





SHOX mutations have been detected in approximately 70% of Léri-Weill dyschondrosteosis (LWD) and approximately 2.5% of idiopathic short stature (ISS) cases, suggesting the implication of other genes or loci. The recent identification of NPR2 mutations in ISS suggested that NPR2 mutations may also be involved in disproportionate short stature.
The objective of the study was to investigate whether NPR2 mutations can account for a proportion of the cases referred for LWD and ISS in whom no SHOX mutation was detected.
We undertook NPR2 mutation screening in 173 individuals referred for suspected LWD and 95 for ISS, with no known defect in SHOX or its enhancers. Intracellular localization and natriuretic peptide precursor C-dependent guanylate cyclase activity were determined for the identified NPR2 variants.
Eight NPR2 variants were identified in nine individuals, seven referred for suspected LWD and two for ISS. Six were demonstrated to affect NPR-B cell trafficking and/or its ability to synthesize cyclic GMP (cGMP) under response to natriuretic peptide precursor C/brain natriuretic peptide stimulation. All pathogenic mutations were detected in the suspected LWD referral group (∼3%). Interestingly, one of these patients is currently being treated with recombinant human GH and in contrast to previous reports is showing a positive response to the treatment.
NPR2 mutations account for approximately 3% of patients with disproportionate short stature and/or clinical or radiographic indicators of SHOX deficiency and in whom no SHOX defect has been identified. However, no patient has yet presented with Madelung deformity. Thus, NPR2 should be screened in the SHOX-negative LWD referrals.
Léri-Weill dyschondrosteosis (LWD; Mendelian Inheritance in Man (MIM) 127300) is a skeletal dysplasia characterized by disproportionate short stature due to mesomelic limb shortening and the characteristic Madelung deformity, the bowing and shortening of the radius, the distal dislocation of the ulna, and pyramidal configuration of the carpal bones. Other minor abnormalities may include high-arched palate, cubitus valgus, short fourth metacarpal, micrognathia, and muscular hypertrophy (1). Most LWD cases are caused by heterozygous mutations in SHOX (short stature homeobox containing gene) or its regulatory regions, located on the pseudoautosomal region 1 (PAR1) (2–7). Heterozygous SHOX mutations are also observed in a small proportion (∼2%-5%) of patients with idiopathic short stature (ISS; MIM 300582) (1, 8, 9). The penetrance of SHOX haploinsufficiency is high, but its clinical expression is very variable, with some LWD individuals having heights within the normal range and others lacking mesomelic shortening of the limbs or Madelung deformity (10). The clinical characteristics become more pronounced with age and are often more severe in females. Auxological analysis of the body proportions, the presence of minor abnormalities, and subtle radiographic signs can often be hard to detect; therefore, genetic analysis is required for confirmation.
Defects in both copies of SHOX are associated with the more severe skeletal dysplasia, Langer mesomelic dysplasia (LMD; MIM 249700), with disproportionate extreme short stature [−5.5 to −7.2 SD score (SDS)] and marked mesomelic and rhizomelic limb shortening (11–14).
As in the case of SHOX, homozygous mutations in NPR2 (natriuretic peptide receptor B/guanylate cyclase B) cause a severe dysplasia: acromesomelic dysplasia, type Maroteaux (AMDM; MIM 602875), which is phenotypically similar to LMD in that patients have severe short stature (<−5 SDS in adults), shortening of the extremities, bowing of the forearm, and shortening and widening of metacarpals and phalanges (15, 16). Furthermore, the mouse model carrying a Npr2 homozygous mutation also exhibited disproportionate dwarfism, short limbs, especially short bowed radius, short tail, and domed skull (17–19). Many NPR2 mutations reported to date were shown to cause the retention of natriuretic peptide receptor B (NPR-B) in the endoplasmic reticulum (ER), promoting a reduced enzymatic activity due to impaired cellular trafficking (20). Interestingly, heterozygous NPR2 mutation carrier parents were suggested to be shorter (21), with heights 5.7 cm shorter than population-matched controls (16). This was subsequently confirmed in a large multigenerational family with a mean −1.4 SDS height reduction in 16 carriers as compared with 23 noncarriers (22). However, no difference was observed for the sitting height to height (SH/H) ratio between the NPR2 mutation carriers and noncarriers (22). More recently, NPR2 heterozygous missense mutations have been also identified in 2%–6% of ISS patients (23–25), suggesting that NPR2 mutations may account for a significant proportion of the short stature population.
NPR2 codes for NPR-B, a transmembrane receptor related with endochondral bone growth (16, 22). NPR-B presents a modular structure as follows: 1) an extracelullar ligand-binding domain that is able to bind to the natriuretic peptides natriuretic peptide precursor C (CNP), brain natriuretic peptide (BNP), and natriuretic peptide A (ANP); 2) a transmembrane region; 3) an intracellular kinase homology domain; and 4) a carboxyl-terminal guanylate cyclase domain, which, under response to natriuretic peptide stimulation, produces cyclic GMP (cGMP) from GTP (26). Natriuretic peptides participate in the regulation of endochondral bone ossification (CNP, BNP) and cardiovascular functions (BNP, ANP, CNP), binding to NPR-B with different efficiencies (CNP ≫ ANP ≥ BNP) (27, 28).
Although mutations in SHOX or its enhancers have been detected in approximately 70% of LWD cases (8, 29, 30), the molecular defect in the remaining approximately 30% is unknown, suggesting that other genes or loci could be implicated in this syndrome. In this study, we set out to determine whether heterozygous NPR2 mutations could account for a proportion of patients referred for possible LWD and in whom no mutation was detected in SHOX or its enhancer regions. Similarly, we studied a cohort of ISS patients with no SHOX defect to investigate whether NPR2 mutations are detected in LWD, ISS, or both pathologies. A total of 173 patients with suspected LWD and 95 with ISS were screened for mutations in NPR2. Functional studies were subsequently performed for the detected variants.
Materials and Methods
Cohort
A total of 173 patients with suspected LWD and 95 patients with ISS, with no detected alteration in SHOX or its regulatory regions, were included in the study. A suspected LWD diagnosis included two or more of the following features: 1) short stature less than −2 SDS (31); 2) Madelung deformity; 3) mesomelic shortening of the arms, determined visually; 4) presence of one or more secondary clinical characteristics of SHOX deficiency, as described previously (1). Arm span to height (A/H) and SH/H ratios were classified as abnormal if less than 0.965 and greater than 0.555, respectively, according to phenotyping scoring data determined for SHOX deficiency in 1608 unrelated individuals (1). ISS was defined as a height less than −2 SDS with normal GH or IGF levels and proportionate growth. A summary of the sex, age, anthropometric, and clinical details of the two clinical cohorts is shown in Supplemental Table 1. All subjects had been extensively analyzed for mutations in SHOX or its enhancers using multiplex ligation-dependent probe amplification (MLPA) (P018E1/F1/G1; MRC Holland), self-designed MLPA assays, high-resolution melting (HRM), and sequencing (3, 4, 29, 32). All participants provided informed consent for the performed studies, and ethical approval was obtained from the respective institutions. Genomic DNA was isolated from peripheral blood (blood kit; QIAGEN; or Chemagic DNA extraction special; Chemagen, PerkinElmer Inc).
