





Abstract
The TSH receptor (TSHR) is considered the main target of stimulatory autoantibodies in the pathogenesis of Graves' ophthalmopathy (GO); however, it has been suggested that stimulatory IGF-1 receptor (IGF-1R) autoantibodies also play a role.
We previously demonstrated that a monoclonal stimulatory TSHR antibody, M22, activates TSHR/IGF-1R cross talk in orbital fibroblasts/preadipocytes obtained from patients with GO (GO fibroblasts [GOFs]). We show that cross talk between TSHR and IGF-1R, not direct IGF-1R activation, is involved in the mediation of GO pathogenesis stimulated by Graves' autoantibodies.
Immunoglobulins were purified from the sera of 57 GO patients (GO-Igs) and tested for their ability to activate TSHR and/or IGF-1R directly and TSHR/IGF-1R cross talk in primary cultures of GOFs. Cells were treated with M22 or GO-Igs with or without IGF-1R inhibitory antibodies or linsitinib, an IGF-1R kinase inhibitor.
Hyaluronan (hyaluronic acid [HA]) secretion was measured as a major biological response for GOF stimulation. IGF-1R autophosphorylation was used as a measure of direct IGF-1R activation. TSHR activation was determined through cAMP production.
A total of 42 out of 57 GO-Ig samples stimulated HA secretion. None of the GO-Ig samples exhibited evidence for IGF-1R autophosphorylation. Both anti-IGF-1R antibodies completely inhibited IGF-1 stimulation of HA secretion. By contrast, only 1 IGF-1R antibody partially blocked HA secretion stimulated by M22 or GO-Igs in a manner similar to linsitinib, whereas the other IGF-1R antibody had no effect on M22 or GO-Ig stimulation. These findings show that the IGF-1R is involved in GO-Igs stimulation of HA secretion without direct activation of IGF-1R.
IGF-1R activation by GO-Igs occurs via TSHR/IGF-1R cross talk rather than direct binding to IGF-1R, and this cross talk is important in the pathogenesis of GO.
Cross talk (or transactivation) between G protein-coupled receptors and receptor tyrosine kinases is a well-established signaling mechanism (1, 2). For example, the receptor tyrosine kinase for IGF-1 (IGF-1 receptor [IGF-1R]) has been shown to cross talk with a number of G protein-coupled receptors leading to cell proliferation or differentiation (3–5). More recently, interactions between TSH receptor (TSHR) and IGF-1R signaling were shown to occur in primary cultures of cells obtained from the retroorbital space of patients with Graves' ophthalmopathy (GO) (6–8). Specifically, we (6) showed there was a synergistic increase in secretion of hyaluronan (hyaluronic acid [HA]), a major component of the pathogenesis of GO (9), and a more than 10-fold increase in sensitivity to (or potency of) TSH when these GO fibroblasts (GOFs) were simultaneously stimulated by TSH and IGF-1. These and other findings have led some investigators to propose that both TSHR and IGF-1R were antigens involved in Graves' disease (GD) and that GD pathogenesis may involve the generation of autoantibodies that bind to and stimulate TSHR (10) and antibodies that bind to and stimulate IGF-1R (11, 12).
The immunoglobulin fractions of sera from GD and GO patients contain antibodies that bind to and activate TSHR production of cAMP (thyroid-stimulating antibodies [TSAbs]). TSAbs are well established as the primary cause of Graves' hyperthyroidism (10) and were shown to be closely associated with clinical activity and clinical severity of GO (13), suggesting their major role in GO pathogenesis (11). It is controversial, however, whether there are antibodies in GO patients's sera (GO-Igs) that directly bind to and activate IGF-1R. It has also been reported that antibodies engineered to inhibit binding to and activation of IGF-1Rs (14–17), one of which is in a clinical trial as therapy for GO (https://clinicaltrials.gov/ct2/show/NCT01868997?term=teprotumumab&rank=2), and a specific IGF-1R kinase inhibitor (6) partially inhibit stimulation of GOFs by TSH and by a monoclonal TSAb, M22, which exhibited a biphasic dose response of GOF stimulation (18). Because neither TSH nor M22 activates IGF-1R autophosphorylation, the major initiating step in the mechanism of IGF-1R signaling (19), we postulated that cross talk between TSHR and IGF-1R is involved in GOF activation by GO-Igs (6). A model of cross talk between TSHR and IGF-1R to explain the biphasic dose response of M22-stimulated HA secretion by GOFs is shown in Figure 1.
