





Abstract
This perspective traces a pipeline of discovery in pituitary medicine over the past 75 years.
To place in context past advances and predict future changes in understanding pituitary pathophysiology and clinical care.
Author's perspective on reports of pituitary advances in the published literature.
Clinical and translational Endocrinology.
Discovery of the hypothalamic-pituitary axis and mechanisms for pituitary control, have culminated in exquisite understanding of anterior pituitary cell function and dysfunction. Challenges facing the discipline include fundamental understanding of pituitary adenoma pathogenesis leading to more effective treatments of inexorably growing and debilitating hormone secreting pituitary tumors as well as medical management of non-secreting pituitary adenomas. Newly emerging pituitary syndromes include those associated with immune-targeted cancer therapies and head trauma.
Novel diagnostic techniques including imaging genomic, proteomic, and biochemical analyses will yield further knowledge to enable diagnosis of heretofore cryptic syndromes, as well as sub classifications of pituitary syndromes for personalized treatment approaches. Cost effective personalized approaches to precision therapy must demonstrate value, and will be empowered by multidisciplinary approaches to integrating complex subcellular information to identify therapeutic targets for enabling maximal outcomes. These goals will be challenging to attain given the rarity of pituitary disorders and the difficulty in conducting appropriately powered prospective trials.
In January 1917, the first issue of Endocrinology: The Bulletin of the Association for the Study of Internal Secretions was published in Glendale, California, to “promote accumulation and diffusion of knowledge of all kinds bearing upon the endocrine glands and their functions” (1). Papers related to pituitary disorders appearing in the first issue included “Prospects for anterior pituitary therapy” and properties of a novel pituitary growth-promoting factor, “tethelin.” Despite a superabundant discovery pipeline for pituitary medicine over subsequent decades, we still grapple with similar questions.
In 1955, Harris published his landmark work “Neural Control of the Pituitary Gland,” in which he postulated that “… nerve fibers from the hypothalamus liberate hormonal substances … carried by portal vessels to excite or inhibit secretion of (anterior pituitary) gland cells …” (2). Barely 15 years later, Nobel Laureates Schally (3) and Guillemin (4) fulfilled Harris's postulates and both isolated the first hypothalamic hormone, thyrotropin releasing hormone. Subsequently, a fascinating repertoire of structurally and functionally confirmed hypothalamic releasing and inhibiting peptides together with their cell-specific anterior pituitary cognate receptors were shown to enable the reciprocal “interplay” among the external environment, brain, pituitary, and target glands. Hypothalamic hormones traverse the hypothalamic portal veins and signal in a highly leveraged fashion via cell-specific anterior pituitary surface receptors to elicit hormone synthesis and secretion to the systemic circulation. Thus, the hypothalamus is the key organ that controls and integrates pituitary function as the vital transducer of brain and peripheral electrical and chemical signals (5).
The knowledge base built by pituitary discovery has defined molecular and cellular underpinnings for our understanding of cell-specific pituitary development, normal and disordered pituitary function, and the phenotypic classification of pituitary masses.
The anterior pituitary gland comprises five distinct cell compartments, each terminally differentiated to express specific hormones as determined by cell-specific transcription factors (6). POUIF-1 determines lactotroph and somatotroph differentiation to express prolactin (PRL) and GH, DAX-1 and STAR proteins determine gonadotroph specification to express FSH and LH, GATA-1 and other proteins determine thyrotroph differentiation to express TSH, and T-Pit drives corticotroph-specific POMC transcription. Inherited mutations of pituitary-specific transcription factors lead to specific hormone deficiencies, as do mutations of more proximal factors (eg, PROP-1), leading to multiple anterior pituitary hormone deficiencies (7–9). The gland also responds in a plastic fashion as determined by both specific cell mass changes as well as by specific hormone gene transcriptional activity (10). Hypothalamic and intrapituitary paracellular signals mediate plastic and regenerative responses by early progenitor or stem cells responding to environmental signals and perturbed peripheral hormonal inputs such as stress, pregnancy, or gland ablation (11, 12). Furthermore, “adult” stem cell populations may continue to enable precursor cell regeneration in the postnatal period, and perhaps throughout the lifespan. In addition to the elegant mapping of cell-specific pituitary specification and differentiation, highly ordered three-dimensional extracellular scaffolding also enables chromatin influences on pituitary response to hormone secretagogues (13).