NPR2 mutation screening
The screening of the coding sequences and intron/exon boundaries of NPR2 (NM_003995.3) was performed using HRM or next-generation sequencing (NGS). Primers were designed with the help of Oligo version 6 software (Molecular Biology Insights, Inc) and SNPCheck version 3 (https://ngrl.manchester.ac.uk/SNPCheckV3/snpcheck.htm). PCR conditions are available in Supplemental Table 2. HRM was performed using a Lightscanner 96 (BioFire Defense). All samples with an abnormal melting curve were subsequently sequenced using the BigDye Terminator version 3.1 kit (Life Technologies, Thermo Scientific Inc) and run on an ABI3730XL sequencer (Applied Biosystems). The NGS approach consisted of a custom-designed skeletal dysplasia panel (SkeletalSeq version 3) of 315 genes/regions using SeqCap EZ library enrichment (Roche Nimblegen) and run on a MiSeq platform (Illumina Inc). All variants were confirmed by direct sequencing, as was family testing.
In silico analysis
Conservation and pathogenicity prediction analysis of the identified NPR2 variants was carried out using Alamut version 2.6–1 software (Interactive Biosoftware). Population frequencies of the detected variants were assessed using the Exome Aggregation Consortium (ExAC) data (http://exac.broadinstitute.org/).
Cell culture
COS-7 and human osteosarcoma (U2OS) cells were maintained in DMEM (Life Technologies), supplemented with 10% fetal bovine serum (Life Technologies) and 1% penicillin/streptomycin (Life Technologies) at 37°C in 5% CO2. Cells were transfected using FuGene 6 (Promega) according to the manufacturer's instructions.
Plasmid construction
The hemagglutinin (HA)-tagged NPR-B wild-type and p.Arg110Cys mutant expression vectors (33) were kindly donated by Dr N. Amano and Dr T. Hasegawa (Keio University School of Medicine, Tokyo, Japan). The HA-tagged expression vectors containing the detected NPR-B mutants were generated by site-directed mutagenesis (QuikChange site-directed mutagenesis kit; GE Healthcare) following the manufacturer's recommendations. Mutagenesis primers can be found in Supplemental Table 3. All expression vectors were verified by sequencing.
Inmunocytochemistry
Inmunocytochemistry was undertaken as previously described (34). Basically, U2OS cells were transiently cotransfected with pHA-NPR-B wild-type or mutants and the ER marker pRED-calreticulin (CALR; Origene Technologies Inc) expression vector. Anti-HA mouse monoclonal (Sigma-Aldrich) and Alexa fluor 488 goat antimouse (Life Technologies), both diluted at 1:2000, were used as primary and secondary antibodies, respectively. Cells were counterstained with diamino-2-phenylindole (DAPI; Sigma-Aldrich) and mounted with DePeX mounting medium (BDH, Merck Chemicals Ltd). NPR-B, CALR, and DAPI intracellular localizations were visualized using a Leica DM5500 B fluorescent microscope (Leica Microsystems GmbH). Two biological replicates were undertaken.
Enzyme-linked immunosorbent assay
ELISAs were carried out as previously described with a few modifications (23). COS-7 cells were seeded on 12-well plates. After 24 hours, cells were transiently transfected with pHA-NPR-B wild-type (WT), NPR-B mutants, or empty vector (EV). After 48 hours after transfection, cells were incubated with 100 nM of CNP-22 (Bachem) in DMEM for 20 minutes. Subsequently 0.1 M HCl was added to stop endogenous phosphodiesterase activity, stabilize the released cGMP, and lyse the cells. Cell lysates were centrifuged at 600 × g for 10 minutes, and the supernatants were isolated. The cGMP levels in the supernatant were measured using the cGMP Complete ELISA kit (ENZO Life Sciences Inc). Each mutant was analyzed in triplicate, and at least two biological replicates were performed. Student's t tests, using SPSS version 15.0 for Windows, were carried out to compare the differences between WT and mutants. A value of P < .05 was considered as statistically significant.
Results
NPR2 mutation screening
We analyzed the coding and intron-exon sequences of NPR2 in 173 patients with suspected LWD and 95 with ISS with no known SHOX/PAR1 defect. Eight variants, seven missense and one in-frame codon deletion, were detected in nine individuals, seven LWD referrals, and two ISS (Table 1). Six of the detected variants have not been previously described in AMDM or ISS patients, whereas the p.Arg819Cys mutation has been previously reported as a pathogenic variant (23). The p.Asp256Tyr, p.Val548del, p.Glu991Gly, and p.Arg1020Trp variants have not been observed in the large population exome studies (ExAc), whereas the other four have been observed at extremely low allelic frequencies (Table 1). The conservation of the affected amino acids and the pathogenicity predictions are shown in Table 1. Cosegregation studies were undertaken where possible (n = 5, Supplemental Figure 1). The p.Asp256Tyr (proband 2), p.Val448del (proband 6), p.Arg819Cys (proband 7), and p.Glu991Gly (proband 8) variants cosegregated with the phenotype. The cosegregation analysis of the p.Asn546Tyr (proband 4) variant was inconclusive, whereas the p.Ala164Gly variant (proband 1) did not cosegregate with the short stature phenotype. The phenotypic characteristics of the probands with NPR2 variants and their relatives are shown in Table 2.
Eight NPR2 Variants Observed in Nine Individuals Referred for Suspected LWD (n = 173) and ISS (n = 95), All With No SHOX Defect
| Proband | cDNA | Exon | Amino Acid | Domain | Country | Cosegregationa | SIFT | Mutation Tester | Polyphen |
|---|---|---|---|---|---|---|---|---|---|
| 1 (ISS) | c.491C>G | 1 | p.Ala164Gly | Ligand binding | Brazil | No | 0.05 | 0.925 | 0.065 |
| Tol. | Polymorphism | Benign | |||||||
| 2 (LWD) | c.766G>T | 2 | p.Asp256Tyr | Ligand binding | Spain | Yes | 0 | 1 | 0.962 |
| Del. | Disease causing | Damaging | |||||||
| 3 (LWD) | c.1262C>T | 6 | p.Thr421Met | Ligand binding | Spain | ND | 0 | 0.883 | 0.090 |
| Del. | Disease causing | Benign | |||||||
| 4 (LWD) | c.1636A>T | 10 | p.Asn546Tyr | Kinase homology | Brazil | NI | 0 | 0.999 | 0.885 |
| Del. | Disease causing | Damaging | |||||||
| 5 (ISS) | c.1636A>T | 10 | p.Asn546Tyr | Kinase homology | Spain | ND | 0 | 0.999 | 0.885 |
| Del. | Disease causing | Damaging | |||||||
| 6 (LWD) | c.1641_1643del | 10 | p.Val548del | Kinase homology | Spain | Yes | NA | NA | NA |
| 7 (LWD) | c.2455C>Tb | 16 | p.Arg819Cys | Inter-domains | Spain | Yes | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging | |||||||
| 8 (LWD) | c.2972A>G | 20 | p.Glu991Gly | Guanylate cyclase | Spain | Yes | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging | |||||||
| 9 (LWD) | c.3058C>T | 21 | p.Arg1020Trp | Guanylate cyclase | Spain | ND | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging |
| Proband | cDNA | Exon | Amino Acid | Domain | Country | Cosegregationa | SIFT | Mutation Tester | Polyphen |
|---|---|---|---|---|---|---|---|---|---|
| 1 (ISS) | c.491C>G | 1 | p.Ala164Gly | Ligand binding | Brazil | No | 0.05 | 0.925 | 0.065 |
| Tol. | Polymorphism | Benign | |||||||
| 2 (LWD) | c.766G>T | 2 | p.Asp256Tyr | Ligand binding | Spain | Yes | 0 | 1 | 0.962 |
| Del. | Disease causing | Damaging | |||||||
| 3 (LWD) | c.1262C>T | 6 | p.Thr421Met | Ligand binding | Spain | ND | 0 | 0.883 | 0.090 |
| Del. | Disease causing | Benign | |||||||
| 4 (LWD) | c.1636A>T | 10 | p.Asn546Tyr | Kinase homology | Brazil | NI | 0 | 0.999 | 0.885 |
| Del. | Disease causing | Damaging | |||||||
| 5 (ISS) | c.1636A>T | 10 | p.Asn546Tyr | Kinase homology | Spain | ND | 0 | 0.999 | 0.885 |
| Del. | Disease causing | Damaging | |||||||
| 6 (LWD) | c.1641_1643del | 10 | p.Val548del | Kinase homology | Spain | Yes | NA | NA | NA |
| 7 (LWD) | c.2455C>Tb | 16 | p.Arg819Cys | Inter-domains | Spain | Yes | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging | |||||||
| 8 (LWD) | c.2972A>G | 20 | p.Glu991Gly | Guanylate cyclase | Spain | Yes | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging | |||||||
| 9 (LWD) | c.3058C>T | 21 | p.Arg1020Trp | Guanylate cyclase | Spain | ND | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging |
Abbreviations: NA, not applicable; ND, not determined; NI, noninformative; SIFT, Sorting Intolerant From Tolerant; Del, deleterious; Tol, tolerated. The protein domain, cosegregation analysis, and pathogenicity predictions are indicated for each variant. All variants were localized at highly conserved amino acids. Frequencies of the variant in the large sequencing project data [ExAC: 60 706 European (non-Finnish and Finnish), African, Asian, and Latino individuals]. Variants c.766G>T, c.1636A>T, c.2972A>G, and c.3058C>T have not been observed. Variant c.491C>G (rs62637657) has been observed in 3 of 66 522 European (non-Finnish) and 1 of 10 378 African American chromosomes (total 4 of 121 138). Variant c.1262C>T (rs140361919) has been observed in 5 of 66 712 European non-Finnish, 4 of 10 402 African, and 1 of 16 510 Asian chromosomes (total 10 of 121 322). Variant c.2455C>T has been observed in 2 of 66 740 European non-Finnish and 1 of 11 574 Latino chromosomes (total of all studied populations 3 of 121 406).