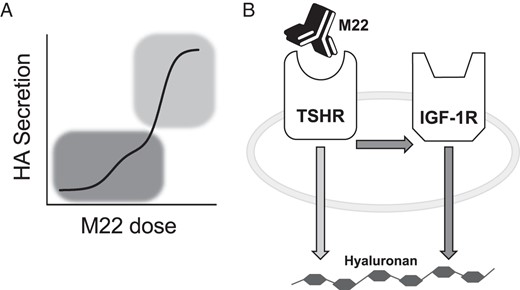
IGF-1R-dependent vs IGF-1R-independent TSHR stimulation of HA secretion in GOFs.
A, Biphasic M22 dose response of HA secretion from primary cultures of 3 GOF strains (n = 132). Only the high-potency, low M22 dose phase (dark gray) was dependent on the presence, not activation, of IGF-1R. The low-potency, high M22 concentration phase (light gray) was not affected by IGF-1R (6). B, Proposed model of receptor cross talk. After stimulation of GOFs with low doses of M22, TSHR commandeers molecular machinery associated with IGF-1R to up-regulate HA secretion. When stimulated with high doses of M22, TSHR up-regulates HA secretion through an IGF-1R-independent pathway.
The principle aim of this study was to determine whether direct binding to and activation of both TSHR and IGF-1R was required for full activation of GOFs or whether cross talk between TSHR and IGF-1R was initiated by binding to TSHR only. Using primary cultures of GOFs, we tested the ability of 2 IGF-1R blocking antibodies to inhibit the secretion of HA in response to stimulation by M22. We found that only 1 IGF-1R antibody had an effect on TSHR/IGF-1R cross talk, and used these differences to establish an experimental system where we could distinguish between IGF-1R activation resulting from binding to IGF-1R or TSHR-stimulated cross talk. We then applied our model to cross talk initiated by GO-Igs purified from 57 patients. We found no evidence for IGF-1R-stimulating antibodies in GO-Ig fractions although GO-Igs were capable of activating TSHR/IGF-1R cross talk. We conclude that IGF-1R activation by GO-Igs occurs via TSHR/IGF-1R cross talk after binding to TSHR, not by direct binding to/activation of IGF-1R, and that this cross talk is important in the pathogenesis of GO.
Materials and Methods
Patients and study approval
A total of 50 serum samples (GO Mainz [GOM]1–GOM50) from patients with phenotypically overt and clinically active GO (Supplemental Table 1) according to the classification of the Consensus Statement of the European Group on GO (20) was collected at the joint thyroid-eye-clinic of the Johannes Gutenberg University Medical Center. The collection of patients' serum samples has been approved by the Ethical Committee of the Medical Chamber of the State Rhineland-Palatinate, Germany and by the institutional review board (IRB) of the Johannes Gutenberg University Medical Center. Written informed consent was received from all patients with GO before blood withdrawal. Six patients with clinically active GO (Supplemental Table 1) (GO Bethesda [GOB]1, GOB8, GOB9, GOB10, GOB12, GOB13) and 1 patient with a history of exposure keratopathy associated with lid retraction (GOB11) were identified out of a cohort of patients with GD at the Diabetes, Endocrinology, and Obesity Branch, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health (NIH). Blood samples from these patients (clinical trial identifier, NCT00001159), and 10 euthyroid healthy volunteers (NCT00428987) were obtained under National Institute of Diabetes and Digestive and Kidney Diseases/National Institute of Arthritis and Musculoskeletal and Skin Diseases IRB-approved protocols after informed consent was obtained. Retroorbital adipose tissue was generously supplied by Dr Neil Miller, Dr Prem Subramanian, Dr Nicholas Mahoney, and Dr Shannath Merbs (Johns Hopkins School of Medicine, Baltimore, MD). Use of human tissues was approved by the Johns Hopkins and National Institute of Diabetes and Digestive and Kidney Diseases IRBs.