The pituitary gland functions to regulate appropriate growth and development; sexual function and fertility; temperature, fluid, and energy and appetite control; stress response maintenance; and overall sustaining of a metabolically homeostatic cell and whole body milieu. In fact, our current knowledge of pituitary medicine certainly fulfills the seminal vision of Sajous for endocrinologists to understand how ductless glands regulate the “very life process itself” (14). Achieving this exquisite control of multiple systems is enabled by several fundamental principles that govern control of the hypothalamic-pituitary unit. These include timed pulsatility of pituitary hormone secretion, robust feedback mechanisms protecting against hormone over- or underproduction, and highly differentiated transcriptional control governing production of each unique pituitary hormone (10). Disordered pituitary hormone excess is also associated with dysregulated feedback control. Rarely, excess pituitary hormone signaling with no underlying pituitary lesion may be encountered, as exemplified by TSH or LH activing receptor mutations. Pituitary hormone loss is usually associated with hereditary or acquired or compressed disrupted hypothalamic or pituitary hormone production, or rarely by disrupted peripheral pituitary trophic hormone signaling, as exemplified by rare pituitary hormone resistance syndromes (15). For example, a familial syndrome associated with mutant inactivated PRL receptors leads to hyperprolactinemia and, intriguingly, menstrual disturbances also occur (16). Thus, integrated pituitary function control is subserved by three tiers: hypothalamus, intrapituitary signals, and peripheral hormone feedback loops (10). The net impact of this complex regulation is to enable production of appropriately abundant and uniquely timed and sized secretory pulses.
Recent diagnostic advances, which impact diagnosis and therapy, include (17) the following.
- •
Development of highly sensitive and precise hormone and receptor assay technologies.
- •
Striking magnetic resonance imaging (MRI) advances enabling meticulous visualization of pituitary lesions and associated morphologic and localization characteristics.
- •
Subcellular immunodetection and in situ techniques have empowered the pathology diagnostic armamentarium.
- •
Receptor-targeted medical therapies and availability of recombinant pituitary and hypothalamic hormones have revolutionized diagnosis and treatment of pituitary disorders.
- •
Development of sophisticated microscopic and endoscopic surgical techniques and more focused high-energy radiotherapeutic approaches have enabled resection or ablation of discreet pituitary lesions.
Emerging Challenges and Opportunities
Pituitary tumor pathogenesis
Pituitary adenomas are characterized by increased cell replication coupled with induced pituitary hormone gene transcription and subsequent hormone hypersecretion. Several rare familial syndromes comprising pituitary tumors have been phenotyped and genotyped, including MEN1 (Online Mendelian Inheritance in Man [OMIM] 131100) with mutated MEN1, Carney complex (OMIM 160980) with mutated PRKAR1A, and familial isolated pituitary adenomas (OMIM 102200) with mutated AIP (18–20). However, these comprise less than 5% of all encountered adenomas.
Comprehensive classification of sporadic nonfamilial adenomas includes defining the cell type, characterizing the unique cell-specific transcription factor driving hormone overexpression, and identifying hormone(s) being overexpressed (Table 1). Several features characterize our understanding of pituitary neoplasms. They are overwhelmingly benign (21), and more than 95% are sporadic and nonfamilial (22). Although hyperplasia, microadenomas, and macro-adenomas are well characterized, whether or not, and how, they sequentially progress remains unclear (Figure 1). Adenomas occur at an increasing annual incidence, estimated at 3.13 (95% confidence interval, 3.07–3.20) per 100 000 population in 2009 (23). This trend is likely due to enhanced awareness, improved imaging and biochemical diagnostic tools, and perhaps a change in tumor biology resulting from environmental factors (24–26).