Cosegregation details are shown in Supplemental Figure 1.
This mutation has been previously described in one Brazilian family and its pathogenicity has been previously demonstrated (23).
Eight NPR2 Variants Observed in Nine Individuals Referred for Suspected LWD (n = 173) and ISS (n = 95), All With No SHOX Defect
| Proband | cDNA | Exon | Amino Acid | Domain | Country | Cosegregationa | SIFT | Mutation Tester | Polyphen |
|---|---|---|---|---|---|---|---|---|---|
| 1 (ISS) | c.491C>G | 1 | p.Ala164Gly | Ligand binding | Brazil | No | 0.05 | 0.925 | 0.065 |
| Tol. | Polymorphism | Benign | |||||||
| 2 (LWD) | c.766G>T | 2 | p.Asp256Tyr | Ligand binding | Spain | Yes | 0 | 1 | 0.962 |
| Del. | Disease causing | Damaging | |||||||
| 3 (LWD) | c.1262C>T | 6 | p.Thr421Met | Ligand binding | Spain | ND | 0 | 0.883 | 0.090 |
| Del. | Disease causing | Benign | |||||||
| 4 (LWD) | c.1636A>T | 10 | p.Asn546Tyr | Kinase homology | Brazil | NI | 0 | 0.999 | 0.885 |
| Del. | Disease causing | Damaging | |||||||
| 5 (ISS) | c.1636A>T | 10 | p.Asn546Tyr | Kinase homology | Spain | ND | 0 | 0.999 | 0.885 |
| Del. | Disease causing | Damaging | |||||||
| 6 (LWD) | c.1641_1643del | 10 | p.Val548del | Kinase homology | Spain | Yes | NA | NA | NA |
| 7 (LWD) | c.2455C>Tb | 16 | p.Arg819Cys | Inter-domains | Spain | Yes | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging | |||||||
| 8 (LWD) | c.2972A>G | 20 | p.Glu991Gly | Guanylate cyclase | Spain | Yes | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging | |||||||
| 9 (LWD) | c.3058C>T | 21 | p.Arg1020Trp | Guanylate cyclase | Spain | ND | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging |
| Proband | cDNA | Exon | Amino Acid | Domain | Country | Cosegregationa | SIFT | Mutation Tester | Polyphen |
|---|---|---|---|---|---|---|---|---|---|
| 1 (ISS) | c.491C>G | 1 | p.Ala164Gly | Ligand binding | Brazil | No | 0.05 | 0.925 | 0.065 |
| Tol. | Polymorphism | Benign | |||||||
| 2 (LWD) | c.766G>T | 2 | p.Asp256Tyr | Ligand binding | Spain | Yes | 0 | 1 | 0.962 |
| Del. | Disease causing | Damaging | |||||||
| 3 (LWD) | c.1262C>T | 6 | p.Thr421Met | Ligand binding | Spain | ND | 0 | 0.883 | 0.090 |
| Del. | Disease causing | Benign | |||||||
| 4 (LWD) | c.1636A>T | 10 | p.Asn546Tyr | Kinase homology | Brazil | NI | 0 | 0.999 | 0.885 |
| Del. | Disease causing | Damaging | |||||||
| 5 (ISS) | c.1636A>T | 10 | p.Asn546Tyr | Kinase homology | Spain | ND | 0 | 0.999 | 0.885 |
| Del. | Disease causing | Damaging | |||||||
| 6 (LWD) | c.1641_1643del | 10 | p.Val548del | Kinase homology | Spain | Yes | NA | NA | NA |
| 7 (LWD) | c.2455C>Tb | 16 | p.Arg819Cys | Inter-domains | Spain | Yes | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging | |||||||
| 8 (LWD) | c.2972A>G | 20 | p.Glu991Gly | Guanylate cyclase | Spain | Yes | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging | |||||||
| 9 (LWD) | c.3058C>T | 21 | p.Arg1020Trp | Guanylate cyclase | Spain | ND | 0 | 1 | 1.000 |
| Del. | Disease causing | Damaging |
Abbreviations: NA, not applicable; ND, not determined; NI, noninformative; SIFT, Sorting Intolerant From Tolerant; Del, deleterious; Tol, tolerated. The protein domain, cosegregation analysis, and pathogenicity predictions are indicated for each variant. All variants were localized at highly conserved amino acids. Frequencies of the variant in the large sequencing project data [ExAC: 60 706 European (non-Finnish and Finnish), African, Asian, and Latino individuals]. Variants c.766G>T, c.1636A>T, c.2972A>G, and c.3058C>T have not been observed. Variant c.491C>G (rs62637657) has been observed in 3 of 66 522 European (non-Finnish) and 1 of 10 378 African American chromosomes (total 4 of 121 138). Variant c.1262C>T (rs140361919) has been observed in 5 of 66 712 European non-Finnish, 4 of 10 402 African, and 1 of 16 510 Asian chromosomes (total 10 of 121 322). Variant c.2455C>T has been observed in 2 of 66 740 European non-Finnish and 1 of 11 574 Latino chromosomes (total of all studied populations 3 of 121 406).
Cosegregation details are shown in Supplemental Figure 1.
This mutation has been previously described in one Brazilian family and its pathogenicity has been previously demonstrated (23).