Materials
3-Isobutyl-1-methylxanthine was purchased from Sigma-Aldrich. Human/mouse polyclonal IGF-1R antibody AF305 and (R)-(+)-trans-4-(1-aminoethyl)-N-(4-pyridyl)-cyclohexanecarboxamide dihydrochloride (Y-27632) were purchased from R&D Systems. Recombinant human IGF-1 was purchased from Peprotech. IGF-1R mouse monoclonal antibody 1H7 was purchased from Abd Serotec. Thyroid-stimulating human monoclonal autoantibody (M22) was purchased from Kronus. IGF-1R receptor kinase inhibitor linsitinib was purchased from Selleckchem. HA ELISA kits were purchased from Corgenix. 1 000 000 HA were purchased from Lifecore Biomedical.
Isolation and culture of primary GOFs
Tissue was obtained from GO patients who underwent orbital decompression surgery. Orbital fibroblasts were isolated as previously described (6). Briefly, tissue explants were minced and plated in culture dishes containing complete growth media comprised of high-glucose DMEM with fetal bovine serum (FBS) (10% vol/vol), penicillin (100 U/mL), and streptomycin (100 μg/mL). Resulting monolayer outgrowths of adherent fibroblasts/preadipocytes were serially passaged with trypsin/EDTA and cultured in F-media designed to prevent the dedifferentiation of isolated cells. These media were comprised of DMEM with FBS (10% vol/vol), penicillin (100 U/mL), streptomycin (100 μg/mL), L-glutamine (2mM), Ham's F-12 nutrient mixture (25% vol/vol), hydrocortisone (25 ng/mL), epithelial growth factor (0.125 ng/mL), insulin (5 μg/mL), cholera toxin (11.7nM), gentamicin (10 μg/mL), Fungizone (250 ng/mL), and Y-27632 (5μM). Cells were maintained in a humidified 7% CO2 incubator at 37°C. Strains were used at passage 3–5. To ensure the same initial state for all experiments, cells were grown to confluence in F-media for approximately 1 week before serum starvation, pretreatment, and/or stimulation.
Purification of GO-Igs from whole serum
Antibodies were isolated from whole serum by thiophilic affinity chromatography. Briefly, a 2-mL gravity flow column was packed with Thiophillic-Superflow resin and equilibrated according to manufacturer's directions. Samples were diluted 1:10 in sample buffer (50mM sodium phosphate and 0.55M sodium sulfate; pH 7.0) and applied to the column. Unbound proteins were washed with equilibrium buffer (50mM sodium phosphate and 0.5M sodium sulfate; pH 7.0). GO-Igs were eluted with 2–3 column volumes of elution buffer (20mM sodium phosphate and 20% glycerol; pH 7.0). Eluent samples were combined and concentrated to their original volume using a Spin-X UF Concentrator then dialyzed with Tube-O-Dialyzer (catalog number 786-619; G-Biosciences) in Hanks' balanced salt solution (HBSS) containing 10mM HEPES (pH 7.4). Final protein concentration was measured with the Pierce BCA Protein Assay. This method likely resulted in a heterogeneous mixture of stimulatory, inhibitory, and blocking TSHR antibodies. High concentrations (∼4 mg/mL) of purified GO-Igs were used to ensure enough stimulatory antibodies were present to induce a detectable HA response.
Induction of HA secretion by GO-Igs and inhibition of cross talk
GOFs were grown to confluence and then pretreated with hyaluronidase (1 U/mL in HBSS) for 1 hour at 37°C. Under serum-deprived conditions, HA production is severely reduced (21), so addition of media with 10% FBS was necessary to permit stimulation of HA. After the digestion of preexisting HA, GOFs treated with M22, IGF-1, or purified GO-Igs in F-media and incubated for 5 days in 7% CO2 at 37°C. For experiments inhibiting cross talk, cells were pretreated with Lins, AF305, or 1H7 in F-media at 37°C for 1 hour before stimulation. Conditioned media were collected and stored at −20°C. Conditioned media were assayed using a modified Corgenix HA ELISA kit as previously described (6).