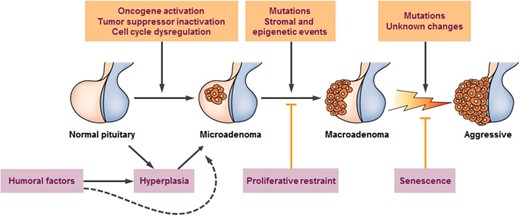
Cascade of pituitary tumorigenesis.
Pituitary hyperplasia is usually reversible, as exemplified by pregnancy. Pituitary carcinoma is exceedingly rare. (From Melmed S. Nat Rev Endocrinol 2011;7:257–266.).
Pituitary Adenomas
| Cell Type Adenoma Type | Population Prevalence (Total/10e) | Transcription Factor Expression | Differentiated Gene Expression | Clinical Features |
|---|---|---|---|---|
| Lactotroph | 45–50 | Pit-1 | PRL | Galactorrhea and hypogonadism |
| Gonadotroph | 15–20 | SF-1, GATA-2 | FSH and/or LH and/or α subunit | Silent, incidental, or pituitary failure Rarely hypergonadism |
| Somatotroph | 10 | Pit-1 | ||
| Sparsely granulated | GH | Acromegaly or gigantism | ||
| Densely granulated | GH | Acromegaly | ||
| Combined GH and PRL cells | GH and prolactin | Acromegaly or gigantism | ||
| Mixed GH and PRL | GH and prolactin | Hypogonadism and acromegaly | ||
| Mammosomatotroph | GH and prolactin | Acromegaly or gigantism | ||
| Acidophil stem cell | Prolactin and GH | Hypogonadism and acromegaly | ||
| Silent | GH | Hypopituitarism | ||
| Corticotroph | 5 | T-Pit | POMC/ACTH | |
| Cushing | ACTH | Cushing disease with hypercortisolism | ||
| Silent | ACTH | Hypopituitarism | ||
| Nelson | ACTH | Pituitary hyperplasia | ||
| Thyrotroph | <1 | Pit-1 | TSH | Hyperthyroxinemia |
| Plurihormonal | Unknown | All | GH, PRL, ACTH, glycoprotein subunit | Mixed |
| Cell Type Adenoma Type | Population Prevalence (Total/10e) | Transcription Factor Expression | Differentiated Gene Expression | Clinical Features |
|---|---|---|---|---|
| Lactotroph | 45–50 | Pit-1 | PRL | Galactorrhea and hypogonadism |
| Gonadotroph | 15–20 | SF-1, GATA-2 | FSH and/or LH and/or α subunit | Silent, incidental, or pituitary failure Rarely hypergonadism |
| Somatotroph | 10 | Pit-1 | ||
| Sparsely granulated | GH | Acromegaly or gigantism | ||
| Densely granulated | GH | Acromegaly | ||
| Combined GH and PRL cells | GH and prolactin | Acromegaly or gigantism | ||
| Mixed GH and PRL | GH and prolactin | Hypogonadism and acromegaly | ||
| Mammosomatotroph | GH and prolactin | Acromegaly or gigantism | ||
| Acidophil stem cell | Prolactin and GH | Hypogonadism and acromegaly | ||
| Silent | GH | Hypopituitarism | ||
| Corticotroph | 5 | T-Pit | POMC/ACTH | |
| Cushing | ACTH | Cushing disease with hypercortisolism | ||
| Silent | ACTH | Hypopituitarism | ||
| Nelson | ACTH | Pituitary hyperplasia | ||
| Thyrotroph | <1 | Pit-1 | TSH | Hyperthyroxinemia |
| Plurihormonal | Unknown | All | GH, PRL, ACTH, glycoprotein subunit | Mixed |
Annual incidence appears to be increasing (see text). All tumor types exhibit a pituitary mass, and which may expand causing local compressive signs. Glycoprotein refers to intact FSH or LH, or respective α or β glycoprotein subunits. Although null-cell tumors do not express hormone genes, they likely arise from the gonadotroph lineage. (Modified from Melmed S. J Clin Invest 2003;112:1603–1618.).
