





Congenital generalized lipodystrophy (CGL) is a rare autosomal recessive disorder characterized by near-total lack of body fat.
We aimed to study natural history and disease burden of various subtypes of CGL.
We attempted to ascertain nearly all patients with CGL in Turkey.
This was a nationwide study.
Participants included 33 patients (22 families) with CGL and 30 healthy controls.
We wanted to ascertain genotypes by sequencing of the known genes. Whole-body magnetic resonance imaging was used to investigate the extent of fat loss. Metabolic abnormalities and end-organ complications were measured on prospective follow-up.
Analysis of the AGPAT2 gene revealed four previously reported and four novel mutations (CGL1; c.144C>A, c.667_705delinsCTGCG, c.268delC, and c.316+1G>T). Analysis of the BSCL2 gene revealed four different homozygous and one compound heterozygous possible disease-causing mutations (CGL2), including four novel mutations (c.280C>T, c.631delG, c.62A>T, and c.465-468delGACT). Two homozygous PTRF mutations (c.481-482insGTGA and c.259C>T) were identified (CGL4). Patients with CGL1 had preservation of adipose tissue in the palms, soles, scalp, and orbital region, and had relatively lower serum adiponectin levels as compared to CGL2 patients. CGL4 patients had myopathy and other distinct clinical features. All patients developed various metabolic abnormalities associated with insulin resistance. Hepatic involvement was more severe in CGL2. End-organ complications were observed at young ages. Two patients died at age 62 years from cardiovascular events.
CGL patients from Turkey had both previously reported and novel mutations of the AGPAT2, BSCL2, and PTRF genes. Our study highlights the early onset of severe metabolic abnormalities and increased risk of end-organ complications in patients with CGL.
We report 33 patients with CGL from a nationwide study from Turkey. Our study highlights the early onset of severe metabolic abnormalities and end-organ complications in CGL.
Congenital generalized lipodystrophy (CGL) is an autosomal recessive disorder characterized by near-total lack of body fat (1). Mutations in the 1-acylglycerol-3-phosphate O-acyltransferase 2 (AGPAT2) gene cause CGL, type 1 (CGL1) (2). AGPAT2 is a key enzyme required for the formation of phosphatidic acid, an important intermediate in the synthesis of triacylglycerol and glycerophospholipids (3). Mutations in the Berardinelli-Seip congenital lipodystrophy 2 (BSCL2) gene are responsible for CGL, type 2 (CGL2) (4). This gene encodes the multipass transmembrane protein seipin that localizes to the endoplasmic reticulum and may be important for lipid droplet morphology (5). Caveolin 1 (CAV1), which encodes a major fatty acid-binding protein on the plasma membranes of the adipocytes that translocates to lipid droplets from the plasma membrane in response to free fatty acids, is mutated in CGL, type 3 (CGL3) (6–8). Finally, mutations in the polymerase I and transcript release factor (PTRF) gene that encodes a caveolar-associated protein essential for formation of caveolae and proper localization, has been shown to cause CGL, type 4 (CGL4) (9, 10).
Standard care in CGL involves achieving metabolic control that is mainly based on the management of diabetes, hypertriglyceridemia, and hepatic steatosis. Recently, metreleptin, a recombinant analog of human leptin, has been approved as an adjunct to diet as replacement therapy to treat the complications of leptin deficiency in patients with generalized lipodystrophy (11). Metreleptin has been demonstrated to improve metabolic abnormalities in patients with CGL (12, 13). It has orphan drug designation in the European Union. However, the drug is not available in Turkey and Middle Eastern countries. This provides a unique opportunity to us in Turkey to study natural history and disease burden of various subtypes of CGL for which there is paucity of data. Therefore, we attempted to ascertain nearly all patients with CGL in this nationwide study. We report on clinical characteristics and mutational analysis of 33 patients from 22 families. All patients were prospectively followed-up for metabolic abnormalities and end-organ complications.
Materials and Methods
Study population
The Turkish Lipodystrophy Study Group (TuLip) is committed to improving knowledge on lipodystrophy syndromes by bringing together physicians taking care of such patients. It registers patients with lipodystrophy and provides genetic testing and serum adipocytokine assays in Turkey. The TuLip registry, at present, includes 106 non-HIV-associated lipodystrophy patients including 33 CGL patients (22 families). The patients included in this study were ascertained during a period of 15 years. The median follow-up was 60 months (3–180 months). All regions of Turkey were covered. The main diagnostic criterion for CGL was the presence of generalized fat loss at birth or early stages of life. The diagnosis of CGL was supported by other clinical characteristics such as overly muscular appearance, prominent superficial veins, pseudoacromegaloid features, and metabolic abnormalities associated with insulin resistance. A control group consisted of 30 age- and gender-matched subjects. A similar proportion of pediatric controls were included. The study was approved by the Dokuz Eylul University Ethics Review Panel. Written informed consent was obtained from all of the patients and control subjects or their parents.
Mutation analyses
Genomic DNA was isolated from peripheral blood cells using standard techniques. Mutation analyses were performed by bidirectional sequencing of the coding exons and the exon-intron boundaries of the AGPAT2, BSCL2, CAV1, and PTRF genes. PCR primers used to amplify the regions of interests are available upon request. All 32 subjects for whom DNA was available were first sequenced for AGPAT2 and BSCL2, because previous studies (14–16) reported that CGL1 and 2 are the most prevalent subtypes. However, the PTRF was the first gene to screen in patients with coexisting myopathy and cardiac arrhythmias (9, 14). The PTRF and CAV1 genes were also sequenced in patients who were negative for disease causing mutations in the AGPAT2 and BSCL2 genes. Sequencing was performed with MiSeq V2 chemistry on MiSeq instrument (Illumina California). Analysis was performed with IGV software. PolyPhen-2 (17), SIFT (18), MutationTaster-2 (19), and Human Splicing Finder (20) were used to classify novel variants.
Follow-up protocol
Prospective follow-up data were collected by the members of TuLip in several centers. According to the follow-up protocol of TuLip, patients had detailed serum biochemistry and urinalysis for protein content at the time of diagnosis for CGL, after which they had these tests on a regular basis approximately every 6 months. Hepatic steatosis was assessed by high-resolution ultrasound with convex transducers (frequency bandwidth, 3–6 MHz). Serum alanine aminotransferase, aspartate aminotransferase, and γ-glutamyl transpeptidase levels were measured. Diabetes was defined according to the recommendations of American Diabetes Association (21). Hypertriglyceridemia and low level of high-density lipoprotein (HDL) cholesterol were defined according to the National Cholesterol Education Program Adult Treatment Panel III guidelines (22). Age-specific thresholds were used for children and adolescents (23, 24).
Final data collection
The registry was first reviewed retrospectively. Then, patients were asked to come for a visit to remeasure their serum biochemistry, collect serum for leptin/adiponectin assays, and update their clinical findings at the time of data collection. Blood was taken from the cannulated antecubital vein between 8:00 am and 9:00 am after a 10-hour overnight fasting. Blood glucose, HbA1c, insulin, triglyceride, and cholesterol levels were measured by standardized methods with appropriate quality control and quality assurance procedures. Homeostasis model assessment (HOMA) score, a method used to quantify insulin resistance and β-cell function, was calculated as fasting serum insulin (μIU/ml) × fasting plasma glucose (mmol/liter)/22.5. Serum leptin and adiponectin levels were measured with ELISA according to the manufacturer's instructions (Boster; Leptin: EK0439, sensitivity: <8 pg/ml; Adiponectin: EK0595, sensitivity: <60 pg/ml). Blood samples for leptin and adiponectin assays were transferred into tubes suitable for serum separation. Tubes were centrifuged at 3000 × g for 10 minutes. Serum samples were extracted, aliquoted, and stored at –80 C until analysis.
Magnetic resonance imaging studies
Whole-body magnetic resonance imaging (MRI) was performed to study the extent of fat loss and to search for residual fat depots. MRI studies were completed in 12 patients (7 with CGL1, 3 with CGL2, and 2 with CGL4). The MRI scans were acquired using a 1.5T MRI system with a six multichannel body coil (Gyroscan Intera, release 8.1; Philips Medical Systems). Hepatic steatosis was assessed by conventional MRI and magnetic resonance spectroscopy. Conventional MRI and magnetic resonance spectroscopy were performed by using a 1.5-T MRI device with a phased-array coil.
Statistical analysis
Statistical analysis was performed using Statistical Package of Social Science (SPSS Inc), version 15.0, for Windows. Data are expressed as median (25th–75th percentiles). Mann-Whitney U test was used for comparison of variables. Categorical variables were compared by the χ2 test. A P value less than .05 was accepted as statistically significant.