Measurement of IGF-1R activation
Cells were grown to confluence and serum starved in DMEM with 0.1% BSA for 18 hours. Cells were then treated with M22, IGF-1, or GO-Igs for 30 minutes. Lysates were prepared using the Bio-Plex Pro cell signaling kit (catalog number 171-304006M; Bio-Rad). Phosphorylated IGF-1R and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels were measured with a Bio-Plex MAGPIX multiplex reader (catalog number 171-015001; Bio-Rad) using the Phospho-IGF-1R (Tyr1131) set (catalog number 171-V50009M; Bio-Rad) and total Human GAPDH set (catalog number 171V60019M; Bio-Rad) according to the manufacturer's directions.
Measurement of cAMP production
Human embryonic kidney (HEK)293 cells overexpressing human TSHR (HEK-TSHR) were stepped-down for 30 minutes in 10mM HEPES in HBSS then treated with GO-Igs in the presence of 3-isobutyl-1-methylxanthine for 30 minutes. Concentration of cAMP was measured using the cAMP-Screen Chemiluminescent Immunoassay (Life Technologies) according to manufacturer's directions.
Statistics
Statistical analysis was performed using GraphPad Prism version 5.04 Windows (GraphPad Software). P < .05 by Student's t tests were used determine significance of treatment vs control. Linear regression was used to determine correlation between HA secretion and cAMP production. P < .05 by the F test was considered significantly different from a 0 slope. One outlier was identified using the extreme Studentized deviate method and removed from further analysis.
Results
Figure 2 compares the effects of 2 anti-IGF-1R antibodies, AF305 and 1H7, with those of Lins on HA secretion stimulated by IGF-1, and by low and high doses of M22. Both these antibodies were developed to inhibit binding to and activation of IGF-1R by IGF-1, and, like Lins, completely inhibited IGF-1 induction of HA. In contrast, the effects on M22-stimulated HA secretion depended on the dose of M22. None of these compounds had a significant effect on basal HA secretion. As shown previously, Lins completely inhibited stimulation by the low dose of M22 (97 ± 12.9% inhibition) and had a small inhibitory effect (32 ± 6.7% inhibition) on stimulation by high dose M22. AF305 had no inhibitory effect at either dose of M22, whereas 1H7 behaved similarly to Lins. 1H7 partially inhibited a low dose of M22 (52 ± 10.9%) and a high dose of M22 (29 ± 9.4% inhibition). An antagonist of TSHR signaling (NCGC00229600) (6) completely inhibited low and high doses of M22 (102 ± 14.8% and 98 ± 6.1% inhibition, respectively). Because M22 does not bind directly to IGF-1R, these data provide further evidence in support of the idea that TSHR/IGF-1R cross talk plays a role in M22 stimulation of HA secretion. Importantly, AF305 does not inhibit cross talk, whereas 1H7 does.
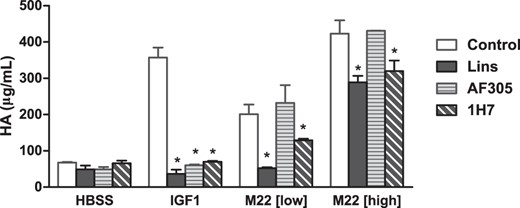
Partial inhibition of TSHR-stimulated cross talk with 1H7, not AF305.
Primary cultures of GOFs were incubated for 1 hour in Lins (10μM), AF305 (30nM), or 1H7 (30nM), then stimulated with IGF-1 (100 ng/mL), a low dose of M22 (42 ng/mL), or a high dose of M22 (100 ng/mL) in the continued presence of the IGF-1R inhibitors. Secreted HA was measured in the conditioned media after 5 days. All results shown are expressed as mean ± range. Data in the control group are from 4 independent experiments. Data from the Lins, AF305, and 1H7 groups are from 2 independent experiments. *, P < .01.