Anthropometric and Phenotypic Characteristics of the Probands and Their Family Members
| NPR-B Mutation/Variant | Family Member | Referral LWD/ISS | Gender (M/F) | Age, y | Mutation/Variant (Yes/No) | Anthropometric Measurements (SDS) | Dis. or Prop. | Other Clinical Characteristics |
|---|---|---|---|---|---|---|---|---|
| p.Ala164Gly | Proband 1 | ISS | M | 1.3 | Y | H: 72 (−2.5) | Prop | ND |
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| Mother | F | Adult | N | H: 144 (−3.0) A/H: ND SH/H: ND | Prop | ND | ||
| p.Asp256Tyr | Proband 2 | LWD | M | 6.6 | Y | H: 108 (−2.5) A/H: 1.00 SH/H: 0.52 | Prop | High-arched palate, cubitus valgus, muscular hypertrophy cone-shaped epiphysis |
| Brother | M | Adult | Y | H: 155 (−3.5) | ND | ND | ||
| A/H: 1.04 | ||||||||
| SH/H: ND | ||||||||
| Father | M | Adult | Y | H: 157 (−2.9) | ND | Cone-shaped epiphysis | ||
| A/H: 1.04 | ||||||||
| SH/H: 0.51 | ||||||||
| p.Thr421Met | Proband 3 | LWD | M | 18.5 | Y | H: 156 (−3.2) A/H: ND SH/H: ND | Dis | Short forearms, peculiar phenotype with long narrow face |
| p.Asn546Tyr | Proband 4 | LWD | M | 12.5 | Y | H: 128 (−2.8) | Prop | Delayed bone age (1 y) |
| A/H: 0.99 | ||||||||
| SH/H: 0.56 | ||||||||
| Mother | F | Adult | N | H: 148 (−2.4) | Dis | ND | ||
| A/H: ND | ||||||||
| SH/H: 0.57 | ||||||||
| p.Asn546Tyr | Proband 5 | ISS | M | 10.3 | Y | H: 124 (−3.1) A/H: 0.94 SH/H: ND | Dis | Cubitus valgus and muscular hypertrophy |
| Father | M | Adult | ND | H: (3.1) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| p.Val548del | Proband 6 (II.1) | LWD | M | 13.8 | Y | H: 140 (−2.9) A/H: 0.973 SH/H: 0.51 | Dis | Bowing of radius, mesomelic shortening of limbs, short fourth metacarpal and delayed bone age (2 y) |
| Father (I.1) | M | Adult | N | H: 168 (−1.5) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| Mother (I.2) | F | Adult | Y | H: 145.6 (−2.7) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| p.Arg819Cys | Proband 7 | LWD | M | 10.2 | Y | H: 126 (−2.3) | Dis | Muscular hypertrophy |
| A/H: 0.95 | ||||||||
| SH/H: 0.54 | ||||||||
| Mother | F | Adult | Y | H: 144 (−3.0) | Dis | ND | ||
| A/H: 0.94 | ||||||||
| SH/H: 0.56 | ||||||||
| p.Glu991Gly | Proband 8 (II.1) | LWD | F | 6.4a | Y | H: 107 (−2.3)a A/H: ND* SH/H: ND* | Prop | High-arched palate, short fifth phalange, muscular hypertrophy, dental malpositioning, cone-shaped epiphysis |
| Brother (II.2) | M | 7.8 | Y | H: 120 (−2.3) A/H: 0.98 SH/H: 0.51 | Prop | High-arched palate, short fifth phalange, muscular hypertrophy, and severely delayed bone age (2 y) | ||
| Sister (II.3) | F | 6.8 | Y | H: 123.8 (−0.7) A/H: 0.973 SH/H: 0.51 | Prop | Short fourth to fifth metacarpals | ||
| Father (I.1) | M | Adult | N | H: 169.8 (−1.2) A/H: ND SH/H: ND | Prop | ND | ||
| Mother (I.2) | F | Adult | Y | H: 155.5 (−1.4) A/H: ND SH/H: ND | Prop | High arched palate, bowing of radius, ankylosing spondylitis | ||
| p.Arg1020Trp | Proband 9 | LWD | F | 9.0 | Y | H: 124.5 (−1.5) A/H: 0.956 SH/H: ND | Dis | Mesomelic limb shortening; lower leg bowing; limited extension of the elbows; brachydactyly, especially of fifth digit; flattened forehead; hypertelorism and strabismus; high-arched palate; dental malpositioning |
| Mother | F | Adult | ND | H:155 (−1.1) | Dis | Deafness | ||
| A/H: 0.965 | ||||||||
| SH/H: ND |
| NPR-B Mutation/Variant | Family Member | Referral LWD/ISS | Gender (M/F) | Age, y | Mutation/Variant (Yes/No) | Anthropometric Measurements (SDS) | Dis. or Prop. | Other Clinical Characteristics |
|---|---|---|---|---|---|---|---|---|
| p.Ala164Gly | Proband 1 | ISS | M | 1.3 | Y | H: 72 (−2.5) | Prop | ND |
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| Mother | F | Adult | N | H: 144 (−3.0) A/H: ND SH/H: ND | Prop | ND | ||
| p.Asp256Tyr | Proband 2 | LWD | M | 6.6 | Y | H: 108 (−2.5) A/H: 1.00 SH/H: 0.52 | Prop | High-arched palate, cubitus valgus, muscular hypertrophy cone-shaped epiphysis |
| Brother | M | Adult | Y | H: 155 (−3.5) | ND | ND | ||
| A/H: 1.04 | ||||||||
| SH/H: ND | ||||||||
| Father | M | Adult | Y | H: 157 (−2.9) | ND | Cone-shaped epiphysis | ||
| A/H: 1.04 | ||||||||
| SH/H: 0.51 | ||||||||
| p.Thr421Met | Proband 3 | LWD | M | 18.5 | Y | H: 156 (−3.2) A/H: ND SH/H: ND | Dis | Short forearms, peculiar phenotype with long narrow face |
| p.Asn546Tyr | Proband 4 | LWD | M | 12.5 | Y | H: 128 (−2.8) | Prop | Delayed bone age (1 y) |
| A/H: 0.99 | ||||||||
| SH/H: 0.56 | ||||||||
| Mother | F | Adult | N | H: 148 (−2.4) | Dis | ND | ||
| A/H: ND | ||||||||
| SH/H: 0.57 | ||||||||
| p.Asn546Tyr | Proband 5 | ISS | M | 10.3 | Y | H: 124 (−3.1) A/H: 0.94 SH/H: ND | Dis | Cubitus valgus and muscular hypertrophy |
| Father | M | Adult | ND | H: (3.1) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| p.Val548del | Proband 6 (II.1) | LWD | M | 13.8 | Y | H: 140 (−2.9) A/H: 0.973 SH/H: 0.51 | Dis | Bowing of radius, mesomelic shortening of limbs, short fourth metacarpal and delayed bone age (2 y) |
| Father (I.1) | M | Adult | N | H: 168 (−1.5) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| Mother (I.2) | F | Adult | Y | H: 145.6 (−2.7) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| p.Arg819Cys | Proband 7 | LWD | M | 10.2 | Y | H: 126 (−2.3) | Dis | Muscular hypertrophy |
| A/H: 0.95 | ||||||||
| SH/H: 0.54 | ||||||||
| Mother | F | Adult | Y | H: 144 (−3.0) | Dis | ND | ||
| A/H: 0.94 | ||||||||
| SH/H: 0.56 | ||||||||
| p.Glu991Gly | Proband 8 (II.1) | LWD | F | 6.4a | Y | H: 107 (−2.3)a A/H: ND* SH/H: ND* | Prop | High-arched palate, short fifth phalange, muscular hypertrophy, dental malpositioning, cone-shaped epiphysis |
| Brother (II.2) | M | 7.8 | Y | H: 120 (−2.3) A/H: 0.98 SH/H: 0.51 | Prop | High-arched palate, short fifth phalange, muscular hypertrophy, and severely delayed bone age (2 y) | ||
| Sister (II.3) | F | 6.8 | Y | H: 123.8 (−0.7) A/H: 0.973 SH/H: 0.51 | Prop | Short fourth to fifth metacarpals | ||
| Father (I.1) | M | Adult | N | H: 169.8 (−1.2) A/H: ND SH/H: ND | Prop | ND | ||
| Mother (I.2) | F | Adult | Y | H: 155.5 (−1.4) A/H: ND SH/H: ND | Prop | High arched palate, bowing of radius, ankylosing spondylitis | ||
| p.Arg1020Trp | Proband 9 | LWD | F | 9.0 | Y | H: 124.5 (−1.5) A/H: 0.956 SH/H: ND | Dis | Mesomelic limb shortening; lower leg bowing; limited extension of the elbows; brachydactyly, especially of fifth digit; flattened forehead; hypertelorism and strabismus; high-arched palate; dental malpositioning |
| Mother | F | Adult | ND | H:155 (−1.1) | Dis | Deafness | ||
| A/H: 0.965 | ||||||||
| SH/H: ND |
Abbreviations: Dis, disproportional growth; F, female; H, height; M, male; ND, not documented; Prop, proportional growth; SH, sitting height. Functionally confirmed pathogenic mutations are indicated in bold.