Results
There were 13 males and 20 females with CGL, ranging in age from 9 months to 64 years (Tables 1–3). These patients were ascertained from various regions of Turkey (Supplemental Figure 1). Eighteen of the 22 pedigrees were consanguineous (Supplemental Figures 2–4). Even those without known parental consanguinity had homozygous mutations in the proband suggesting inbreeding or endogamy. The mutations in CGL genes in the patients are reported in Tables 1–3. Analysis of the AGPAT2 gene in 16 patients (10 females and 6 males) from 10 families revealed four previously reported mutations: c.202C>T (p.R68*), c.646A>T (p.K216*), c.662-2A>C (IVS5-2A>C), and c.685G>T (p.E229*) (2, 15, 16, 25), and four novel mutations: c.144C>A (p.C48*), c.667_705delinsCTGCG (p.V223Lfs*19), c.268delC (p.R90Vfs*15), and c.316+1G>T (IVS2+1G>T) (Table 1).
Clinical Characteristics of Patients With AGPAT2 Mutations (CGL1)
| Patient ID Age/Gender | Mutation | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 1.1 30 y/F | c.662-2A>C (IVS5-2A>C) c.662-2A>C (IVS5-2A>C) | Pseudoacromegaly, acanthosis nigricans, PCO, hypertension, diabetes, retinopathy, proteinuria, ESRD (renal transplantation), CAD, diabetic neuropathy, diabetic foot ulcer, toe amputation | Insulin (260 U/day), metformin, fenofibrate, fish oil, zofenopril, ASA, nebivolol, prednisolone, tacrolimus, mycophenolic acid |
| 2.1 30 y/F | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | Pseudoacromegaly, acanthosis nigricans, PCO, bone cysts, splenic artery aneurysm, hypertension diabetes, retinopathy, proteinuria, kidney failure, diabetic neuropathy, recurrent acute pancreatitis | Insulin (140 U/day), gemfibrozil, atorvastatin, furosemide |
| 2.2 31 y/M | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | Pseudoacromegaly, acanthosis nigricans, bone cysts, diabetes, microalbuminuria | Metformin, fenofibrate |
| 3.1 22 y/F | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | PCO, acanthosis nigricans, hypertension, diabetes, proteinuria, diabetic neuropathy, recurrent acute pancreatitis | Insulin (210 U/day), metformin, pioglitazone, gemfibrozil, alfa lipoic acid, ramipril |
| 4.1 62 y/F | c.202C>T (p.R68a) c.202C>T (p.R68a) | Bone cysts, toxic multinodular nodular goiter (received radioiodine), hypertension, diabetes, retinopathy, microalbuminuria, CAD, died at age of 62 | Insulin (160 U/day), ramipril, carvedilol, atorvastatin, fenofibrate |
| 4.2 64 y/M | c.202C>T (p.R68a) c.202C>T (p.R68a) | Bone cysts, FMF, hypertension, diabetes, proteinuria, ESRD (on hemodialysis), diabetic neuropathy | Insulin (30 U/day), fenofibrate, amlodipine, metoprolol, doxazosin, colchicine alfa lipoic acid, sinopryl, erythropoietin |
| 5.1 62 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, seizures, atrial flutter, subclinical hypothyroidism, diabetes, elevated liver enzymes, proteinuria, renal failure, CAD, died at age of 62 | Insulin (60 U/day), fenofibrate, atorvastatin, phenytoin, levetiracetam, warfarin |
| 5.2 21 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, PCO, acanthosis nigricans, diabetes, retinopathy, recurrent acute pancreatitis | Insulin (64 U/day), metformin, fenofibrate, fish oil |
| 5.3 11 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, IFG, IGT, elevated liver enzymes | MNT only |
| 6.1 25 y/F | c.646A>T (p.K216a) c.646A>T (p.K216a) | Diabetes, elevated liver enzymes, retinopathy, proteinuria, diabetic neuropathy, foot ulcer, amputation | Insulin, fenofibrate, diltiazem |
| 7.1 12 y/F | c.667_705delinsCTGCG (p.V223Lfsa19)b c.667_705delinsCTGCG (p.V223Lfsa19)b | Elevated HOMA score | MNT only |
| 7.2 19 y/M | c.667_705delinsCTGCG (p.V223Lfsa19)b c.667_705delinsCTGCG (p.V223Lfsa19)b | IGT | MNT only |
| 8.1 3 y/F | c.268delC (p.R90Vfsa15)b c.268delC (p.R90Vfsa15)b | Elevated HOMA scorea,c | MNT only |
| 9.1 6 y/M | c.316 + 1G>T (IVS2 + 1G>T)b c.316 + 1G>T (IVS2 + 1G>T)b | Pseudoacromegaly, elevated HOMA scorec | MNT only |
| 9.2 11 y/M | c.316 + 1G>T (IVS2 + 1G>T)b c.316 + 1G>T (IVS2 + 1G>T)b | Pseudoacromegaly, acanthosis nigricans, IFG, elevated liver enzymesc | MNT only |
| 10.1 54 mo/M | c.685G>T (p.E229a) c.685G>T (p.E229a) | Pseudoacromegalyc | MNT only |
| Patient ID Age/Gender | Mutation | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 1.1 30 y/F | c.662-2A>C (IVS5-2A>C) c.662-2A>C (IVS5-2A>C) | Pseudoacromegaly, acanthosis nigricans, PCO, hypertension, diabetes, retinopathy, proteinuria, ESRD (renal transplantation), CAD, diabetic neuropathy, diabetic foot ulcer, toe amputation | Insulin (260 U/day), metformin, fenofibrate, fish oil, zofenopril, ASA, nebivolol, prednisolone, tacrolimus, mycophenolic acid |
| 2.1 30 y/F | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | Pseudoacromegaly, acanthosis nigricans, PCO, bone cysts, splenic artery aneurysm, hypertension diabetes, retinopathy, proteinuria, kidney failure, diabetic neuropathy, recurrent acute pancreatitis | Insulin (140 U/day), gemfibrozil, atorvastatin, furosemide |
| 2.2 31 y/M | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | Pseudoacromegaly, acanthosis nigricans, bone cysts, diabetes, microalbuminuria | Metformin, fenofibrate |
| 3.1 22 y/F | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | PCO, acanthosis nigricans, hypertension, diabetes, proteinuria, diabetic neuropathy, recurrent acute pancreatitis | Insulin (210 U/day), metformin, pioglitazone, gemfibrozil, alfa lipoic acid, ramipril |
| 4.1 62 y/F | c.202C>T (p.R68a) c.202C>T (p.R68a) | Bone cysts, toxic multinodular nodular goiter (received radioiodine), hypertension, diabetes, retinopathy, microalbuminuria, CAD, died at age of 62 | Insulin (160 U/day), ramipril, carvedilol, atorvastatin, fenofibrate |
| 4.2 64 y/M | c.202C>T (p.R68a) c.202C>T (p.R68a) | Bone cysts, FMF, hypertension, diabetes, proteinuria, ESRD (on hemodialysis), diabetic neuropathy | Insulin (30 U/day), fenofibrate, amlodipine, metoprolol, doxazosin, colchicine alfa lipoic acid, sinopryl, erythropoietin |
| 5.1 62 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, seizures, atrial flutter, subclinical hypothyroidism, diabetes, elevated liver enzymes, proteinuria, renal failure, CAD, died at age of 62 | Insulin (60 U/day), fenofibrate, atorvastatin, phenytoin, levetiracetam, warfarin |
| 5.2 21 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, PCO, acanthosis nigricans, diabetes, retinopathy, recurrent acute pancreatitis | Insulin (64 U/day), metformin, fenofibrate, fish oil |
| 5.3 11 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, IFG, IGT, elevated liver enzymes | MNT only |
| 6.1 25 y/F | c.646A>T (p.K216a) c.646A>T (p.K216a) | Diabetes, elevated liver enzymes, retinopathy, proteinuria, diabetic neuropathy, foot ulcer, amputation | Insulin, fenofibrate, diltiazem |
| 7.1 12 y/F | c.667_705delinsCTGCG (p.V223Lfsa19)b c.667_705delinsCTGCG (p.V223Lfsa19)b | Elevated HOMA score | MNT only |
| 7.2 19 y/M | c.667_705delinsCTGCG (p.V223Lfsa19)b c.667_705delinsCTGCG (p.V223Lfsa19)b | IGT | MNT only |
| 8.1 3 y/F | c.268delC (p.R90Vfsa15)b c.268delC (p.R90Vfsa15)b | Elevated HOMA scorea,c | MNT only |
| 9.1 6 y/M | c.316 + 1G>T (IVS2 + 1G>T)b c.316 + 1G>T (IVS2 + 1G>T)b | Pseudoacromegaly, elevated HOMA scorec | MNT only |
| 9.2 11 y/M | c.316 + 1G>T (IVS2 + 1G>T)b c.316 + 1G>T (IVS2 + 1G>T)b | Pseudoacromegaly, acanthosis nigricans, IFG, elevated liver enzymesc | MNT only |
| 10.1 54 mo/M | c.685G>T (p.E229a) c.685G>T (p.E229a) | Pseudoacromegalyc | MNT only |
Abbreviations: ASA, acetylsalicylic acid; EPO, erythropoietin; F, female; FMF, familial Mediterranean fever; M, male; MNT, medical nutrition therapy; PCO, polycystic ovaries.