Although we previously showed that M22 did not cause activation/phosphorylation of IGF-1R directly (6), it was still possible that antibodies in the serum of GO patients would. We, therefore, determined whether GO-Igs would stimulate IGF-1R phosphorylation. Figure 3 shows that IGF-1 stimulated a 4.4 ± 0.09-fold increase in phosphorylated IGF-1R, whereas antibodies purified from healthy volunteers (HV-Igs) had minimal effect (1.0 ± 0.11-fold). Stimulation of IGF-1R phosphorylation by GO-Igs (1.1 ± 0.25-fold) was not significantly different than HV-Igs. Thus, we found no evidence for IGF-1R-stimulating antibodies in GO patient's sera (see Discussion).
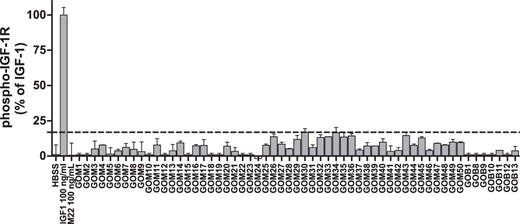
The IGF-1R is not activated by purified GO-Igs.
Primary cultures of GOFs were serum starved for 18 hours, then stimulated for 30 minutes with HBSS, IGF-1 (100 ng/mL), M22 (100 ng/mL), HV-Igs, or GO-Igs (3.5–4.5 mg/mL, labeled GOM or GOB). Levels of phosphorylated IGF-1R were normalized to total GAPDH. Dotted line represents the mean + 3SD of 10 HV-Igs samples (n = 20). Results are normalized to controls and expressed as mean ± range (n = 2 per condition). IGF-1R phosphorylations by 1H7 (30nM), AF305 (30nM), or Goat-Igs (30nM) were not significantly different from HBSS (data not shown).
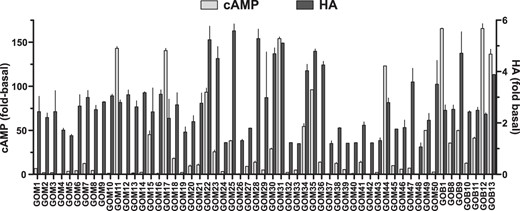
We next compared the GO-Igs as TSAbs, by measuring stimulation of cAMP production in HEK293 cells overexpressing TSHRs (HEK-TSHR cells), with their activities to stimulate HA secretion by GOFs. We used HEK-TSHR cells to measure cAMP stimulation because engineered cells like HEK-TSHR cells are used routinely in clinical laboratories to measure TSAb activity. Figure 4 illustrates the characterization of 57 patient sera studied in this way. Fifty had TSAb activity; 28 had modest activity (fold basal = 2–10), and 22 had high TSAb activity (fold basal >10). Only 7 had no TSAb activity. Of the 57 patient sera, 42 exhibited activity to increase HA secretion by GOFs as defined by fold basal more than or equal to 2.0, whereas none of the HV-Igs significantly increased HA secretion (data not shown).
GOF activation by purified GO-Igs.
Purified GO-Igs were tested for their ability to stimulate TSHR and activate orbital fibroblasts. HEK-TSHR cells were stimulated with GO-Igs (3.4–4.5 ng/mL) for 30 minutes. cAMP was measured by ELISA. Results are normalized to HBSS control and expressed as mean (n = 2 per condition). GOFs were stimulated with GO-Igs (3.5–4.5 mg/mL) for 5 days, and secreted HA was measured as previously described. Samples whose fold basal more than or equal to 2 were defined as stimulatory in following experiments. Experiments were conducted in 2 GOF strains. HA results are expressed as mean ± SEM (n = 4 per condition).
The role of cAMP signaling in GO-Ig stimulation of HA secretion by GOFs is controversial (22–25). To attempt to better understand this relationship, we compared GO-Ig-stimulated HA secretion with that of cAMP (Figure 5). There was an overall correlation between these 2 parameters (P < .001, F test). However, this correlation does not signify a causal relationship between cAMP and HA production. A slope of 0.72 ± 0.16 and R2 = 0.17 imply a weak relationship at best. Importantly, there were 8 samples that exhibited no or only very minor TSAb activity (<2-fold stimulation) but high activities to stimulate HA production.