Pituitary Adenomas
| Cell Type Adenoma Type | Population Prevalence (Total/10e) | Transcription Factor Expression | Differentiated Gene Expression | Clinical Features |
|---|---|---|---|---|
| Lactotroph | 45–50 | Pit-1 | PRL | Galactorrhea and hypogonadism |
| Gonadotroph | 15–20 | SF-1, GATA-2 | FSH and/or LH and/or α subunit | Silent, incidental, or pituitary failure Rarely hypergonadism |
| Somatotroph | 10 | Pit-1 | ||
| Sparsely granulated | GH | Acromegaly or gigantism | ||
| Densely granulated | GH | Acromegaly | ||
| Combined GH and PRL cells | GH and prolactin | Acromegaly or gigantism | ||
| Mixed GH and PRL | GH and prolactin | Hypogonadism and acromegaly | ||
| Mammosomatotroph | GH and prolactin | Acromegaly or gigantism | ||
| Acidophil stem cell | Prolactin and GH | Hypogonadism and acromegaly | ||
| Silent | GH | Hypopituitarism | ||
| Corticotroph | 5 | T-Pit | POMC/ACTH | |
| Cushing | ACTH | Cushing disease with hypercortisolism | ||
| Silent | ACTH | Hypopituitarism | ||
| Nelson | ACTH | Pituitary hyperplasia | ||
| Thyrotroph | <1 | Pit-1 | TSH | Hyperthyroxinemia |
| Plurihormonal | Unknown | All | GH, PRL, ACTH, glycoprotein subunit | Mixed |
| Cell Type Adenoma Type | Population Prevalence (Total/10e) | Transcription Factor Expression | Differentiated Gene Expression | Clinical Features |
|---|---|---|---|---|
| Lactotroph | 45–50 | Pit-1 | PRL | Galactorrhea and hypogonadism |
| Gonadotroph | 15–20 | SF-1, GATA-2 | FSH and/or LH and/or α subunit | Silent, incidental, or pituitary failure Rarely hypergonadism |
| Somatotroph | 10 | Pit-1 | ||
| Sparsely granulated | GH | Acromegaly or gigantism | ||
| Densely granulated | GH | Acromegaly | ||
| Combined GH and PRL cells | GH and prolactin | Acromegaly or gigantism | ||
| Mixed GH and PRL | GH and prolactin | Hypogonadism and acromegaly | ||
| Mammosomatotroph | GH and prolactin | Acromegaly or gigantism | ||
| Acidophil stem cell | Prolactin and GH | Hypogonadism and acromegaly | ||
| Silent | GH | Hypopituitarism | ||
| Corticotroph | 5 | T-Pit | POMC/ACTH | |
| Cushing | ACTH | Cushing disease with hypercortisolism | ||
| Silent | ACTH | Hypopituitarism | ||
| Nelson | ACTH | Pituitary hyperplasia | ||
| Thyrotroph | <1 | Pit-1 | TSH | Hyperthyroxinemia |
| Plurihormonal | Unknown | All | GH, PRL, ACTH, glycoprotein subunit | Mixed |
Annual incidence appears to be increasing (see text). All tumor types exhibit a pituitary mass, and which may expand causing local compressive signs. Glycoprotein refers to intact FSH or LH, or respective α or β glycoprotein subunits. Although null-cell tumors do not express hormone genes, they likely arise from the gonadotroph lineage. (Modified from Melmed S. J Clin Invest 2003;112:1603–1618.).
Elucidating cellular mechanisms underlying the pathogenesis of pituitary adenomas has been constrained by several factors (17), as follows.
- •
Inaccessibility of the pituitary gland to convenient tissue biopsy of mass lesions.
- •
No functional human hormone-secreting hypothalamic or pituitary cell lines are available to readily study pathogenetic signaling pathways.
- •
Very limited amounts of surgically resected diseased tissue available for ex vivo study.
- •
Low prevalence of pituitary adenomas preclude easy design of single-center prospective trials for answering challenging questions for diagnosis or therapy of these disorders.