Patients without hepatic steatosis.
Novel mutations.
Patients without hypertriglyceridemia.
Clinical Characteristics of Patients With AGPAT2 Mutations (CGL1)
| Patient ID Age/Gender | Mutation | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 1.1 30 y/F | c.662-2A>C (IVS5-2A>C) c.662-2A>C (IVS5-2A>C) | Pseudoacromegaly, acanthosis nigricans, PCO, hypertension, diabetes, retinopathy, proteinuria, ESRD (renal transplantation), CAD, diabetic neuropathy, diabetic foot ulcer, toe amputation | Insulin (260 U/day), metformin, fenofibrate, fish oil, zofenopril, ASA, nebivolol, prednisolone, tacrolimus, mycophenolic acid |
| 2.1 30 y/F | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | Pseudoacromegaly, acanthosis nigricans, PCO, bone cysts, splenic artery aneurysm, hypertension diabetes, retinopathy, proteinuria, kidney failure, diabetic neuropathy, recurrent acute pancreatitis | Insulin (140 U/day), gemfibrozil, atorvastatin, furosemide |
| 2.2 31 y/M | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | Pseudoacromegaly, acanthosis nigricans, bone cysts, diabetes, microalbuminuria | Metformin, fenofibrate |
| 3.1 22 y/F | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | PCO, acanthosis nigricans, hypertension, diabetes, proteinuria, diabetic neuropathy, recurrent acute pancreatitis | Insulin (210 U/day), metformin, pioglitazone, gemfibrozil, alfa lipoic acid, ramipril |
| 4.1 62 y/F | c.202C>T (p.R68a) c.202C>T (p.R68a) | Bone cysts, toxic multinodular nodular goiter (received radioiodine), hypertension, diabetes, retinopathy, microalbuminuria, CAD, died at age of 62 | Insulin (160 U/day), ramipril, carvedilol, atorvastatin, fenofibrate |
| 4.2 64 y/M | c.202C>T (p.R68a) c.202C>T (p.R68a) | Bone cysts, FMF, hypertension, diabetes, proteinuria, ESRD (on hemodialysis), diabetic neuropathy | Insulin (30 U/day), fenofibrate, amlodipine, metoprolol, doxazosin, colchicine alfa lipoic acid, sinopryl, erythropoietin |
| 5.1 62 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, seizures, atrial flutter, subclinical hypothyroidism, diabetes, elevated liver enzymes, proteinuria, renal failure, CAD, died at age of 62 | Insulin (60 U/day), fenofibrate, atorvastatin, phenytoin, levetiracetam, warfarin |
| 5.2 21 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, PCO, acanthosis nigricans, diabetes, retinopathy, recurrent acute pancreatitis | Insulin (64 U/day), metformin, fenofibrate, fish oil |
| 5.3 11 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, IFG, IGT, elevated liver enzymes | MNT only |
| 6.1 25 y/F | c.646A>T (p.K216a) c.646A>T (p.K216a) | Diabetes, elevated liver enzymes, retinopathy, proteinuria, diabetic neuropathy, foot ulcer, amputation | Insulin, fenofibrate, diltiazem |
| 7.1 12 y/F | c.667_705delinsCTGCG (p.V223Lfsa19)b c.667_705delinsCTGCG (p.V223Lfsa19)b | Elevated HOMA score | MNT only |
| 7.2 19 y/M | c.667_705delinsCTGCG (p.V223Lfsa19)b c.667_705delinsCTGCG (p.V223Lfsa19)b | IGT | MNT only |
| 8.1 3 y/F | c.268delC (p.R90Vfsa15)b c.268delC (p.R90Vfsa15)b | Elevated HOMA scorea,c | MNT only |
| 9.1 6 y/M | c.316 + 1G>T (IVS2 + 1G>T)b c.316 + 1G>T (IVS2 + 1G>T)b | Pseudoacromegaly, elevated HOMA scorec | MNT only |
| 9.2 11 y/M | c.316 + 1G>T (IVS2 + 1G>T)b c.316 + 1G>T (IVS2 + 1G>T)b | Pseudoacromegaly, acanthosis nigricans, IFG, elevated liver enzymesc | MNT only |
| 10.1 54 mo/M | c.685G>T (p.E229a) c.685G>T (p.E229a) | Pseudoacromegalyc | MNT only |
| Patient ID Age/Gender | Mutation | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 1.1 30 y/F | c.662-2A>C (IVS5-2A>C) c.662-2A>C (IVS5-2A>C) | Pseudoacromegaly, acanthosis nigricans, PCO, hypertension, diabetes, retinopathy, proteinuria, ESRD (renal transplantation), CAD, diabetic neuropathy, diabetic foot ulcer, toe amputation | Insulin (260 U/day), metformin, fenofibrate, fish oil, zofenopril, ASA, nebivolol, prednisolone, tacrolimus, mycophenolic acid |
| 2.1 30 y/F | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | Pseudoacromegaly, acanthosis nigricans, PCO, bone cysts, splenic artery aneurysm, hypertension diabetes, retinopathy, proteinuria, kidney failure, diabetic neuropathy, recurrent acute pancreatitis | Insulin (140 U/day), gemfibrozil, atorvastatin, furosemide |
| 2.2 31 y/M | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | Pseudoacromegaly, acanthosis nigricans, bone cysts, diabetes, microalbuminuria | Metformin, fenofibrate |
| 3.1 22 y/F | c.144C>A (p.C48a)b c.144C>A (p.C48a)b | PCO, acanthosis nigricans, hypertension, diabetes, proteinuria, diabetic neuropathy, recurrent acute pancreatitis | Insulin (210 U/day), metformin, pioglitazone, gemfibrozil, alfa lipoic acid, ramipril |
| 4.1 62 y/F | c.202C>T (p.R68a) c.202C>T (p.R68a) | Bone cysts, toxic multinodular nodular goiter (received radioiodine), hypertension, diabetes, retinopathy, microalbuminuria, CAD, died at age of 62 | Insulin (160 U/day), ramipril, carvedilol, atorvastatin, fenofibrate |
| 4.2 64 y/M | c.202C>T (p.R68a) c.202C>T (p.R68a) | Bone cysts, FMF, hypertension, diabetes, proteinuria, ESRD (on hemodialysis), diabetic neuropathy | Insulin (30 U/day), fenofibrate, amlodipine, metoprolol, doxazosin, colchicine alfa lipoic acid, sinopryl, erythropoietin |
| 5.1 62 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, seizures, atrial flutter, subclinical hypothyroidism, diabetes, elevated liver enzymes, proteinuria, renal failure, CAD, died at age of 62 | Insulin (60 U/day), fenofibrate, atorvastatin, phenytoin, levetiracetam, warfarin |
| 5.2 21 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, PCO, acanthosis nigricans, diabetes, retinopathy, recurrent acute pancreatitis | Insulin (64 U/day), metformin, fenofibrate, fish oil |
| 5.3 11 y/F | c.685G>T (p.E229a) c.685G>T (p.E229a) | Bone cysts, IFG, IGT, elevated liver enzymes | MNT only |
| 6.1 25 y/F | c.646A>T (p.K216a) c.646A>T (p.K216a) | Diabetes, elevated liver enzymes, retinopathy, proteinuria, diabetic neuropathy, foot ulcer, amputation | Insulin, fenofibrate, diltiazem |
| 7.1 12 y/F | c.667_705delinsCTGCG (p.V223Lfsa19)b c.667_705delinsCTGCG (p.V223Lfsa19)b | Elevated HOMA score | MNT only |
| 7.2 19 y/M | c.667_705delinsCTGCG (p.V223Lfsa19)b c.667_705delinsCTGCG (p.V223Lfsa19)b | IGT | MNT only |
| 8.1 3 y/F | c.268delC (p.R90Vfsa15)b c.268delC (p.R90Vfsa15)b | Elevated HOMA scorea,c | MNT only |
| 9.1 6 y/M | c.316 + 1G>T (IVS2 + 1G>T)b c.316 + 1G>T (IVS2 + 1G>T)b | Pseudoacromegaly, elevated HOMA scorec | MNT only |
| 9.2 11 y/M | c.316 + 1G>T (IVS2 + 1G>T)b c.316 + 1G>T (IVS2 + 1G>T)b | Pseudoacromegaly, acanthosis nigricans, IFG, elevated liver enzymesc | MNT only |
| 10.1 54 mo/M | c.685G>T (p.E229a) c.685G>T (p.E229a) | Pseudoacromegalyc | MNT only |
Abbreviations: ASA, acetylsalicylic acid; EPO, erythropoietin; F, female; FMF, familial Mediterranean fever; M, male; MNT, medical nutrition therapy; PCO, polycystic ovaries.
Patients without hepatic steatosis.
Novel mutations.
Patients without hypertriglyceridemia.