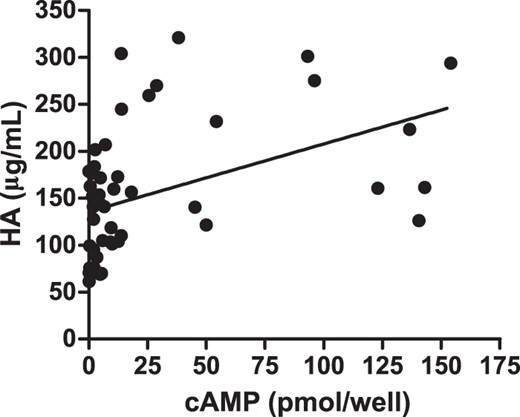
GO-Ig induction of cAMP production vs HA secretion.
Linear regression analysis shows the abilities of GO-Igs to induce cAMP and HA secretion are weakly correlated. Shown are the data from 1 GOF strain expressed as the means of 2 independent experiments. Slope = 0.72 ± 0.16, R2 = 0.17 and significantly deviates from 0 (P < .001, F test).
We next measured the effects of AF305 and 1H7 on stimulated HA secretion. Importantly, there was no consistent effect of AF305 on GO-Ig-stimulated HA secretion, but there was a consistent inhibition by 1H7 (Figure 6). The effects of AF305 and 1H7 on GO-Ig-stimulated HA secretion are compiled in Figure 6B. Analyzing the data of the 43 GO-Ig samples that stimulated HA secretion, AF305 had no effect (109 ± 3.6% of control), whereas consistently 1H7 reduced HA secretion by 60 ± 2.3%. Thus, the effects of AF305 and 1H7 on GO-Ig-stimulated HA secretion were similar to those observed with M22-stimulated HA secretion.
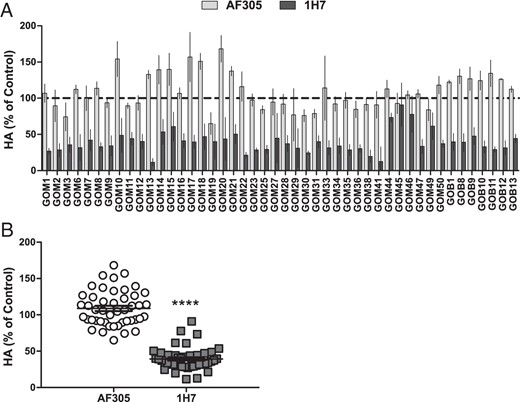
Reduced HA induction by stimulatory GO-Igs by 1H7, not AF305.
A, GOFs were preincubated with AF305 (30nM), or 1H7 (30nM), then treated with stimulatory (HA fold basal ≥2) GO-Igs (3.5–4.5 mg/mL) for 5 days in the continued presence of IGF-1R blocking antibodies. HA was measured as previously described. Experiments were conducted in 2 GOF strains. Data were normalized to their control and expressed as mean ± SEM (n = 4 per condition). B, AF305 has no significant effect on HA induction by GO-Igs, whereas 1H7 reduced HA secretion by 60% (****, P < .0001 vs GO-Ig control by Student's t test). Data represent the averaged mean ± SEM of all stimulatory GO-Ig samples (n = 4 per point, n = 184 per group).