Data at 6.4 years, prior to GH treatment. Further data are available in Supplemental Data.
Anthropometric and Phenotypic Characteristics of the Probands and Their Family Members
| NPR-B Mutation/Variant | Family Member | Referral LWD/ISS | Gender (M/F) | Age, y | Mutation/Variant (Yes/No) | Anthropometric Measurements (SDS) | Dis. or Prop. | Other Clinical Characteristics |
|---|---|---|---|---|---|---|---|---|
| p.Ala164Gly | Proband 1 | ISS | M | 1.3 | Y | H: 72 (−2.5) | Prop | ND |
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| Mother | F | Adult | N | H: 144 (−3.0) A/H: ND SH/H: ND | Prop | ND | ||
| p.Asp256Tyr | Proband 2 | LWD | M | 6.6 | Y | H: 108 (−2.5) A/H: 1.00 SH/H: 0.52 | Prop | High-arched palate, cubitus valgus, muscular hypertrophy cone-shaped epiphysis |
| Brother | M | Adult | Y | H: 155 (−3.5) | ND | ND | ||
| A/H: 1.04 | ||||||||
| SH/H: ND | ||||||||
| Father | M | Adult | Y | H: 157 (−2.9) | ND | Cone-shaped epiphysis | ||
| A/H: 1.04 | ||||||||
| SH/H: 0.51 | ||||||||
| p.Thr421Met | Proband 3 | LWD | M | 18.5 | Y | H: 156 (−3.2) A/H: ND SH/H: ND | Dis | Short forearms, peculiar phenotype with long narrow face |
| p.Asn546Tyr | Proband 4 | LWD | M | 12.5 | Y | H: 128 (−2.8) | Prop | Delayed bone age (1 y) |
| A/H: 0.99 | ||||||||
| SH/H: 0.56 | ||||||||
| Mother | F | Adult | N | H: 148 (−2.4) | Dis | ND | ||
| A/H: ND | ||||||||
| SH/H: 0.57 | ||||||||
| p.Asn546Tyr | Proband 5 | ISS | M | 10.3 | Y | H: 124 (−3.1) A/H: 0.94 SH/H: ND | Dis | Cubitus valgus and muscular hypertrophy |
| Father | M | Adult | ND | H: (3.1) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| p.Val548del | Proband 6 (II.1) | LWD | M | 13.8 | Y | H: 140 (−2.9) A/H: 0.973 SH/H: 0.51 | Dis | Bowing of radius, mesomelic shortening of limbs, short fourth metacarpal and delayed bone age (2 y) |
| Father (I.1) | M | Adult | N | H: 168 (−1.5) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| Mother (I.2) | F | Adult | Y | H: 145.6 (−2.7) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| p.Arg819Cys | Proband 7 | LWD | M | 10.2 | Y | H: 126 (−2.3) | Dis | Muscular hypertrophy |
| A/H: 0.95 | ||||||||
| SH/H: 0.54 | ||||||||
| Mother | F | Adult | Y | H: 144 (−3.0) | Dis | ND | ||
| A/H: 0.94 | ||||||||
| SH/H: 0.56 | ||||||||
| p.Glu991Gly | Proband 8 (II.1) | LWD | F | 6.4a | Y | H: 107 (−2.3)a A/H: ND* SH/H: ND* | Prop | High-arched palate, short fifth phalange, muscular hypertrophy, dental malpositioning, cone-shaped epiphysis |
| Brother (II.2) | M | 7.8 | Y | H: 120 (−2.3) A/H: 0.98 SH/H: 0.51 | Prop | High-arched palate, short fifth phalange, muscular hypertrophy, and severely delayed bone age (2 y) | ||
| Sister (II.3) | F | 6.8 | Y | H: 123.8 (−0.7) A/H: 0.973 SH/H: 0.51 | Prop | Short fourth to fifth metacarpals | ||
| Father (I.1) | M | Adult | N | H: 169.8 (−1.2) A/H: ND SH/H: ND | Prop | ND | ||
| Mother (I.2) | F | Adult | Y | H: 155.5 (−1.4) A/H: ND SH/H: ND | Prop | High arched palate, bowing of radius, ankylosing spondylitis | ||
| p.Arg1020Trp | Proband 9 | LWD | F | 9.0 | Y | H: 124.5 (−1.5) A/H: 0.956 SH/H: ND | Dis | Mesomelic limb shortening; lower leg bowing; limited extension of the elbows; brachydactyly, especially of fifth digit; flattened forehead; hypertelorism and strabismus; high-arched palate; dental malpositioning |
| Mother | F | Adult | ND | H:155 (−1.1) | Dis | Deafness | ||
| A/H: 0.965 | ||||||||
| SH/H: ND |
| NPR-B Mutation/Variant | Family Member | Referral LWD/ISS | Gender (M/F) | Age, y | Mutation/Variant (Yes/No) | Anthropometric Measurements (SDS) | Dis. or Prop. | Other Clinical Characteristics |
|---|---|---|---|---|---|---|---|---|
| p.Ala164Gly | Proband 1 | ISS | M | 1.3 | Y | H: 72 (−2.5) | Prop | ND |
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| Mother | F | Adult | N | H: 144 (−3.0) A/H: ND SH/H: ND | Prop | ND | ||
| p.Asp256Tyr | Proband 2 | LWD | M | 6.6 | Y | H: 108 (−2.5) A/H: 1.00 SH/H: 0.52 | Prop | High-arched palate, cubitus valgus, muscular hypertrophy cone-shaped epiphysis |
| Brother | M | Adult | Y | H: 155 (−3.5) | ND | ND | ||
| A/H: 1.04 | ||||||||
| SH/H: ND | ||||||||
| Father | M | Adult | Y | H: 157 (−2.9) | ND | Cone-shaped epiphysis | ||
| A/H: 1.04 | ||||||||
| SH/H: 0.51 | ||||||||
| p.Thr421Met | Proband 3 | LWD | M | 18.5 | Y | H: 156 (−3.2) A/H: ND SH/H: ND | Dis | Short forearms, peculiar phenotype with long narrow face |
| p.Asn546Tyr | Proband 4 | LWD | M | 12.5 | Y | H: 128 (−2.8) | Prop | Delayed bone age (1 y) |
| A/H: 0.99 | ||||||||
| SH/H: 0.56 | ||||||||
| Mother | F | Adult | N | H: 148 (−2.4) | Dis | ND | ||
| A/H: ND | ||||||||
| SH/H: 0.57 | ||||||||
| p.Asn546Tyr | Proband 5 | ISS | M | 10.3 | Y | H: 124 (−3.1) A/H: 0.94 SH/H: ND | Dis | Cubitus valgus and muscular hypertrophy |