Clinical Characteristics of Patients With BSCL2 Mutations (CGL2)
| Patient ID Age/Gender | Mutations | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 11.1 25 y/F | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Pseudoacromegaly, acanthosis nigricans, PCO, bone cysts, mild mental retardation, diabetes, retinopathy, microalbuminuria | Insulin (110 U/day), metformin, fenofibrate |
| 11.2 19 y/M | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Pseudoacromegaly, acanthosis nigricans, bone cysts, mild mental retardation, diabetes, elevated liver enzymes, proteinuria | Metformin |
| 12.1 11 y/M | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Elevated HOMA score, elevated liver enzymes, cirrhosis | Metformin, MCT oil, ursodeoxycholic acid |
| 13.1 11 y/F | c.631delG (p.V211a) c.631delG (p.V211a) | Elevated HOMA score, elevated liver enzymes | Metformin |
| 14.1 25 y/F | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Pseudoacromegaly, acanthosis nigricans, PCO, mild mental retardation, diabetes, elevated liver enzymes, cirrhosis, retinopathy, proteinuria, ESRD (on hemodialysis), diabetic neuropathy, diabetic foot ulcer | Insulin (72 U/day), fenofibrate, propranolol, furosemide, EPO, calcium, calcitriol |
| 14.2 19 y/M | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, cardiomyopathy, hypertension, diabetes, elevated liver enzymes | Metformin, gliclazide |
| 14.3 16 y/M | c.280C>T (p.Q94a)b c.280C>T (p.Q94*)b | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, cardiomyopathy, diabetes, elevated liver enzymes, microalbuminuria | Insulin (54 U/day), gemfibrozil, ramipril |
| 15.1 11 mo/F | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Elevated HOMA score, elevated liver enzymes | Metformin |
| 16.1 64 mo/M | c.62A>T (p.Q21 liter)b c.133G>A (p.G45S) | Mild mental retardation, elevated liver enzymes | MNT only |
| 16.2 37 mo/F | c.62A>T (p.Q21 liter)b c.133G>A (p.G45S) | Mild mental retardationa | MNT only |
| 17.1 9 mo/M | c.465-468delGACT (p.T156RfsX8)b c.465-468delGACT (p.T156RfsX8)b | Acanthosis nigricans, elevated HOMA score, elevated liver enzymes | MNT only |
| Patient ID Age/Gender | Mutations | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 11.1 25 y/F | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Pseudoacromegaly, acanthosis nigricans, PCO, bone cysts, mild mental retardation, diabetes, retinopathy, microalbuminuria | Insulin (110 U/day), metformin, fenofibrate |
| 11.2 19 y/M | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Pseudoacromegaly, acanthosis nigricans, bone cysts, mild mental retardation, diabetes, elevated liver enzymes, proteinuria | Metformin |
| 12.1 11 y/M | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Elevated HOMA score, elevated liver enzymes, cirrhosis | Metformin, MCT oil, ursodeoxycholic acid |
| 13.1 11 y/F | c.631delG (p.V211a) c.631delG (p.V211a) | Elevated HOMA score, elevated liver enzymes | Metformin |
| 14.1 25 y/F | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Pseudoacromegaly, acanthosis nigricans, PCO, mild mental retardation, diabetes, elevated liver enzymes, cirrhosis, retinopathy, proteinuria, ESRD (on hemodialysis), diabetic neuropathy, diabetic foot ulcer | Insulin (72 U/day), fenofibrate, propranolol, furosemide, EPO, calcium, calcitriol |
| 14.2 19 y/M | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, cardiomyopathy, hypertension, diabetes, elevated liver enzymes | Metformin, gliclazide |
| 14.3 16 y/M | c.280C>T (p.Q94a)b c.280C>T (p.Q94*)b | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, cardiomyopathy, diabetes, elevated liver enzymes, microalbuminuria | Insulin (54 U/day), gemfibrozil, ramipril |
| 15.1 11 mo/F | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Elevated HOMA score, elevated liver enzymes | Metformin |
| 16.1 64 mo/M | c.62A>T (p.Q21 liter)b c.133G>A (p.G45S) | Mild mental retardation, elevated liver enzymes | MNT only |
| 16.2 37 mo/F | c.62A>T (p.Q21 liter)b c.133G>A (p.G45S) | Mild mental retardationa | MNT only |
| 17.1 9 mo/M | c.465-468delGACT (p.T156RfsX8)b c.465-468delGACT (p.T156RfsX8)b | Acanthosis nigricans, elevated HOMA score, elevated liver enzymes | MNT only |
Abbreviations: EPO, erythropoietin; F, female; MCT, medium-chain triglycerides; M, male; MNT, medical nutrition therapy; PCO, polycystic ovaries.
Patients without hepatic steatosis.
Indicates novel mutations.
Clinical Characteristics of Patients With BSCL2 Mutations (CGL2)
| Patient ID Age/Gender | Mutations | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 11.1 25 y/F | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Pseudoacromegaly, acanthosis nigricans, PCO, bone cysts, mild mental retardation, diabetes, retinopathy, microalbuminuria | Insulin (110 U/day), metformin, fenofibrate |
| 11.2 19 y/M | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Pseudoacromegaly, acanthosis nigricans, bone cysts, mild mental retardation, diabetes, elevated liver enzymes, proteinuria | Metformin |
| 12.1 11 y/M | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Elevated HOMA score, elevated liver enzymes, cirrhosis | Metformin, MCT oil, ursodeoxycholic acid |
| 13.1 11 y/F | c.631delG (p.V211a) c.631delG (p.V211a) | Elevated HOMA score, elevated liver enzymes | Metformin |
| 14.1 25 y/F | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Pseudoacromegaly, acanthosis nigricans, PCO, mild mental retardation, diabetes, elevated liver enzymes, cirrhosis, retinopathy, proteinuria, ESRD (on hemodialysis), diabetic neuropathy, diabetic foot ulcer | Insulin (72 U/day), fenofibrate, propranolol, furosemide, EPO, calcium, calcitriol |
| 14.2 19 y/M | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, cardiomyopathy, hypertension, diabetes, elevated liver enzymes | Metformin, gliclazide |
| 14.3 16 y/M | c.280C>T (p.Q94a)b c.280C>T (p.Q94*)b | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, cardiomyopathy, diabetes, elevated liver enzymes, microalbuminuria | Insulin (54 U/day), gemfibrozil, ramipril |
| 15.1 11 mo/F | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Elevated HOMA score, elevated liver enzymes | Metformin |
| 16.1 64 mo/M | c.62A>T (p.Q21 liter)b c.133G>A (p.G45S) | Mild mental retardation, elevated liver enzymes | MNT only |
| 16.2 37 mo/F | c.62A>T (p.Q21 liter)b c.133G>A (p.G45S) | Mild mental retardationa | MNT only |
| 17.1 9 mo/M | c.465-468delGACT (p.T156RfsX8)b c.465-468delGACT (p.T156RfsX8)b | Acanthosis nigricans, elevated HOMA score, elevated liver enzymes | MNT only |
| Patient ID Age/Gender | Mutations | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 11.1 25 y/F | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Pseudoacromegaly, acanthosis nigricans, PCO, bone cysts, mild mental retardation, diabetes, retinopathy, microalbuminuria | Insulin (110 U/day), metformin, fenofibrate |
| 11.2 19 y/M | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Pseudoacromegaly, acanthosis nigricans, bone cysts, mild mental retardation, diabetes, elevated liver enzymes, proteinuria | Metformin |
| 12.1 11 y/M | c.630 + 1G>A (IVS4 + 1G>A) c.630 + 1G>A (IVS4 + 1G>A) | Elevated HOMA score, elevated liver enzymes, cirrhosis | Metformin, MCT oil, ursodeoxycholic acid |
| 13.1 11 y/F | c.631delG (p.V211a) c.631delG (p.V211a) | Elevated HOMA score, elevated liver enzymes | Metformin |
| 14.1 25 y/F | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Pseudoacromegaly, acanthosis nigricans, PCO, mild mental retardation, diabetes, elevated liver enzymes, cirrhosis, retinopathy, proteinuria, ESRD (on hemodialysis), diabetic neuropathy, diabetic foot ulcer | Insulin (72 U/day), fenofibrate, propranolol, furosemide, EPO, calcium, calcitriol |
| 14.2 19 y/M | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, cardiomyopathy, hypertension, diabetes, elevated liver enzymes | Metformin, gliclazide |
| 14.3 16 y/M | c.280C>T (p.Q94a)b c.280C>T (p.Q94*)b | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, cardiomyopathy, diabetes, elevated liver enzymes, microalbuminuria | Insulin (54 U/day), gemfibrozil, ramipril |
| 15.1 11 mo/F | c.280C>T (p.Q94a)b c.280C>T (p.Q94a)b | Elevated HOMA score, elevated liver enzymes | Metformin |
| 16.1 64 mo/M | c.62A>T (p.Q21 liter)b c.133G>A (p.G45S) | Mild mental retardation, elevated liver enzymes | MNT only |
| 16.2 37 mo/F | c.62A>T (p.Q21 liter)b c.133G>A (p.G45S) | Mild mental retardationa | MNT only |
| 17.1 9 mo/M | c.465-468delGACT (p.T156RfsX8)b c.465-468delGACT (p.T156RfsX8)b | Acanthosis nigricans, elevated HOMA score, elevated liver enzymes | MNT only |
Abbreviations: EPO, erythropoietin; F, female; MCT, medium-chain triglycerides; M, male; MNT, medical nutrition therapy; PCO, polycystic ovaries.