Discussion
In this study, we addressed an important but still controversial aspect of the pathogenesis of GO. Namely, whether the TSHR is the primary (and perhaps sole) target for stimulatory autoantibodies or whether autoantibodies that stimulate the IGF-1R are involved also. Antibodies that inhibit binding of radioactive IGF-1 to receptors on GOFs have been found in the serum of patients with GD (17, 26), and evidence has been presented that inhibition of IGF-1R signaling partially inhibits stimulation of GOFs by GO-Igs (6). However, the authors of 2 recent reports in which direct activation of IGF-1Rs by Graves' antibodies was measured have come to different conclusions (27, 28). One report (27), studying sera or GO-Igs from 108 GO patients, found that 14% of GO patients and 11% of normal subjects had antibodies that bound to IGF-1R. All of these antibodies inhibited IGF-1R activation by IGF-1 but none of the 10 samples tested stimulated IGF-1R. These authors concluded their data “do not support the hypothesis that the [IGF-1R-stimulating antibodies] contribute to GO pathogenesis.” Another study (28), of sera from 70 patients with GO, found IGF-1R-stimulating activity increased with age and in a small number of patients, depletion of immunoglobulins was followed by a small decrease in IGF-1R-stimulating activity. These authors suggested that “in a subset of patients with GO, [immunoglobulins] may have IGF-1R stimulating activities.” However, these authors used an engineered cell line overexpressing IGF-1Rs to measure IGF-1R stimulating antibodies and the effects were minimal. Thus, at present a role for IGF-1R stimulating antibodies in the sera of GO patients has not been definitively shown. This is an important issue when considering treatment of GO, because the effect of drug therapy would likely be more efficacious if the therapy were directed at the primary target of GO-Ig activation.
We report studies designed to gain additional evidence in support of our model of the mechanism of autoantibody action (Figure 1). We used the monoclonal stimulatory antibody M22 in initial experiments because interpretation of pharmacological findings is simpler when the agonist is a single molecular species like M22 rather than a polyclonal pool of GO-Igs that likely contain several activating and blocking antibodies. Moreover, our previous studies (6) allowed us to appreciate the biphasic nature of the stimulatory M22 dose response of HA secretion and therefore tease apart different mechanisms mediating the 2 phases of M22 stimulation. The difference between the 2 IGF-1R antibodies seen after M22 stimulation was initially surprising given their presumably similar functions. Both AF305 and 1H7 are reported IGF-1R inhibitory antibodies and completely blocked IGF-1-stimulated HA secretion (Figure 2). AF305's inability to inhibit M22-induced HA secretion is further evidence that M22 does not bind to IGF-1R. The partial efficacy of 1H7, on the other hand, is indicative of IGF-1R's secondary role in TSHR-induced HA secretion. We previously showed that approximately 30% of M22's effect is mediated through an IGF-1R dependent pathway. Similar to Linsitinib, 1H7 likely targets this mechanism of M22 stimulation by preventing the participation of IGF-1R in TSHR-stimulated cross talk. This contrasts complete inhibition by a TSHR antagonist, which we have previously demonstrated was capable of inhibiting both IGF-1R-dependent and IGF-1R-independent pathways stimulated by M22. Based on these data, we concluded that the effects of AF305 and 1H7 support the model proposed in Figure 1, and these 2 antibodies can be used in tandem to develop an experimental protocol to directly determine whether GO-Igs acted by binding to IGF-1R or whether the involvement of IGF-1R was mediated solely by cross talk with TSHR.
We showed that GO-Igs do not stimulate IGF-1R phosphorylation/activation in GOFs (Figure 3). We also sought to confirm these results in HEK293 cells, which do not express TSHR and therefore are not affected by TSHR/IGF-1R cross talk. The apparent subtle increase in IGF-1R phosphorylation after GO-Ig treatment was not significantly higher than what was observed with HV-Igs, 1H7, or AF305 (Supplemental Figure 1). These findings are different than those reported by Varewijck et al (28), in which stimulation of phosphorylation by GO-Igs was found in HEK293 cells overexpressing IGF-1Rs, but similar to those of Minich et al (27), who found no increase in IGF-1R phosphorylation in HepG2 cells. We suggest the findings in HEK293 cells are nonspecific and do not represent the effects of specific IGF-1R-stimulating antibodies. Indeed, we are not aware of any report definitively demonstrating IGF-1R-stimulating antibodies in humans. Moreover, we suggest that circulating IGF-1R-stimulating antibodies would have widespread and profound effects on many tissues within the body.
