





The objective is to formulate clinical practice guidelines for treating Cushing's syndrome.
Participants include an Endocrine Society-appointed Task Force of experts, a methodologist, and a medical writer. The European Society for Endocrinology co-sponsored the guideline.
The Task Force used the Grading of Recommendations, Assessment, Development, and Evaluation system to describe the strength of recommendations and the quality of evidence. The Task Force commissioned three systematic reviews and used the best available evidence from other published systematic reviews and individual studies.
The Task Force achieved consensus through one group meeting, several conference calls, and numerous e-mail communications. Committees and members of The Endocrine Society and the European Society of Endocrinology reviewed and commented on preliminary drafts of these guidelines.
Treatment of Cushing's syndrome is essential to reduce mortality and associated comorbidities. Effective treatment includes the normalization of cortisol levels or action. It also includes the normalization of comorbidities via directly treating the cause of Cushing's syndrome and by adjunctive treatments (eg, antihypertensives). Surgical resection of the causal lesion(s) is generally the first-line approach. The choice of second-line treatments, including medication, bilateral adrenalectomy, and radiation therapy (for corticotrope tumors), must be individualized to each patient.
Summary of Recommendations
1. Treatment goals for Cushing's syndrome
1.1 In patients with overt Cushing's syndrome (CS), we recommend normalizing cortisol levels or action at its receptors to eliminate the signs and symptoms of CS and treating comorbidities associated with hypercortisolism. (1|⊕⊕⊕○)
1.2 We recommend against treatment to reduce cortisol levels or action if there is not an established diagnosis of CS. (1|⊕○○○)
1.3 We suggest against treatments designed to normalize cortisol or its action when there is only borderline biochemical abnormality of the hypothalamic-pituitary-adrenal (HPA) axis without any specific signs of CS. The benefit of treating to normalize cortisol is not established in this setting. (2|⊕○○○)
2. Optimal adjunctive management
2.1 We recommend providing education to patients and their family/caretaker(s) about their disease, treatment options, and what to expect after remission. (Ungraded best practice statement)
2.2 We recommend that all patients receive monitoring and adjunctive treatment for cortisol-dependent comorbidities (psychiatric disorders, diabetes, hypertension, hypokalemia, infections, dyslipidemia, osteoporosis, and poor physical fitness). (Ungraded best practice statement)
2.3 We recommend that a multidisciplinary team, including an experienced endocrinologist, takes patient values and preferences into consideration and provides education about the treatment options to the patient. (Ungraded best practice statement)
2.4 We suggest evaluating CS patients for risk factors of venous thrombosis. (2|⊕⊕○○)
2.5 In patients with CS undergoing surgery, we suggest perioperative prophylaxis for venous thromboembolism. (2|⊕⊕○○)
2.6 We recommend that clinicians discuss and offer age-appropriate vaccinations to CS patients—particularly influenza, Herpes zoster, and pneumococcal vaccinations—due to an increased risk of infection. (Ungraded best practice statement)
3. First-line treatment options
3.1 We recommend initial resection of primary lesion(s) underlying Cushing's disease (CD), ectopic and adrenal (cancer, adenoma, and bilateral disease) etiologies, unless surgery is not possible or is unlikely to significantly reduce glucocorticoid excess (Figure 1). (1|⊕⊕⊕⊕)
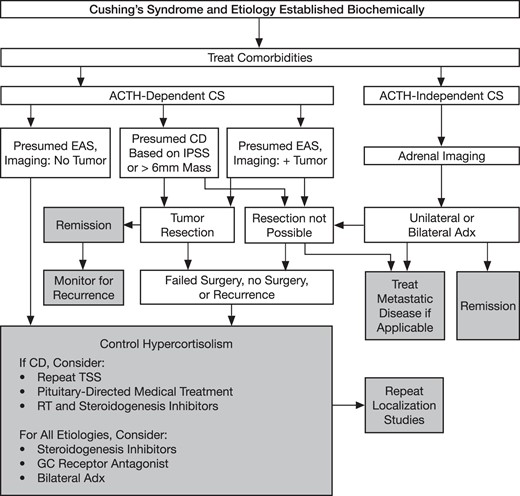
An algorithm for the treatment of CS.