- •
Because pituitary tumors are rare, usually not immediately fatal, and are chronic and insidious with a considerable comorbidity burden, funding agencies generally do not recognize the priority of supporting translational research or large-scale, appropriately powered multicenter trials.
Multiple factors act to dysregulate both pituitary cell proliferation and hormone gene transcription. Classic pituitary adenoma oncogene mutations have not been encountered, except for activating GNAS1 mutations that are observed in up to 40% of GH-secreting adenomas, and constitutively induce cAMP to enhance GH gene transcription (27). The observed widespread loss of heterozygosity may be reflective of a selective growth advantage conferred by attenuation of a putative growth suppressor(s) (22, 28–30). Nevertheless, whole genome sequencing and analysis of somatic copy numbers in 12 somatotroph cell adenomas yielded no recurrent classic oncogene mutations (31). Similarly, in 24 nonfunctioning adenomas, whole exome sequencing did not yield mutated gene targets (32).
Mitogenic factors leading to cell transformation include the hypothalamic releasing hormones (including GHRH, CRH) as well as growth factors (including epidermal growth factor [EGF] and fibroblast growth factor), and peripheral hormones (including sex steroids (33).
Consistent with these findings, EGF receptor signaling has been shown to drive POMC transcription and ACTH production as well as corticotroph tumor growth in Cushing disease (34). A mechanism underlying the pathogenesis of Cushing disease with increased EGF receptor abundance was identified as gain-of-function somatic mutations in the USP8 deubiquitinase gene seen in 36% of ACTH-secreting adenomas (35). This mutation leads to attenuated EGF receptor degradation, and the subsequent enhanced growth factor signaling results in ACTH overproduction (36, 37). These findings have prompted trials of tyrosine-kinase inhibitors targeting the EGF receptor in patients with Cushing disease and prolactinomas (38).
Cell-specific CDK inhibitors and other cell-cycle protein disruptions lead to chromosomal instability and cellular senescence (39). Current evidence indicates that pathogenesis of these tumors involves cell-cycle disruption leading to DNA damage with premature cell-cycle arrest and cellular senescence, coupled with enhanced hormone gene transcription (40). Epigenetic and posttranscriptional events including demethylation and ubiquitination also determine growth factor receptor abundance and hormone gene expression. Thus, complex proliferative constraints including overexpressed CDK inhibitors and cellular senescence safeguard against development of true malignancy, whereas hypothalamic somatostatin and dopamine also constrain hormone hypersecretion. These findings pose the tantalizing question as to whether elucidation of pituitary adenoma signaling defects may in fact shed light on development of malignancy in other organs.
Elucidating genomic, proteomic, transcriptomic, and likely microbiomic landscapes and unraveling specific genetic causes for pituitary adenomas, isolated familial or nonfamilial hypothalamic and pituitary hormone deficits as well as complex syndromes including gigantism and hypogonadism will require more sophisticated cell, zebrafish, and murine disease models faithfully recapitulating human disease phenotypes, as has been exemplified for Kallman syndrome (41) and Cushing disease (42). Exploitation of whole genome and whole exome sequencing, and elegant computational modeling and bioinformatics analyses will certainly yield discovery of novel subcellular pathway disruptions. An exemplar of this approach has been the discovery of X-linked acrogigantism, a previously unrecognized form of very early-onset gigantism. In studying 43 patients with gigantism, a distinct pituitary adenoma or hyperplasia-associated GH hypersecretion phenotype was observed. These patients experience rapid growth acceleration arising at a mean age of 1 year; genetic analysis revealed Xq26.3 microduplications, associated with overexpressed mutant GPR101, a G protein–coupled receptor, within the pituitary lesion. Although mutant GPR101 elicits GH secretion in vitro, the specific ligand for this receptor is as yet elusive and remains to be discovered (43). In another example of applying genomic analyses, familial acromegaly with younger age of onset for GH oversecretion predominantly by macro-adenomas has been associated with germline AIP mutations (44). This low penetrance autosomal dominant predisposition has very rarely been observed in sporadic acromegaly (six of 148 patients), and unknown familial associations could also account for the mutant gene (45). Guidelines for cost-effective follow-up of unaffected carriers would be especially important as the presence of the mutation is very rare, polymorphisms are as yet not fully defined, and, when present, a mutation does not appear to confer unique clinical differences (46).