Patients without hepatic steatosis.
Indicates novel mutations.
Clinical Characteristics of Subjects With PTRF Mutations (CGL4), Mutation-Negative Patients, and a CGL Patient With no DNA Available
| Patient ID Age/Gender | Mutations | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 18.1 | c.481_482insGTGA (p.K161Sfs*51) | Myopathy, percussion-induced muscle mounding, exercise-induced atrial and ventricular arrhythmias, atlantoaxial instability, scoliosis, tight heel cords, gastrointestinal dysmotility, IFG, IGTa | Propranolol, verapamil, flecainide |
| 16 y/F | c.481_482insGTGA (p.K161Sfs*51) | ||
| 19.1 | c.259C>T (p.Gln87a) | Myopathy, percussion-induced muscle mounding, exercise-induced atrial and ventricular arrhythmias, atlantoaxial instability, scoliosis, gastrointestinal dysmotility, IFG | Propranolol |
| 13 y/F | c.259C>T (p.Gln87a) | ||
| 20.1 12 y/M | Negative | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, elevated HOMA score, elevated liver enzymes, proteinuria, recurrent acute pancreatitis | MNT only |
| 21.1 24 y/F | Negative | Acanthosis nigricans, PCO, retroverted uterus, diabetes, elevated liver enzymes | Metformin |
| 21.2 22 y/F | Negative | Acanthosis nigricans, PCO, elevated HOMA score | Metformin |
| 22.1 29 y/F | No DNA available | Diabetes, elevated liver enzymes, retinopathy, microalbuminuria | Metformin, insulin (72 U/day), fenofibrate, ramipril |
| Patient ID Age/Gender | Mutations | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 18.1 | c.481_482insGTGA (p.K161Sfs*51) | Myopathy, percussion-induced muscle mounding, exercise-induced atrial and ventricular arrhythmias, atlantoaxial instability, scoliosis, tight heel cords, gastrointestinal dysmotility, IFG, IGTa | Propranolol, verapamil, flecainide |
| 16 y/F | c.481_482insGTGA (p.K161Sfs*51) | ||
| 19.1 | c.259C>T (p.Gln87a) | Myopathy, percussion-induced muscle mounding, exercise-induced atrial and ventricular arrhythmias, atlantoaxial instability, scoliosis, gastrointestinal dysmotility, IFG | Propranolol |
| 13 y/F | c.259C>T (p.Gln87a) | ||
| 20.1 12 y/M | Negative | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, elevated HOMA score, elevated liver enzymes, proteinuria, recurrent acute pancreatitis | MNT only |
| 21.1 24 y/F | Negative | Acanthosis nigricans, PCO, retroverted uterus, diabetes, elevated liver enzymes | Metformin |
| 21.2 22 y/F | Negative | Acanthosis nigricans, PCO, elevated HOMA score | Metformin |
| 22.1 29 y/F | No DNA available | Diabetes, elevated liver enzymes, retinopathy, microalbuminuria | Metformin, insulin (72 U/day), fenofibrate, ramipril |
Abbreviations: F, female; M, male; MNT, medical nutrition therapy; PCO, polycystic ovaries.
Patients without hepatic steatosis.
Clinical Characteristics of Subjects With PTRF Mutations (CGL4), Mutation-Negative Patients, and a CGL Patient With no DNA Available
| Patient ID Age/Gender | Mutations | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 18.1 | c.481_482insGTGA (p.K161Sfs*51) | Myopathy, percussion-induced muscle mounding, exercise-induced atrial and ventricular arrhythmias, atlantoaxial instability, scoliosis, tight heel cords, gastrointestinal dysmotility, IFG, IGTa | Propranolol, verapamil, flecainide |
| 16 y/F | c.481_482insGTGA (p.K161Sfs*51) | ||
| 19.1 | c.259C>T (p.Gln87a) | Myopathy, percussion-induced muscle mounding, exercise-induced atrial and ventricular arrhythmias, atlantoaxial instability, scoliosis, gastrointestinal dysmotility, IFG | Propranolol |
| 13 y/F | c.259C>T (p.Gln87a) | ||
| 20.1 12 y/M | Negative | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, elevated HOMA score, elevated liver enzymes, proteinuria, recurrent acute pancreatitis | MNT only |
| 21.1 24 y/F | Negative | Acanthosis nigricans, PCO, retroverted uterus, diabetes, elevated liver enzymes | Metformin |
| 21.2 22 y/F | Negative | Acanthosis nigricans, PCO, elevated HOMA score | Metformin |
| 22.1 29 y/F | No DNA available | Diabetes, elevated liver enzymes, retinopathy, microalbuminuria | Metformin, insulin (72 U/day), fenofibrate, ramipril |
| Patient ID Age/Gender | Mutations | Clinical Features, Metabolic Abnormalities, and End-Organ Complications | Current Treatment |
|---|---|---|---|
| 18.1 | c.481_482insGTGA (p.K161Sfs*51) | Myopathy, percussion-induced muscle mounding, exercise-induced atrial and ventricular arrhythmias, atlantoaxial instability, scoliosis, tight heel cords, gastrointestinal dysmotility, IFG, IGTa | Propranolol, verapamil, flecainide |
| 16 y/F | c.481_482insGTGA (p.K161Sfs*51) | ||
| 19.1 | c.259C>T (p.Gln87a) | Myopathy, percussion-induced muscle mounding, exercise-induced atrial and ventricular arrhythmias, atlantoaxial instability, scoliosis, gastrointestinal dysmotility, IFG | Propranolol |
| 13 y/F | c.259C>T (p.Gln87a) | ||
| 20.1 12 y/M | Negative | Pseudoacromegaly, acanthosis nigricans, mild mental retardation, elevated HOMA score, elevated liver enzymes, proteinuria, recurrent acute pancreatitis | MNT only |
| 21.1 24 y/F | Negative | Acanthosis nigricans, PCO, retroverted uterus, diabetes, elevated liver enzymes | Metformin |
| 21.2 22 y/F | Negative | Acanthosis nigricans, PCO, elevated HOMA score | Metformin |
| 22.1 29 y/F | No DNA available | Diabetes, elevated liver enzymes, retinopathy, microalbuminuria | Metformin, insulin (72 U/day), fenofibrate, ramipril |
Abbreviations: F, female; M, male; MNT, medical nutrition therapy; PCO, polycystic ovaries.
Patients without hepatic steatosis.
Analysis of the BSCL2 gene in 11 patients (5 females and 6 males) from 7 families determined 4 different homozygous and 1 compound heterozygous possible disease-causing mutations (Table 2). Four novel BSCL2 mutations were identified: c.280C>T (p.Q94*), c.631delG (p.V211*), c.62A>T (p.Q21L) and c.465-468delGACT (p.T156Rfs*8). The BSCL2 c.630+1G>A (IVS4+1G>A) and c.133G>A (p.G45S) mutations have been previously reported (4).
Two patients were identified as having CGL4 caused by PTRF mutations (Table 3). The PTRF c.481-482insGTGA (p.K161Sfs*51) homozygous mutation has been previously reported (10). The PTRF mutation c.259C>T (p.Gln87*) mutation has recently been reported in a single Turkish patient from Netherlands (26).
All sequence variants were absent in 100 unrelated Turkish subjects (data not shown), suggesting their pathogenic role in our patients. All novel nonsense variants [c.144C>A (p.C48*) in the AGPAT2 gene and c.280C>T (p.Q94*) in the BSCL2 gene] and insertion/deletions [c.667_705delinsCTGCG (p.V223Lfs*19) and c.268delC (p.R90Vfs*15) in the AGPAT2 gene; and c.631delG (p.V211*) and c.465-468delGACT (p.T156RfsX8) in the BSCL2 gene] were predicted to be “disease causing” by MutationTaster-2 in silico analysis software (probability value was 1 for all the mutations, which indicated high security of the predictions). The c.316+1G>T (IVS2+1G>T) mutation in the AGPAT2 gene was predicted to be “disease causing” by Human Splicing Finder (score, 87.34) and MutationTaster-2 (score, 0.74). The c.62A>T (p.Q21L) mutation in the BSCL2 gene was predicted to “deleterious” by SIFT software (score, 0.02) and “possibly damaging” by PolyPhen-2 software (score, 0.557). None of these novel mutations has been reported in the ExAC and 1000G databases.