Derived from Nieman LK, Biller BM, Finding, JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:1526–1540. (17)
3.1a We recommend unilateral resection by an experienced adrenal surgeon for all cases of benign unilateral disease. (1|⊕⊕⊕○)
3.1b We recommend localizing and resecting ectopic ACTH-secreting tumors with node dissection as appropriate. (1|⊕⊕⊕⊕)
3.1c We recommend transsphenoidal selective adenomectomy (TSS) by an experienced pituitary surgeon as the optimal treatment for CD in pediatric and adult patients. (1|⊕⊕⊕⊕)
3.1ci We recommend measuring serum sodium several times during the first 5–14 days after transsphenoidal surgery. (1|⊕⊕○○)
3.1cii We recommend assessing free T4 and prolactin within 1–2 weeks of surgery, to evaluate for overt hypopituitarism. (1|⊕⊕○○)
3.1ciii We recommend obtaining a postoperative pituitary magnetic resonance imaging (MRI) scan within 1–3 months of successful TSS. (Ungraded best practice statement)
3.1d We recommend surgical resection of bilateral adrenal disorders (1|⊕⊕○○) and suggest medical therapy to block aberrant hormone receptors for bilateral macronodular adrenal hyperplasia (BMAH) (2|⊕⊕○○).
4. Remission and recurrence after surgical tumor resection
4.1 We suggest an individualized management approach based on whether the postoperative serum cortisol values categorize the patient's condition as hypocortisolism, hypercortisolism, or eucortisolism. (Ungraded best practice statement)
4.2 We recommend additional treatments in patients with persistent overt hypercortisolism. (1|⊕⊕⊕⊕)
4.3 We recommend measuring late-night salivary or serum cortisol in patients with eucortisolism after TSS, including those cases where eucortisolism was established by medical treatment before surgery. (1|⊕⊕○○)
4.4 We recommend using tests to screen for hypercortisolism to assess for recurrence in patients with ACTH-dependent CS. (1|⊕⊕⊕○)
5. Glucocorticoid replacement and discontinuation, and resolution of other hormonal deficiencies
5.1 We recommend that hypocortisolemic patients receive glucocorticoid replacement and education about adrenal insufficiency after surgical remission. (1|⊕⊕⊕⊕)
5.2 We recommend follow-up morning cortisol and/or ACTH stimulation tests or insulin-induced hypoglycemia to assess the recovery of the HPA axis in patients with at least one intact adrenal gland, assuming there are no contraindications. We also recommend discontinuing glucocorticoid when the response to these test(s) is normal. (1|⊕⊕⊕○)
5.3 We recommend re-evaluating the need for treatment of other pituitary hormone deficiencies in the postoperative period. (1|⊕⊕⊕○)
6. Second-line therapeutic options
6.1 In patients with ACTH-dependent CS who underwent a noncurative surgery or for whom surgery was not possible, we suggest a shared decision-making approach because there are several available second-line therapies (eg, repeat transsphenoidal surgery, radiotherapy, medical therapy, and bilateral adrenalectomy). (2|⊕⊕○○)
6.1a We suggest bilateral adrenalectomy for occult or metastatic ectopic ACTH secretion (EAS) or as a life-preserving emergency treatment in patients with very severe ACTH-dependent disease who cannot be promptly controlled by medical therapy. (2|⊕⊕⊕○)
6.1b We recommend regularly evaluating for corticotrope tumor progression using pituitary MRIs and ACTH levels in patients with known CD who undergo bilateral adrenalectomy and in patients who undergo this procedure for presumed occult EAS (because some of the latter have a pituitary and not ectopic tumor). (1|⊕⊕⊕○)
6.2 Repeat transsphenoidal surgery
6.2 We suggest repeat transsphenoidal surgery, particularly in patients with evidence of incomplete resection, or a pituitary lesion on imaging. (2|⊕⊕○○)
6.3 Radiation therapy/radiosurgery for CD
6.3 We recommend confirming that medical therapy is effective in normalizing cortisol before administering radiation therapy (RT)/radiosurgery for this goal because this will be needed while awaiting the effect of radiation. (1|⊕○○○)
6.3a We suggest RT/radiosurgery in patients who have failed TSS or have recurrent CD. (2|⊕⊕○○)
