone level of 15–18 ng/dL. He did not respond to kisspeptin. Subject 6 received kisspeptin while he was simultaneously being treated with testosterone. Despite testosterone levels squarely within the normal male range, subject 6 did not respond to kisspeptin at any dose. Therefore, the ability to respond to kisspeptin in men with reversal with relapse was not facilitated by the restoration of a normal sex-steroid milieu.
We present an integrated model that suggests dynamic changes in kisspeptin responsiveness may be responsible for both reversal and puberty. Future studies examining kisspeptin responsiveness in children across puberty, and testing very high doses of kisspeptin in IHH patients may provide additional information for this model. Clinically, this study demonstrates the ability of exogenous kisspeptin to stimulate GnRH-induced LH secretion in those patients with sustained reversal and an active GnRH pulse generator, and raises the possibility that kisspeptin could be used as an adjunctive tool to assess for reversal in the clinical setting.
Acknowledgments
We thank the research subjects, members of the Massachusetts General Hospital Reproductive Endocrine Unit for discussions and reading of the manuscript, staff of the Harvard Catalyst Clinical Research Center and NIH Clinical Research Center for assistance with the frequent sampling studies, the Massachusetts General Hospital Clinical Laboratory Research Core, and Patrick Sluss for assistance with assays. We also thank physicians who referred research subjects: Maria Yialamas, Leena Nahata, Joshua Safer, and Ronald Lechan. S.B.S. is a Robert and Laura Reynolds Research Scholar.
This work was supported by grants R01 HD043341 and P50 HD028138 from the Eunice Kennedy Shriver National Institute for Child Health and Human Development (NICHD) and the Harvard Catalyst Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health Awards UL1 RR 025758 and UL1 TR000170 and financial contributions from Harvard University and its affiliated academic health care centers). S.B.S. was supported by the NIH NICHD Grant K24 HD067388. Y.-M.C. was supported by a Doris Duke Clinical Scientist Development Award (Grant 2013110). M.F.L. was supported by the NIH NICHD Grant F32 HD078083 and by a Postdoctoral Fellowship Award for Clinical Research from the Massachusetts General Hospital Executive Committee on Research Fund for Medical Discovery. A.D. was supported by the NIH Intramural Research Program of NICHD.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AUC
area under the curve
- IHH
idiopathic hypogonadotropic hypogonadism
- IPI
interpulse interval
- RSV
rare sequence variant
- TV
testicular volume.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}