Although fat loss was generalized in CGL1 patients, some fat was noted in the palms, soles, and scalp. Orbital fat was also preserved. However, no bone marrow fat was observed (Figure 1, C and D). Fat loss was generalized in CGL2 patients and also involved the palms, soles, scalp, orbital region, and bone marrow (Figures 1, E and F). On the other hand, despite marked loss of subcutaneous, intraabdominal, and intrathoracic fat, bone marrow fat was preserved in CGL4 patients (Figure 1G). Mild mental retardation was remarkable in patients with CGL2. Hypertrophic cardiomyopathy was evident in several patients with CGL2 (Table 2). Radiolucent bone lesions (cysts) were detected both in patients with CGL1 and CGL2 (Tables 1 and 2, Figures 2, D—F). CGL4 was associated with myopathy (increased serum creatine kinase levels), percussion-induced muscle mounding, exercise-induced atrial and ventricular arrhythmias, atlantoaxial instability, scoliosis, tight heel cords from shortening of the Achilles tendon, and gastrointestinal dysmotility (Table 3, Figure 2G).
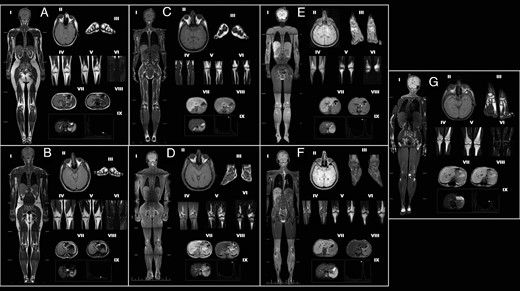
MRI scans and magnetic resonance spectroscopy findings of the controls and patients with CGL in each subgroup.
(A) Female control, 28 year-old healthy woman with normal fat distribution. (B) Male control, 26 year-old healthy man with normal fat distribution. (C) CGL1: patient 3.1, 22-year-old female; AGPAT2 c.144C>A (p.C48*). (D) CGL1: patient 2.2, 31-year-old male; AGPAT2 c.144C>A (p.C48*). (E) CGL2: patient 11.1, 25-year-old female; BSCL2 c.630+1G>A (IVS4+1G>A). (F) CGL2: patient 11.2, 19-year-old male; BSCL2 c.630+1G>A (IVS4+1G>A). (G) CGL4: patient 18.1, 16-year-old female; PTRF c.481_482insGTGA (p.K161Sfs*51). Whole-body T1-weighted imaging (I). Orbital fat, axial T1 (II). Soles, coronal T1 (III). Patellar region MRIs, coronal T1 (IV); coronal T2 (V); and fat suppression coronal T2 (VI) showing loss of bone marrow fat in CGL1 and CGL2. Bone marrow fat is preserved in CGL4. Bone cysts are visible in patients 2.2, 11.1, and 11.2. Dual-phase T1-weighted images (VII and VIII) show signal loss on out of phase images (VII) in affected patients, which is in consistent with variable degree of hepatic steatosis. magnetic resonance spectroscopy spectra of the affected patients indicate variable degree of hepatic steatosis (IX, arrows).
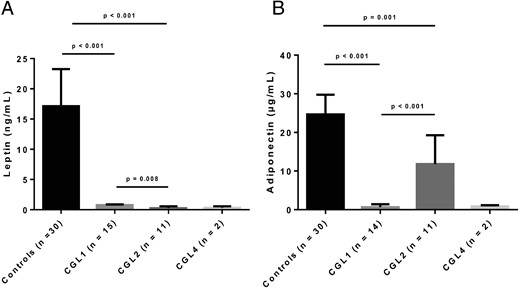
Comparison of serum leptin (A) and adiponectin levels (B) of patients with CGL1, CGL2, and CGL4.
Metabolic parameters of patients with CGL and healthy controls are shown in the Supplemental Table. More than one-half of the patients with CGL had severe hypertriglyceridemia despite lipid-lowering therapy. The median age at the onset of hypertriglyceridemia was 11 years (range, 2 months–46 years). Table 4 shows the comparison of metabolic characteristics of patients with CGL1 and CGL2. Hypertriglyceridemia was detected at a younger age in patients with CGL2. Four patients had recovered from several pancreatitis episodes (patients 2.1, 3.1, 5.2, and 20.1; Tables 1 and 3). All patients with an available serum leptin level had severe hypoleptinemia. Adiponectin levels were also markedly low, except in those with CGL2 who had moderately reduced levels of adiponectin. Leptin levels were lower in CGL2 compared to those with CGL1 (Figure 2A), whereas adiponectin levels were significantly higher (Figure 2B).
Comparison of Metabolic Characteristics in Patients With CGL1 and CGL2
| CGL1 (n = 16) | CGL2 (n = 11) | P Value | |
|---|---|---|---|
| Age | 22 (11–31) | 11 (3–19) | .054 |
| Gender (F/M) | 10/6 (F: 62.5%) | 5/6 (F: 45.5%) | .452 |
| Diabetes | 9 (56.3%) | 5 (45.5%) | .704 |
| The median age when diabetes develops | 17 (12–37) | 15 (7.5–17.5) | .19 |
| Hypertriglyceridemia | 12 (75%) | 11 (100%) | .123 |
| The median age when hypertriglyceridemia detected | 14 (10–26) | 1 (0.5–11) | .003 |
| Low HDL cholesterol | 15 (93.8%) | 10 (90.9%) | .786 |
| Hepatic steatosis | 15 (93.8%) | 10 (90.9%) | .786 |
| The median age when hepatic steatosis detected | 16 (10–29) | 3.5 (0.5–13) | .007 |
| Elevated liver enzymes | 4 (25%) | 9 (81.8%) | .006 |
| Cirrhosis | 0 | 2 (18.2%) | .138 |
| Pancreatitis | 3 (18.8%) | 0 | .248 |
| Retinopathy | 5 (31.3%) | 2 (18.2%) | .662 |
| Nephropathy | 8 (50%) | 4 (36.4%) | .696 |
| Neuropathy | 5 (31.3%) | 1 (9.1%) | .35 |
| Atherosclerosis | 3 (18.8%) | 0 | .248 |
| Glucose (mmol/liter) | 7.11 (5.09–12.25) | 5.11 (4.56–8) | .076 |
| HbA1c (%) | 7 (5.5–9.7) | 5.9 (4.9–8.2) | .251 |
| HOMA score | 8.11 (3.47–16.39) | 12.46 (3.75–29.42) | .331 |
| Triglyceride (mmol/liter) | 6.01 (1.86–13.81) | 4.58 (2.88–7.64) | .882 |
| Total cholesterol (mmol/liter) | 4.42 (3.83–6.01) | 4.24 (3.64–5.92) | .748 |
| LDL cholesterol (mmol/liter) | 2.16 (1.24–2.73) | 2.53 (1.73–3.93) | .183 |
| HDL cholesterol (mmol/liter) | 0.75 (0.65–0.95) | 0.7 (0.41–0.86) | .277 |
| ALT (IU/liter) | 28 (23–41) | 75 (60–109) | .003 |
| AST (IU/liter) | 22 (17–33) | 43 (29–85) | .013 |
| GGT (IU/liter) | 24 (14–70) | 85 (53–178) | .007 |
| Leptin (ng/ml) | 0.74 (0.4–0.87) | 0.26 (0.1–0.57) | .008 |
| Adiponectin (μg/ml) | 0.67 (0.39–1.43) | 11.77 (6.81–19.29) | <.001 |
| CGL1 (n = 16) | CGL2 (n = 11) | P Value | |
|---|---|---|---|
| Age | 22 (11–31) | 11 (3–19) | .054 |
| Gender (F/M) | 10/6 (F: 62.5%) | 5/6 (F: 45.5%) | .452 |
| Diabetes | 9 (56.3%) | 5 (45.5%) | .704 |
| The median age when diabetes develops | 17 (12–37) | 15 (7.5–17.5) | .19 |
| Hypertriglyceridemia | 12 (75%) | 11 (100%) | .123 |
| The median age when hypertriglyceridemia detected | 14 (10–26) | 1 (0.5–11) | .003 |
| Low HDL cholesterol | 15 (93.8%) | 10 (90.9%) | .786 |
| Hepatic steatosis | 15 (93.8%) | 10 (90.9%) | .786 |
| The median age when hepatic steatosis detected | 16 (10–29) | 3.5 (0.5–13) | .007 |
| Elevated liver enzymes | 4 (25%) | 9 (81.8%) | .006 |
| Cirrhosis | 0 | 2 (18.2%) | .138 |
| Pancreatitis | 3 (18.8%) | 0 | .248 |
| Retinopathy | 5 (31.3%) | 2 (18.2%) | .662 |
| Nephropathy | 8 (50%) | 4 (36.4%) | .696 |
| Neuropathy | 5 (31.3%) | 1 (9.1%) | .35 |
| Atherosclerosis | 3 (18.8%) | 0 | .248 |
| Glucose (mmol/liter) | 7.11 (5.09–12.25) | 5.11 (4.56–8) | .076 |
| HbA1c (%) | 7 (5.5–9.7) | 5.9 (4.9–8.2) | .251 |
| HOMA score | 8.11 (3.47–16.39) | 12.46 (3.75–29.42) | .331 |
| Triglyceride (mmol/liter) | 6.01 (1.86–13.81) | 4.58 (2.88–7.64) | .882 |
| Total cholesterol (mmol/liter) | 4.42 (3.83–6.01) | 4.24 (3.64–5.92) | .748 |
| LDL cholesterol (mmol/liter) | 2.16 (1.24–2.73) | 2.53 (1.73–3.93) | .183 |
| HDL cholesterol (mmol/liter) | 0.75 (0.65–0.95) | 0.7 (0.41–0.86) | .277 |
| ALT (IU/liter) | 28 (23–41) | 75 (60–109) | .003 |
| AST (IU/liter) | 22 (17–33) | 43 (29–85) | .013 |
| GGT (IU/liter) | 24 (14–70) | 85 (53–178) | .007 |
| Leptin (ng/ml) | 0.74 (0.4–0.87) | 0.26 (0.1–0.57) | .008 |
| Adiponectin (μg/ml) | 0.67 (0.39–1.43) | 11.77 (6.81–19.29) | <.001 |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT: γ-glutamyl transferase; LDL, low-density lipoprotein; NASH, nonalcoholic steatohepatitis.