| Father | M | Adult | ND | H: (3.1) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| p.Val548del | Proband 6 (II.1) | LWD | M | 13.8 | Y | H: 140 (−2.9) A/H: 0.973 SH/H: 0.51 | Dis | Bowing of radius, mesomelic shortening of limbs, short fourth metacarpal and delayed bone age (2 y) |
| Father (I.1) | M | Adult | N | H: 168 (−1.5) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| Mother (I.2) | F | Adult | Y | H: 145.6 (−2.7) | ND | ND | ||
| A/H: ND | ||||||||
| SH/H: ND | ||||||||
| p.Arg819Cys | Proband 7 | LWD | M | 10.2 | Y | H: 126 (−2.3) | Dis | Muscular hypertrophy |
| A/H: 0.95 | ||||||||
| SH/H: 0.54 | ||||||||
| Mother | F | Adult | Y | H: 144 (−3.0) | Dis | ND | ||
| A/H: 0.94 | ||||||||
| SH/H: 0.56 | ||||||||
| p.Glu991Gly | Proband 8 (II.1) | LWD | F | 6.4a | Y | H: 107 (−2.3)a A/H: ND* SH/H: ND* | Prop | High-arched palate, short fifth phalange, muscular hypertrophy, dental malpositioning, cone-shaped epiphysis |
| Brother (II.2) | M | 7.8 | Y | H: 120 (−2.3) A/H: 0.98 SH/H: 0.51 | Prop | High-arched palate, short fifth phalange, muscular hypertrophy, and severely delayed bone age (2 y) | ||
| Sister (II.3) | F | 6.8 | Y | H: 123.8 (−0.7) A/H: 0.973 SH/H: 0.51 | Prop | Short fourth to fifth metacarpals | ||
| Father (I.1) | M | Adult | N | H: 169.8 (−1.2) A/H: ND SH/H: ND | Prop | ND | ||
| Mother (I.2) | F | Adult | Y | H: 155.5 (−1.4) A/H: ND SH/H: ND | Prop | High arched palate, bowing of radius, ankylosing spondylitis | ||
| p.Arg1020Trp | Proband 9 | LWD | F | 9.0 | Y | H: 124.5 (−1.5) A/H: 0.956 SH/H: ND | Dis | Mesomelic limb shortening; lower leg bowing; limited extension of the elbows; brachydactyly, especially of fifth digit; flattened forehead; hypertelorism and strabismus; high-arched palate; dental malpositioning |
| Mother | F | Adult | ND | H:155 (−1.1) | Dis | Deafness | ||
| A/H: 0.965 | ||||||||
| SH/H: ND |
Abbreviations: Dis, disproportional growth; F, female; H, height; M, male; ND, not documented; Prop, proportional growth; SH, sitting height. Functionally confirmed pathogenic mutations are indicated in bold.
Data at 6.4 years, prior to GH treatment. Further data are available in Supplemental Data.
Functional characterization of NPR-B variants
To functionally evaluate the pathogenicity of the identified variants, we first determined whether they affected NPR-B cell trafficking. For this purpose, we transiently transfected the WT and variant cDNAs and subcloned them into an HA-expression vector in human osteosarcoma cells (U2OS). Their intracellular localization was determined by immunocytochemistry using antibodies against NPR-B and the ER marker, CALR. The NPR-B variants p.Ala164Gly, p.Asn546Tyr, p.Val548del, and p.Glu991Gly were located at the cell surface, whereas three mutants, p.Asp256Tyr, p.Thr421Met, and p.Arg1020Trp, colocalized with the ER (Figure 1).
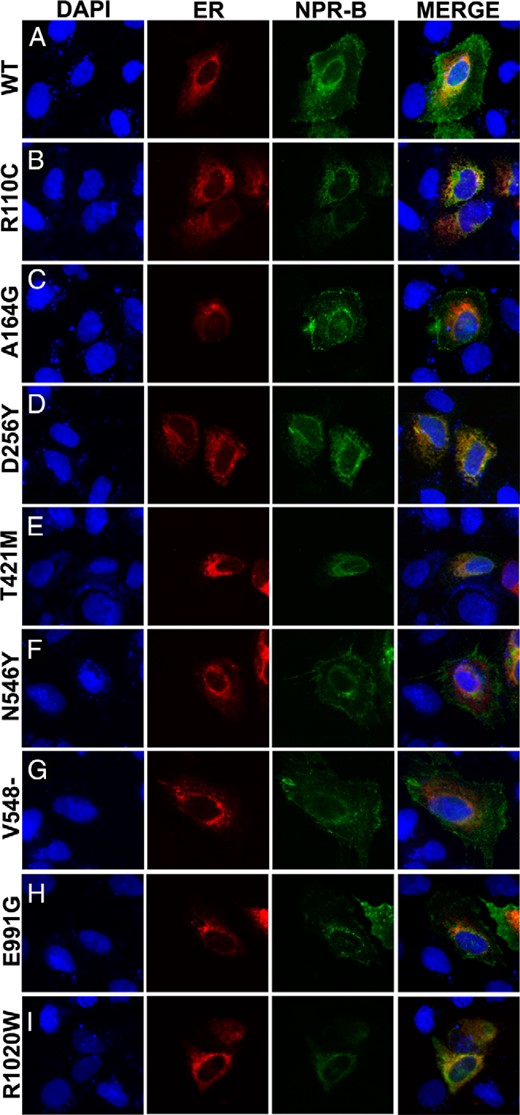
Intracelullar localization of the NPR-B mutants.
U2OS cells were transfected with NPR-B WT (A), p.Arg110C (R110C) mutant (B) as an ER retention control (24), or each mutant identified in our cohort (C–I). Intracellular localization of NPR-B (green), ER marker CALR (red), and nuclei with DAPI (blue) are shown. The three fluorescent images are merged in the fourth column. Images are at ×20 magnification. The NPR-B mutants, p.Ala164Gly (A164G), p.Asn546Tyr (N546Y), p.Val548del (V548del), and p.Glu991Gly (E991G), all showed cell surface localization, whereas the p.Asp256Tyr (N256Y), p.Thr421Met (T421M), and p.Arg1020Trp (R1020W) mutants were retained in the ER. Identical results were obtained in two independent experiments.
Second, we analyzed the receptor activity of all identified variants by assessing the CNP-dependent guanylate cyclase activity. Four of the analyzed mutants, p.Asp256Tyr, p.Val548del, p.Glu991Gly, and p.Arg1020Trp, showed a total absence of cGMP synthesis, similar to that of unstimulated wild type and the control mutant p.Arg110Cys (Figure 2A), whereas p.Thr421Met showed a 61% activity decrease. The p.Ala164Gly and p.Asn546Tyr mutants showed only moderate reductions in cGMP synthesis, 19% and 31%, respectively (Figure 2A). Because we had detected the variants in a heterozygous state, we also analyzed whether they had a dominant-negative effect by cotransfecting each mutant with NPR-B WT at a ratio of 1:1. We observed a highly significant reduction of CNP-stimulated cGMP synthesis as compared with cells cotransfected with WT and EV with six of the NPR-B mutants tested, indicating a dominant-negative effect. However, the p.Ala164Gly and p.Asn546Tyr mutants did not show a significant reduction when cotransfected with the WT (Figure 2B), suggesting that they did not have a dominant-negative effect in the heterozygous state. Thus, functional analysis confirmed that six of the NPR2 mutations, including the previously reported mutation, were pathogenic mutations (Table 3).