Although pituitary carcinoma is exceedingly rare (21), the World Health Organization has classified the more commonly encountered atypical pituitary tumors as those large invasive tumors with a Ki67 index greater than 3%, or p53 immunopositivity (47). These tumors are now recognized in 15% of resected lesions (48) and usually do not progress to true malignancy. Management of these often recurring aggressively growing lesions is challenging, and multiple surgeries with or without adjuvant radiation is usually required. Temozolomide therapy is a valuable option for uncontrolled adenoma growth or invasiveness, and tumor DNA methyltransferase expression below 50% may predict enhanced likelihood of responsiveness, as evidenced in a study of 24 patients (49). More rigorous subclassifications of atypical adenomas including morphometric (size), invasive, aggressiveness, and subcellular markers will enable an evidence-based assessment of therapeutic outcomes.
Medical therapy for pituitary tumors
Because exact mechanisms linking pituitary cell proliferation with hormone hypersecretion are yet enigmatic, therapeutic pituitary tumor targets are still imprecisely identified. Importantly, some tumors do retain inherent physiologic suppressive properties, including inhibition of by dopamine or somatostatin. We have witnessed striking advances in development and implementation of specific receptor-targeted therapies for prolactinomas, acromegaly, and Cushing disease. However, except for prolactinomas, overall efficacy of available therapeutic molecules remains quite modest. There have been few prospective attempts at precisely defining individual characteristics of patients destined to respond favorably to long-term medical treatments.
Fundamental knowledge of somatostatin receptor subtype signaling has enabled application of highly selective somatostatin receptor ligand (SRL) therapies with high SST2 affinity to be particularly effective at controlling somatotroph tumor GH hypersecretion (50–52). In contrast, pasireotide, which recognizes both SST2 and SST5 with high affinity, has been shown effective in controlling ACTH hypersecretion in patients with mild Cushing disease associated with corticotroph tumors (53). SST5 signaling may also confer added benefit for treating acromegaly resistant to SST2-selective SRLs (54). Recent reports of long-term biochemical outcomes for SRLs in acromegaly show less robust responses than prior reported experience. In fact, overall SRL efficacy for acromegaly assessed in a meta-analysis appeared to be 55% or lower (54, 55), less compelling than more favorable initial reports (56). Unanswered questions for acromegaly medications that require elucidation include a rigorous prospective assessment of overall individual drug efficacy as well as predictive determinants for both efficacy and drug resistance, respectively. Clearly, our currently assumed efficacy assumptions require revisiting. The role of surgical debulking in enabling subsequent SRL responsiveness and the role of perioperative SRLs in enhancing surgical outcomes also require an appropriately powered prospective study.
Combination “cocktail” therapies using two different drug classes (ie, SRLs and GH receptor antagonist), although not yet an Food and Drug Administration–approved approach, target both pituitary and peripheral hormone receptor to inhibit both pituitary GH secretion and peripheral GH action. This approach exhibits evidence for high biochemical efficacy in acromegaly. Thus, when weekly pegvisomant injections (mean dose, 80 mg) were added to monthly SRL injections, IGF I normalized in 97% of patients after a mean 4.9 years of follow-up (57). Optimal dose titrations, side effect profiles, and long-term outcomes are yet to be determined.
Cushing disease remains a formidable clinical challenge with extensive morbidity and high mortality (58, 59). Pasireotide was the first drug approved for targeting ACTH hypersecretion in Cushing disease. However, efficacy is limited to those with mild disease and an important drawback for SST5-targeted therapy is suppression of insulin secretion with resultant hyperglycemia (53). The impact of pasireotide therapy on Cushing disease comorbidities and mortality will require long-term prospective surveillance.