Data are presented as median (25th–75th percentiles).
Comparison of Metabolic Characteristics in Patients With CGL1 and CGL2
| CGL1 (n = 16) | CGL2 (n = 11) | P Value | |
|---|---|---|---|
| Age | 22 (11–31) | 11 (3–19) | .054 |
| Gender (F/M) | 10/6 (F: 62.5%) | 5/6 (F: 45.5%) | .452 |
| Diabetes | 9 (56.3%) | 5 (45.5%) | .704 |
| The median age when diabetes develops | 17 (12–37) | 15 (7.5–17.5) | .19 |
| Hypertriglyceridemia | 12 (75%) | 11 (100%) | .123 |
| The median age when hypertriglyceridemia detected | 14 (10–26) | 1 (0.5–11) | .003 |
| Low HDL cholesterol | 15 (93.8%) | 10 (90.9%) | .786 |
| Hepatic steatosis | 15 (93.8%) | 10 (90.9%) | .786 |
| The median age when hepatic steatosis detected | 16 (10–29) | 3.5 (0.5–13) | .007 |
| Elevated liver enzymes | 4 (25%) | 9 (81.8%) | .006 |
| Cirrhosis | 0 | 2 (18.2%) | .138 |
| Pancreatitis | 3 (18.8%) | 0 | .248 |
| Retinopathy | 5 (31.3%) | 2 (18.2%) | .662 |
| Nephropathy | 8 (50%) | 4 (36.4%) | .696 |
| Neuropathy | 5 (31.3%) | 1 (9.1%) | .35 |
| Atherosclerosis | 3 (18.8%) | 0 | .248 |
| Glucose (mmol/liter) | 7.11 (5.09–12.25) | 5.11 (4.56–8) | .076 |
| HbA1c (%) | 7 (5.5–9.7) | 5.9 (4.9–8.2) | .251 |
| HOMA score | 8.11 (3.47–16.39) | 12.46 (3.75–29.42) | .331 |
| Triglyceride (mmol/liter) | 6.01 (1.86–13.81) | 4.58 (2.88–7.64) | .882 |
| Total cholesterol (mmol/liter) | 4.42 (3.83–6.01) | 4.24 (3.64–5.92) | .748 |
| LDL cholesterol (mmol/liter) | 2.16 (1.24–2.73) | 2.53 (1.73–3.93) | .183 |
| HDL cholesterol (mmol/liter) | 0.75 (0.65–0.95) | 0.7 (0.41–0.86) | .277 |
| ALT (IU/liter) | 28 (23–41) | 75 (60–109) | .003 |
| AST (IU/liter) | 22 (17–33) | 43 (29–85) | .013 |
| GGT (IU/liter) | 24 (14–70) | 85 (53–178) | .007 |
| Leptin (ng/ml) | 0.74 (0.4–0.87) | 0.26 (0.1–0.57) | .008 |
| Adiponectin (μg/ml) | 0.67 (0.39–1.43) | 11.77 (6.81–19.29) | <.001 |
| CGL1 (n = 16) | CGL2 (n = 11) | P Value | |
|---|---|---|---|
| Age | 22 (11–31) | 11 (3–19) | .054 |
| Gender (F/M) | 10/6 (F: 62.5%) | 5/6 (F: 45.5%) | .452 |
| Diabetes | 9 (56.3%) | 5 (45.5%) | .704 |
| The median age when diabetes develops | 17 (12–37) | 15 (7.5–17.5) | .19 |
| Hypertriglyceridemia | 12 (75%) | 11 (100%) | .123 |
| The median age when hypertriglyceridemia detected | 14 (10–26) | 1 (0.5–11) | .003 |
| Low HDL cholesterol | 15 (93.8%) | 10 (90.9%) | .786 |
| Hepatic steatosis | 15 (93.8%) | 10 (90.9%) | .786 |
| The median age when hepatic steatosis detected | 16 (10–29) | 3.5 (0.5–13) | .007 |
| Elevated liver enzymes | 4 (25%) | 9 (81.8%) | .006 |
| Cirrhosis | 0 | 2 (18.2%) | .138 |
| Pancreatitis | 3 (18.8%) | 0 | .248 |
| Retinopathy | 5 (31.3%) | 2 (18.2%) | .662 |
| Nephropathy | 8 (50%) | 4 (36.4%) | .696 |
| Neuropathy | 5 (31.3%) | 1 (9.1%) | .35 |
| Atherosclerosis | 3 (18.8%) | 0 | .248 |
| Glucose (mmol/liter) | 7.11 (5.09–12.25) | 5.11 (4.56–8) | .076 |
| HbA1c (%) | 7 (5.5–9.7) | 5.9 (4.9–8.2) | .251 |
| HOMA score | 8.11 (3.47–16.39) | 12.46 (3.75–29.42) | .331 |
| Triglyceride (mmol/liter) | 6.01 (1.86–13.81) | 4.58 (2.88–7.64) | .882 |
| Total cholesterol (mmol/liter) | 4.42 (3.83–6.01) | 4.24 (3.64–5.92) | .748 |
| LDL cholesterol (mmol/liter) | 2.16 (1.24–2.73) | 2.53 (1.73–3.93) | .183 |
| HDL cholesterol (mmol/liter) | 0.75 (0.65–0.95) | 0.7 (0.41–0.86) | .277 |
| ALT (IU/liter) | 28 (23–41) | 75 (60–109) | .003 |
| AST (IU/liter) | 22 (17–33) | 43 (29–85) | .013 |
| GGT (IU/liter) | 24 (14–70) | 85 (53–178) | .007 |
| Leptin (ng/ml) | 0.74 (0.4–0.87) | 0.26 (0.1–0.57) | .008 |
| Adiponectin (μg/ml) | 0.67 (0.39–1.43) | 11.77 (6.81–19.29) | <.001 |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT: γ-glutamyl transferase; LDL, low-density lipoprotein; NASH, nonalcoholic steatohepatitis.
Data are presented as median (25th–75th percentiles).
Sixteen patients with CGL developed diabetes during follow-up. The median age at the onset of diabetes was 16.5 years (range, 5–45 years). Diabetes developed at a slightly younger age in patients with CGL2, although the difference was not statistically significant (Table 4). Eight patients with diabetes had HbA1c values exceeding 8% and five of them had HbA1c exceeding 10% despite various glucose-lowering treatments. Insulin dose ranged from 30 U/d to 260 U/d. Five additional patients had impaired fasting glucose (IFG) or impaired glucose tolerance (IGT). Twelve patients had normal glucose tolerance; all were younger than age 16 years. HOMA scores were elevated in patients with CGL (median, 8.53 [25th–75th percentiles, 3.84–21.58]). Even CGL patients without diabetes had elevated HOMA scores (median, 6.44 [25–75 percentiles, 2.77–9.59]), except three very young patients with CGL1/CGL2 who had HOMA scores within normal limits (patient 10.1: CGL1, 54 months old; and patients 16.1 and 16.2: CGL2, 64 and 37 months old, respectively). Metabolic abnormalities were moderate, but present in patients with CGL4. Patient 18.1 had no metabolic abnormality apart from low HDL cholesterol when she was first diagnosed with CGL4 5 years ago (10). Thereafter, she developed hypertriglyceridemia at age 15, and IFG and IGT at age 16. Patient 19.1 also had IFG, low HDL cholesterol, hypertriglyceridemia, and hepatic steatosis.