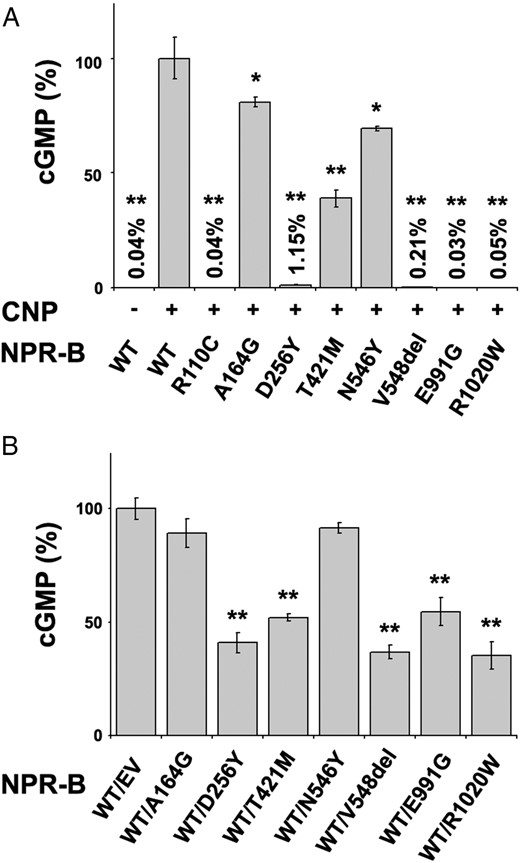
CNP-dependent guanylate cyclase activity of WT NPR-B and mutants.
A, Guanylate cyclase activity of COS-7 cells transfected with NPR-B WT or mutants (A164G, D256Y, T421M, N546Y, V548del, E991G, and R1020W) and the positive control (R110C), stimulated (+) or unstimulated (−) with CNP. CNP-dependent receptor activity is expressed as a percentage relative to CNP-stimulated NPR2-B WT. All mutants showed a reduction in CNP-dependent cGMP synthesis, to different degrees. B, cGMP activity of COS-7 cells transfected with a 1:1 ratio of NPR-B WT and EV or mutants and treated with CNP. CNP-stimulated cGMP values are expressed as a percentage relative to CNP-stimulated WT/EV. Five mutants showed a dominant-negative effect. Bars are SD of the mean. Each assay was undertaken in triplicate and two biological replicates were performed. Significant P values are indicated with asterisks (*, P < .05; **, P < .01).
Functional Characterization of the NPR-B Mutants
| NPR-B Mutant | Retained in the ER | cGMP Synthesis (% of WT) | Dominant-Negative Effect on cGMP Synthesis | Pathogenic Summary |
|---|---|---|---|---|
| A164G | − | 81 | − | Nonpathogenic |
| D256Y | + | 1.15 | + | Pathogenic |
| T421M | + | 39 | + | Pathogenic |
| N546Y | − | 69 | − | Nonpathogenic |
| V548del | − | 0.20 | + | Pathogenic |
| R819Ca | − | ∼2 | + | Pathogenic |
| E991G | − | 0.03 | + | Pathogenic |
| R1020W | + | 0.05 | + | Pathogenic |
| NPR-B Mutant | Retained in the ER | cGMP Synthesis (% of WT) | Dominant-Negative Effect on cGMP Synthesis | Pathogenic Summary |
|---|---|---|---|---|
| A164G | − | 81 | − | Nonpathogenic |
| D256Y | + | 1.15 | + | Pathogenic |
| T421M | + | 39 | + | Pathogenic |
| N546Y | − | 69 | − | Nonpathogenic |
| V548del | − | 0.20 | + | Pathogenic |
| R819Ca | − | ∼2 | + | Pathogenic |
| E991G | − | 0.03 | + | Pathogenic |
| R1020W | + | 0.05 | + | Pathogenic |
Cellular localization studies were undertaken using immunocytochemistry in U2OS cells, and cGMP synthesis capacity was assessed by ELISA.
Functional studies were previously reported (23).
Functional Characterization of the NPR-B Mutants
| NPR-B Mutant | Retained in the ER | cGMP Synthesis (% of WT) | Dominant-Negative Effect on cGMP Synthesis | Pathogenic Summary |
|---|---|---|---|---|
| A164G | − | 81 | − | Nonpathogenic |
| D256Y | + | 1.15 | + | Pathogenic |
| T421M | + | 39 | + | Pathogenic |
| N546Y | − | 69 | − | Nonpathogenic |
| V548del | − | 0.20 | + | Pathogenic |
| R819Ca | − | ∼2 | + | Pathogenic |
| E991G | − | 0.03 | + | Pathogenic |
| R1020W | + | 0.05 | + | Pathogenic |
| NPR-B Mutant | Retained in the ER | cGMP Synthesis (% of WT) | Dominant-Negative Effect on cGMP Synthesis | Pathogenic Summary |
|---|---|---|---|---|
| A164G | − | 81 | − | Nonpathogenic |
| D256Y | + | 1.15 | + | Pathogenic |
| T421M | + | 39 | + | Pathogenic |
| N546Y | − | 69 | − | Nonpathogenic |
| V548del | − | 0.20 | + | Pathogenic |
| R819Ca | − | ∼2 | + | Pathogenic |
| E991G | − | 0.03 | + | Pathogenic |
| R1020W | + | 0.05 | + | Pathogenic |
Cellular localization studies were undertaken using immunocytochemistry in U2OS cells, and cGMP synthesis capacity was assessed by ELISA.
Functional studies were previously reported (23).
Discussion
A total of eight NPR2 variants were identified in nine individuals, seven of the 173 patients referred for suspected LWD and two from the cohort of 95 ISS, all of whom had been previously excluded for SHOX/PAR1 defects. First, the pathogenicity of the identified NPR2 variants was assessed using in silico prediction tools. Seven of the eight variants were predicted to be pathogenic, whereas only the p.Ala164Gly mutation was classified as nonpathogenic. Only one of the identified variants, p.Arg819Cys, had been previously reported in an ISS patient and functionally demonstrated to be pathogenic (23). Therefore, functional studies were undertaken for the characterization of the additional seven NPR2 variants identified.
Six of the NPR2 variants were confirmed to be pathogenic mutations, and three were shown to cause the retention of NPR-B in the ER, resulting in a reduced enzymatic guanylate cyclase activity due to impaired cellular trafficking. However, the p.Thr421Met mutant behaved differently from the other two ER-retained mutants, showing 39% of WT cGMP activity, suggesting that some residual trafficking may still exist, despite not being observed in the immunochemical assay. Three NPR2 mutations did not affect the ability to translocate to the plasma membrane but showed reduced cGMP synthesis. All six mutations were shown to have a dominant-negative effect over the WT form. Only three NPR-B mutants forms showed impaired cellular trafficking, a frequency similar to that previously observed in short-stature individuals (three of five), although very different from that reported in AMDM patients, in whom most NPR2 missense mutants (11 of 12) were retained in the ER and showed reduced enzymatic activity (20, 23, 24). Although the cellular localization has been studied only in a total of 23 NPR-B mutants so far (20, 23, 24), there is no apparent correlation between the domain location of the mutations and their ability to translocate to the plasma membrane, whereas all mutants caused a reduced capacity to synthesize cGMP.