Nonfunctioning pituitary adenomas represent the most significant challenge for medical treatment because they are not responsive to available therapies and are resistant to all medical treatments. It has become apparent that nonfunctioning adenomas are also heterogenous in terms of tumor biology, aggressiveness, and recurrence rates (60). Nonsecreting “silent” adenomas often grow more readily and are increasingly recognized as awareness increases and appropriate immunostaining for intracellular hormone markers is being performed on surgical samples. Thus, more aggressively growing and recurring silent corticotroph adenomas comprising both gonadotroph and corticotroph elements require more vigilant follow-up for postoperative recurrence. The most appropriate postoperative management strategies for recurring tumors are yet to be definitively developed.
Several novel acromegaly therapies are emerging. In a phase III trial, oral octreotide capsules were shown to effectively suppress GH and IGF-I levels when patients were switched from injectable SRLs (61). In a phase II trial, antisense oligonucleotides directed against the GH receptor have been shown effective in suppressing IGF I levels (62). Emerging therapies that exploit discovery of novel preclinical targets include signaling pathway inhibitors (63), cell-cycle regulators (64), vascular EGF regulators, microRNA (65), and gene manipulations (29).
Pituitary failure
Although hormone replacement is not usually determined by etiology of the disorder, significant advances have been made in our understanding of mechanisms underlying congenital and acquired causes of attenuated pituitary reserve function including head, trauma, and stroke (66). Discovery of mutated cell-specific pituitary hormone transcription factors have led to the pathogenetic elucidation of many heretofore enigmatic causes of pediatric growth, reproductive, adrenal, and thyroid failure (67). As more of these genetic causes are unraveled, it is likely that the diagnosis of “isolated “ or “idiopathic” cryptic causes of pituitary failure will no longer be applicable.
Hypopituitarism is well managed by replacing attenuated peripheral steroid and/or thyroid hormones, and direct GH replacement for the adult GH deficiency phenotype leads to sustained benefits for patients with proven hypothalamic-pituitary disease and associated with one or more additional pituitary hormone deficits (68–72). Documenting adult GH reserve and subsequent GH replacement should be restricted to those with unequivocal compromised pituitary function (ie, have undergone pituitary surgery or radiation, have a demonstrable sellar lesion, or have experienced significant brain trauma). There is no scientific justification for administering GH to elderly patients with age-associated attenuated GH or IGF I levels, nor in using GH for athletic doping, because these practices are illegal, costly, and fraught with side effects, with no ethical justification (72).
The advent of successful c immune checkpoint blockade for melanoma and other tumors, has resulted in a new emerging pituitary syndrome, hypophysitis, in a subset of these patients (73). Hormone-expressing anterior pituitary cells have been shown to coexpress CTLA-4 and are targets for complement activation with subsequent type II hypersensitive inflammatory cascades resulting in hypophysitis (74). Thus, ipilimumab, a CTLA-4 antibody, as well as PD-1 inhibitors, lead to pituitary failure with compromised adrenal, thyroid, or gonadal failure in about 10% of patients, and associated with reversible visible pituitary mass on MRI (75, 76). Predictive determinants of hypophysitis are unclear and require prospective study. Because cancer mortality outcomes using these therapies have improved so strikingly, it will be important to determine whether initial observations of improved survival conferred by the development of hypophysitis, is in fact a prospective mortality determinant.
Personalized predictive pituitary medicine
Because clinical endocrinology practice moves inexorably toward value-based reimbursements, analyses for treatment cost-effectiveness are required to define optimal control of disease progression for the individual patient. Managing and treating pituitary disorders is inherently expensive because the disorders are chronic, requiring decades of management, imaging, sophisticated histologic, and biochemical testing, and new effective medications are costly. To this end, registry generated real-time clinical information will be a vital resource for physicians. Establishing and sustaining these processes requires enhanced knowledge of disease pathophysiology, therapeutic outcomes, a team-based approach to information integration as well as significant infrastructure (77). A proposed algorithm for evaluating patients with nonfamilial sporadic pituitary adenoma is outlined in Figure 2. Familial pituitary adenoma syndromes associated with MEN1, AIP, CDKN18, and PRKARIA are very rarely encountered and would therefore require unique and highly specialized screening and management approaches (78). The heterogenous biology of sporadic pituitary tumors, even when arising from the same cell type, has become apparent. Accordingly, more rigorously defined pituitary tumor subtype classifications are required to enable application of precise, personalized approaches to therapy. In this vein, acromegaly has been classified to three subtypes based on tumor size hormone secretion, T2-weighted MRI characteristics, somatostatin receptor SSTR2, and hormone granule abundance as well as cell-cycle protein expression (79).