Hepatic steatosis was present in 30 patients (15 with CGL1, 10 with CGL2, 1 with CGL4, 3 mutation negative patients, and 1 patient with no available DNA). The median age at the diagnosis of hepatic steatosis was 12 years (range, 6 months–46 years). Hepatic steatosis was not evident in two very young patients with CGL1/CGL2 (patient 8.1: CGL1, 3 years old; patient 16.2: CGL2, 37 months old). In addition, a 16-year-old patient with CGL4 had no hepatic steatosis, whereas the other patient with CGL4 had mild steatosis of the liver on MRI scans. Although hepatic steatosis was quite common in all subtypes of CGL, hepatic involvement was more severe in patients with CGL2. Hepatic steatosis was detected at a younger age in patients with CGL2 (Table 4). When compared to CGL1, patients with CGL2 had higher levels of alanine aminotransferase, aspartate aminotransferase, and γ-glutamyl transpeptidase. More patients with CGL2 had elevated liver enzymes (Table 4). Liver cirrhosis was also detected in two patients with CGL2 (patients 12.1 and 14.1). There was no history of underlying liver disorder, hepatotoxic agent use, or environmental exposures such as alcohol in these patients.
Eight patients were diagnosed with retinopathy, 14 with proteinuria or microalbuminuria, and 5 with renal failure (Tables 1–3). Patients 1.1 and 4.2 (CGL1) and patient 14.1 (CGL2) developed end-stage renal disease (ESRD). ESRD was treated with hemodialysis in patients 4.2 and 14.1, whereas patient 1.1 underwent a renal transplant from a living unrelated donor following a course of continue ambulatory peritoneal dialysis for 6 months. Diabetic neuropathy was evident in 6 patients. Patient 14.1 (CGL2) recovered from a diabetic foot ulcer. Patient 6.1 with (CGL1) had an amputation because of diabetic foot ulcer. Patient 1.1 (CGL1) developed recurrent neuropathic foot ulcers and underwent a toe amputation. She is still being treated for another neuropathic ulcer under the right metatarsal. Three patients were diagnosed with coronary artery disease (CAD). Two females with CGL1 (patients 4.1 and 5.1) died at 62 years of age, both from myocardial infarction.
There were two mutation-negative families in our registry (patients 20.1, 21.1, and 21.2) with consanguineous parents. Parents of patient 20.1 described the lack of body fat and muscular appearance since birth. He was diagnosed with proteinuria at age 3. The specific diagnosis of CGL was done at age 7 when he was admitted with acute pancreatitis. Hypertriglyceridemia and hepatic steatosis were also remarkable. Both leptin and adiponectin levels were extremely low (Supplemental Table). Genetic testing of candidate genes also including CAV1 revealed no disease-causing mutations. Two sisters in kindred 21 had a milder form of generalized fat loss that became obvious after puberty. The fat loss was particularly severe from the trunk, abdomen, and distal parts of the limbs. Genetic testing for CGL revealed no disease-causing mutations. We also sequenced for mutations in LMNA and PPARG, but there were no disease-causing mutation found. In one kindred (patient 22.1), DNA sample was not available for genetic testing.
Discussion
We report 33 patients with CGL from Turkey with a current population of approximately 78 million. Of these, two patients died recently. Thus, the estimated prevalence of CGL in Turkey is about 1 in 2 million, which is more common compared to its worldwide estimated prevalence of 1:10 million (14).
We confirm the previous reports (9, 10, 14–16, 25, 27) that CGL1 and CGL2 are the most prevalent subtypes of CGL, with CGL4 being extremely rare. We did not find any patient with CGL3 harboring CAV1 mutations. Some of our patients had previously reported variants in AGPAT2 and BSCL2, but we also reported several novel disease-causing variants in both genes. There were only four recurrent mutations in our families; two (c.144C>A and c.685G>T) in AGPAT2 and two (c.630+1G>A and c.280C>T) in BSCL2, in two families each. These data suggest that these variants may have passed on through generations in this region. Eighteen of the 22 families had known parental consanguinity. Of the remaining four pedigrees, two probands had homozygous AGPAT2 mutations, one proband had a homozygous PTRF mutation, and the DNA of the other proband was not available. On the other hand, in pedigree 16, despite parental consanguinity, the affected siblings had compound heterozygous BSCL2 mutations. Overall, these data reveal lack of one or two founder mutations in CGL patients from Turkey as has been noted in patients of Lebanese origin, nearly all of whom have the same c.315_319delGTATC BSCL2 mutation; in patients of African origin who frequently have the c.589-2A>G AGPAT2 variant; and in patients of Portuguese origin who have either the exon 3 and 4 del mutation in AGPAT2 or a c.669insA BSCL2 variant (14, 28).
Although patients with CGL1 and CGL2 mostly had overlapping phenotypic features in our registry, some minor differences may help distinguish CGL1 from CGL2. Despite generalized lack of fat, patients with CGL1 had well preserved adipose tissues in the palms, soles, scalp, and orbital region, as reported in a previous study (29). Although Haghighi et al (27) reported that bone cysts were noticed only in CGL1 patients in a very recent paper, we observed bone cysts both in patients with CGL1 and CGL2, which was in concordance with previous studies (30, 31). On the other hand, mild mental retardation and hypertrophic cardiomyopathy were observed only in patients with CGL2. Patients with CGL2 were younger, and male:female ratio in CGL1 was 6:10 but was 6:5 in CGL2, although these differences were not statistically significant. Interestingly, previous reports have reported similar trends (2, 29, 31). Considering that both CGL1 and CGL2 are autosomal recessive diseases, the likelihood that a child will inherit the mutated gene should be the same in females and males. In that case, because the fat loss is less severe in CGL1 compared to CGL2, diagnosis of CGL1 in males may be either missed or they die in utero. However, there are no reports of excess miscarriages in CGL1 families. Our personal observation also supports the lack of recognition of CGL1 phenotype in young males. For example, the parents of patient 2.1 repeatedly denied the occurrence of CGL in any other sibling. However, 6 years later, examination of her brother revealed CGL, which was confirmed by molecular testing. We also confirmed more severe fat loss and lower serum leptin but higher adiponectin levels in patients with CGL2 compared to those with CGL1 (32).
Our study provides prospective data on the natural course and disease burden of CGL patients especially related to metabolic abnormalities and end-organ complications. Both CGL1 and CGL2 patients develop very similar metabolic abnormalities characterized by poorly controlled diabetes developing during childhood or early adulthood, severe hypertriglyceridemia, and hepatic steatosis. Although hepatic steatosis is common in all subtypes of CGL, our data suggest that the liver is more severely affected in patients with CGL2. Liver enzymes were higher in patients with CGL2 compared to those with CGL1. Poor glycemic control was associated with micro- and macrovascular complications of diabetes. Retinopathy developed at least 3 years after diabetes was diagnosed, and CGL patients with poor glycemic control receiving high/extreme doses of insulin were mostly affected. Proteinuria was strikingly prevalent in the entire cohort, most likely due to diabetic kidney disease.
Among the end-stage organ diseases, cirrhosis was detected in two patients with CGL2 at an early age of 9 and 17 years, and three patients, aged 25–64 years, had ESRD requiring hemodialysis in two patients, and renal transplantation in another. Previously, only two CGL patients have been reported to have had renal transplantation, in one of them it was unsuccessful (33), whereas another one from a living-related haploidentical brother was successful (34). We also report two patients who died of cardiovascular events. The morbidity and mortality in CGL1 and CGL2 patients might be influenced by age, gender, diet, and presumably other genetic variants that may need further study.
Clinical features of patients with CGL4 are very distinctive from those of CGL1 and CGL2. While they suffer from coexisting myopathy, gastrointestinal dysmotility, and skeletal abnormalities, the major cause of premature death seems to catecholaminergic ventricular tachycardia and other arrhythmias, which can cause sudden death during childhood and adolescence (35). Metabolic abnormalities may be less severe in CGL4 patients compared to the two other subtypes (9, 10, 36). The affected members of two families had no mutation in any of the CGL genes, suggesting the possibility of additional loci which may be discovered using whole exome or genome sequencing.
There are a few limitations of our study. Although our study showed that CGL was relatively more prevalent in Turkey when compared to its worldwide estimated prevalence (14, 28), the TuLip registry may not have ascertained all CGL cases. We are aware of at least two additional patients (37). There could be more CGL cases elsewhere in Turkey, especially in the East and Southeast parts of the country where we were not able to ascertain a good number of patients. The existence of undiagnosed cases could also affect prevalence rates as was exemplified in family 2. Another patient with CGL phenotype (family 5), who died 18 years ago, was not included in the analysis, because no clinical data were available on him.
We conclude that the prevalence of CGL in Turkey may be more than what has been previously estimated. CGL patients from Turkey had both previously reported and novel mutations of the AGPAT2, BSCL2, and PTRF genes. Our study highlights the early onset of severe metabolic complications in these patients. These patients also develop end-organ complications resulting in cirrhosis and ESRD requiring organ transplantations and may die of premature cardiovascular disease.
Acknowledgments
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- CAD
coronary artery disease
- CGL
congenital generalized lipodystrophy
- ESRD
end-stage renal disease
- HDL
high-density lipoprotein
- HOMA
homeostasis model assessment
- IFG
impaired fasting glucose
- IGT
impaired glucose tolerance
- MRI
magnetic resonance imaging
- TuLip
Turkish Lipodystrophy Study Group.
References

{kind=link}
{kind=link}