The remaining two NPR-B variants, p.Ala164Gly and p.Asn546Tyr, identified in two ISS patients, did not show any dominant-negative effect when cotransfected with the WT construct. Both were transported to the cell membrane and showed a milder reduction of cGMP synthesis in the homozygous state. This emphasizes the utility of the cotransfection assays for the functional assessment of the dominant-negative effect of novel NPR-B variants on cGMP synthesis capacity, emulating the heterozygous state. Moreover, cosegregation analysis did not confirm cosegregation of the p.Ala164Gly variant with the short-stature phenotype within the examined family or in a second family, in which this variant was recently reported as a variant of unknown significance (25). Hence, in contrast to previous studies (23, 24), we did not find any NPR2 pathogenic defects in patients classified as ISS, probably because we used a more stringent definition of ISS, according to Cohen et al (2006) (35) to determine which patient should be tested for NPR2 mutations.
The previously reported p.Arg819Cys mutation (23) was also identified in proband 7. At age 10 years, he had a height SDS of −2.3, an A/H ratio of 0.95, an SH/H ratio of 0.54, and muscle hypertrophy, ie, disproportionate short stature and shortening of the upper limbs. His carrier mother also has disproportional short stature (−3.0 SDS), and A/H and SH/H ratios of 0.94 and 0.56, respectively, indicative of shortening of the upper and lower limbs. The previously reported Brazilian patient, aged 17 years, had proportional short stature (height −2.4 SDS, SH/H 1.3 SDS) and shortened metacarpals, whereas his mother had normal height (−1.7 SDS) but had an altered SH/H ratio of 0.57, suggesting shortening of the lower limbs. Thus, the phenotypic expression of the p.Arg819Cys mutation is highly variable in the two examined families.
Interestingly, probands 2 and 8 presented with cone-shaped epiphyses, probands 8 and 9 presented with short phalanges, and proband 8 also presented with a broad forehead, all of which are clinical and radiological features described in patients with homozygous NPR2 mutations (AMDM). Dental malpositioning was also described in probands 8 and 9, a feature previously not reported in patients with NPR2 or SHOX mutations, ie, expanding the phenotypic spectrum of clinical features associated with NPR2 mutations, although the implication of other factors is not excluded.
Hence, in this study, disproportionate short stature and/or phenotypic or radiographic indicators of SHOX deficiency were observed in all individuals with pathogenic NPR2 mutations. This observation is not surprising because AMDM patients with NPR2 homozygous mutations have severe disproportional short stature. Nevertheless, due to the high clinical heterogeneity observed in individuals with SHOX haploinsufficiency, it is quite understandable why these patients were referred as suspected LWD cases. Moreover, previous reports have shown that SHOX activates the NPPB promoter, causing an increase in BNP (36), which may either directly influence NPR-B signaling or indirectly increase local CNP levels by saturating the NPR-C receptor. SHOX also activates aggrecan (ACAN) and FGFR3 expression in human U2OS cells (37, 38) and Col2A1 and ACAN in chicken micromasses (39) while down-regulating FGFR3 in chicken micromasses (38). Likewise, CNP enhances the synthesis of collagens (Col2A1) and proteoglycans (aggrecan) and influences cell proliferation, indicating a switch from proliferative to differentiated and hypertrophic chondrocytes, mediated by NPR-B (40). Therefore, SHOX and NPR-B and their downstream targets appear to be implicated in common growth pathways, which could explain why similar phenotypes are observed in individuals with SHOX and NPR2 mutations.
Interestingly, none of the patients with NPR2 mutations presented to date with the Madelung deformity, the most characteristic abnormality of LWD. This absence is a phenotypic feature that may be helpful for the differential diagnosis of disproportionate short stature caused by autosomic dominant NPR2 mutations. Nevertheless, we have to keep in mind that Madelung deformity may not be observed before 6 years of age (41), although the condition can become apparent earlier in some cases. Therefore, we should wait for the prepubertal patients with NPR2 mutations to complete their puberty to definitely confirm the absence of the Madelung deformity. For now, our opinion is that these patients have phenotypic characteristics of the heterozygous form of AMDM, which is similar to that observed in many LWD individuals but it cannot be classified as LWD because no individual has Madelung deformity.
Recombinant human growth hormone (rhGH) is indicated for patients with SHOX haploinsufficiency. Improvements in height (+1.3 SDS) was observed in most individuals and 57% of them achieved a final height greater than −2 SDS (42). In contrast, few patients with NPR2 mutations have been treated with rhGH, showing no or mild improvements on height (23–25). An initial good response observed in proband 8 (Supplemental Figure 2) suggests that rhGH treatment may be beneficial for patients with heterozygous NPR2 mutations if the treatment is started at an early age. Nevertheless, larger prospective studies will be necessary to confirm this.
In summary, in our study 6 of 173 suspected LWD referrals (∼3%) were found to have a heterozygous mutation in NPR2, whereas no mutation was found in the ISS cohort (n = 95). Functional analysis of the identified pathogenic mutations demonstrated a dominant-negative effect on the CNP-dependent guanylate cyclase activity, resulting in a decreased cGMP synthesis, as the likely molecular mechanism implicated in the etiology of disproportionate short stature. Hence, NPR2 mutation screening is recommended in LWD-suspected individuals without Madelung deformity in whom no alteration was detected in SHOX or its enhancers.
Acknowledgments
We thank the Institute of Medical and Molecular Genetics Sequencing Core for the Sanger sequencing and the Bioinformatic Unit for help with the next-generation sequencing data analysis.
This work was supported in part by the following grants: MINECO (SAF2012-30871), Comunidad de Madrid ENDOSCREEN (S2010/BMD-2396), FIS 09/01266 and 12/00649 (to A.C.-B.), and the VII Jose Igea Award by the Spanish Society of Pediatric Endocrinology Foundation. A.H.-O. was a recipient of a Formación profesional investigador (FPI) PhD studentship from the Basque Country, and S.B.-S. a postdoctoral Centro de Investigación Biomédica en Red Enfermedades Raras fellowship. A.B. was a recipient of a postdoctoral fellowship from Lilly (España).
Disclosure Summary: A.B. was a recipient of a postdoctoral fellowship by Lilly (España) but the company did not finance any part of this work. The other authors have nothing to disclose.
Abbreviations
- A/H
arm span to height
- AMDM
acromesomelic dysplasia, type Maroteaux
- ANP
natriuretic peptide A
- BNP
brain natriuretic peptide
- CALR
calreticulin
- cGMP
cyclic GMP
- CNP
natriuretic peptide precursor C
- DAPI
diamino-2-phenylindole
- ER
endoplasmic reticulum
- EV
empty vector
- ExAC
Exome Aggregation Consortium
- HA
hemagglutinin
- HRM
high-resolution melting
- ISS
idiopathic short stature
- LMD
Langer mesomelic dysplasia
- LWD
Léri-Weill dyschondrosteosis
- MIM
Mendelian Inheritance in Man
- NGS
next-generation sequencing
- NPR2
natriuretic peptide receptor B/guanylate cyclase B
- NPR-B
natriuretic peptide receptor B
- PAR1
pseudoautosomal region 1
- rhGH
recombinant human GH
- SDS
SD score
- SH/H
sitting height to height ratio
- SHOX
short stature homeobox containing gene
- WT
wild type.

{kind=link}
{kind=link}