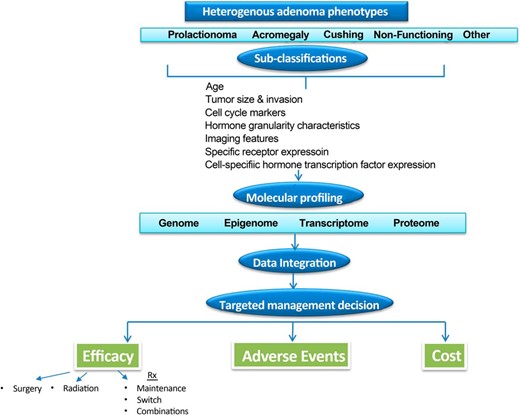
A personalized predictive approach to nonfamilial pituitary tumor management.
The most significant challenge for diagnosis of a pituitary mass is the inaccessibility of the gland for tissue biopsy. This constraint highlights the growing opportunity of applying novel imaging techniques for differential diagnosis of pituitary lesions, reliable markers of treatment responsiveness and outcomes, and application of image-guided cellular therapies. More precise imaging techniques will be particularly important for informing anatomical localization and hormone functionality of pituitary lesions. Enhanced imaging technologies detect novel adenoma biomarkers to inform pituitary mass diagnosis and therapeutic responsiveness. For example, T2-weighted pituitary MRI intensity in acromegaly has been proposed as a marker for SRL responsiveness (80). More precise imaging techniques will also yield important in vivo information on adenoma localization, including heterogeneity of tumor vs normal pituitary signals, and availability of accurate three-dimensionally reconstructed images (81). Diffusion tensor imaging will also be applicable to more accurately determine visual and optic tract damage patterns (82). Molecular imaging techniques will be helpful both for diagnosis as well as for development of receptor-guided therapeutic interventions.
Ideally, pituitary adenoma analysis will not be confined to exploring immunochemical and proliferative hormone markers. Thus, using deeper sequencing techniques, epigenetic approaches, and further elucidation of disrupted signaling pathways governing cell-cycle control, hormone synthesis, and secretion will likely yield novel targets for subcellular small molecule therapies. Especially challenging are understanding factors that determine whether a tumor remains a microadenoma or proceeds to become a macro-adenoma with attendant structural brain invasiveness.
It is likely that integrating results of whole genome, whole exome, and transcriptional sequencing will yield valuable additional information including the presence of gene mutations and copy number alterations, chromosomal changes, and signaling pathway disruptions. Interpretation and clinical application of this large and complex knowledge repository will require endocrinologists to effectively lead tumor boards comprising bioinformaticians, pathologists, neurosurgeons, and radiation oncologists. This approach will require sophisticated system infrastructure support for electronic medical information and algorithm development.
Rigorous validation of the cost-benefit of this comprehensive personalized approach will require prospective clinical trials and outcomes analyses (77). Given the relatively low prevalence of pituitary tumor phenotypes and the insidious and heterogenous natural history of these disorders, prospective and appropriately controlled studies will be difficult to design and implement and will require multicenter collaborations to generate the megadata required for sufficiently powered analyses.
Acknowledgments
Work in the author's laboratory was funded by National Institutes of health grants DK 103198, DK 007770, and CA 075979.
Disclosure Summary: S.M. is a scientific consultant for ISIS and Chiasma, receives educational grants from Novartis, and research grants from Ipsen and Pfizer.
Abbreviations
- EGF
epidermal growth factor
- MRI
magnetic resonance imaging
- OMIM
Online Mendelian Inheritance in Man
- PRL
prolactin
- SRL
somatostatin receptor ligand.

{kind=link}
{kind=link}