Although the vast majority of reports demonstrate that IGF-1R signaling is initiated by IGF-1R autophosphorylation (19), there is a limited body of evidence that IGF-1R signaling may occur in the absence of receptor phosphorylation. IGF-1R activation by IGF-1 has been reported to be mediated by a phosphorylation-independent, β-arrestin-1-mediated pathway in a melanoma cell line (29) and by an anti-IGF-1R antibody in Ewing sarcoma cells in culture (30). Of note, we found that β-arrestin-1 mediates aspects of TSHR signaling in bone precursor cells (31). However, at present, there is no evidence that GO-Igs can signal via this pathway. We suggest that this mechanism is unlikely to account for IGF-1R involvement in GO-Ig stimulation of GOFs as it is likely to be inhibited by AF305.
Of note is our finding that 7 GO-Ig preparations exhibited no TSAb activity but stimulated HA secretion (Figures 4 and 5). Several reports of euthyroid GO patients (32, 33) have been taken as evidence that TSHR may not be the primary antigen in GO. Yet this clinical finding can be alternatively explained by the potential role of TSHR antibodies that may stimulate non-cAMP TSHR signaling pathways. The role of cAMP as a mediator of TSHR-mediated HA secretion in GOFs is controversial. Guo et al (22) found that elevation of cAMP by forskolin and by prostaglandin stimulation increased HA production. Zhang et al (25) found that TSH and TSAbs increased the levels of expression of 2 HA synthases, hyaluronan synthase 1 and hyaluronan synthase 2, and minimally increased HA production. In 2 papers, van Zeijl et al (23, 24) presented evidence that cAMP does not play a major role in GD Ig stimulation of HA production in GOFs and GOFs differentiated in culture to adipocytes. Our experiments were not directed specifically at this issue but our findings that there is a cohort of GO-Igs that have little or no TSAb activities show that cAMP is not a necessary mediator of GO-Ig stimulation of HA secretion. Moreover, we suggest that the GO-Igs that exhibited little or no TSAb activity but stimulated HA secretion were acting as functionally selective agonists at TSHR. Several monoclonal TSAbs have been shown to signal selectively from one or another transduction pathway (34, 35). Therefore, we suggest that GO-Ig stimulation of cAMP-mediated signal transduction is not the primary pathway used by GO-Igs to stimulate HA secretion.
In summary, our findings are consistent with the idea that the primary, if not sole target, of GO-Igs is TSHR. However, inhibition of IGF-1R signaling partially inhibits GO-Ig stimulation of HA secretion by inhibiting cross talk and this may be the mechanism of action of the anti-IGF-1R antibody in clinical trials at present (14), which is likely acting like 1H7. We suggest that a more effective medical approach to the treatment of GO would be by inhibiting TSHR signaling (6) or by a combination therapy with antagonists to both TSHR and IGF-1R that might allow for lower doses of each and thereby decrease the likelihood of adverse effects.
Acknowledgments
We thank Dr Neil Miller, Dr Prem Subramanian, Dr Nicholas Mahoney, and Dr Shannath Merbs (Johns Hopkins School of Medicine, Baltimore, MD) who have kindly provided materials for our experiments. We also thank Ms Anula Bhusry, Dr Mary Walter, and the members of the National Institute of Diabetes and Digestive and Kidney Diseases Clinical Core for processing the blood samples from GO patients.
This work was supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892 (DK011006 to M.C.K.).
R.F.P. is supported by Nova Therapeutics.
Disclosure Summary: Nova Therapeutics LLC holds licenses to intellectual properties applicable in the development of GO therapies. C.C.K., R.F.P., S.N., and M.C.G. have filed a patent pertaining to drug combinations targeting TSHR and IGF-1R. C.B., B.M.-S., B.S.A., M.C.S., and G.J.K. have nothing to disclose.
Abbreviations
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GD
Graves' disease
- GO
Graves' ophthalmopathy
- GOB
GO Bethesda
- GOF
GO fibroblast
- GOM
GO Mainz
- HA
hyaluronic acid
- IGF-1R
IGF-1 receptor
- IRB
institutional review board
- TSAb
thyroid-stimulating antibody
- TSHR
TSH receptor.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}