6.3b We recommend using RT where there are concerns about the mass effects or invasion associated with corticotroph adenomas. (1|⊕⊕⊕○)
6.3c We recommend measuring serum cortisol or urine free cortisol (UFC) off-medication at 6- to 12-month intervals to assess the effect of RT and also if patients develop new adrenal insufficiency symptoms while on stable medical therapy. (1|⊕⊕⊕○)
6.4 Medical treatment
6.4 We recommend steroidogenesis inhibitors under the following conditions: as second-line treatment after TSS in patients with CD, either with or without RT/radiosurgery; as primary treatment of EAS in patients with occult or metastatic EAS; and as adjunctive treatment to reduce cortisol levels in adrenocortical carcinoma (ACC). (1|⊕⊕⊕○)
6.4a We suggest pituitary-directed medical treatments in patients with CD who are not surgical candidates or who have persistent disease after TSS. (2|⊕⊕⊕○)
6.4b We suggest administering a glucocorticoid antagonist in patients with diabetes or glucose intolerance who are not surgical candidates or who have persistent disease after TSS. (2|⊕⊕⊕○)
6.4c We suggest targeted therapies to treat ectopic ACTH syndrome. (2|⊕○○○)
7. Approach for long-term follow-up
7.1 We recommend treating the specific comorbidities associated with CS (eg, cardiovascular risk factors, osteoporosis and psychiatric symptoms) in all patients with CS throughout their lives until resolution (Figure 1). We also recommend testing for recurrence throughout life, except in patients who underwent resection of an adrenal adenoma with a computerized tomography (CT) density of < 10 Hounsfield units. (1|⊕⊕⊕○)
7.2 We recommend educating patients and families about the clinical features of remission. (Ungraded best practice statement)
7.3 In patients with adrenal adenoma, we suggest follow-up tests for the specific comorbidities associated with CS if the adenoma density on CT was < 10 Hounsfield units. (2|⊕⊕○○) For those with higher Hounsfield unit values or pathology consistent with possible carcinoma, we suggest evaluating for malignancy using imaging. (2|⊕○○○)
7.4 We recommend that patients with Carney complex have lifelong follow-up tests for cardiac myxoma and other associated disease (testicular tumors, acromegaly, thyroid lesions). (1|⊕⊕⊕⊕)
8. Special populations/considerations
8.1 We recommend urgent treatment (within 24–72 h) of hypercortisolism if life-threatening complications of CS such as infection, pulmonary thromboembolism, cardiovascular complications, and acute psychosis are present. (1|⊕⊕⊕○). The associated disorder(s) should be addressed as well (eg, anticoagulation, antibiotics).
Developmental Method for Evidence-Based Clinical Practice Guidelines
The Clinical Guidelines Subcommittee of the Endocrine Society deemed the treatment of CS a priority area in need of practice guidelines and appointed a Task Force to formulate evidence-based recommendations. The Task Force followed the approach recommended by the Grading of Recommendations, Assessment, Development, and Evaluation group—an international group with expertise in the development and implementation of evidence-based guidelines (1). A detailed description of the grading scheme has been published elsewhere (2). The Task Force used the best available research evidence to develop the recommendations. The Task Force also used consistent language and graphical descriptions of both the strength of a recommendation and the quality of evidence. In terms of the strength of the recommendation, strong recommendations use the phrase “we recommend” and the number 1, and weak recommendations use the phrase “we suggest” and the number 2. Cross-filled circles indicate the quality of the evidence—⊕○○○ denotes very low quality evidence; ⊕⊕○○, low quality; ⊕⊕⊕○, moderate quality; and ⊕⊕⊕⊕, high quality. The Task Force has confidence that persons who receive care according to its strong recommendations will derive, on average, more good than harm. Weak recommendations require more careful consideration of the person's circumstances, values, and preferences to determine the best course of action. Linked to each recommendation is a description of the evidence and the values that panelists considered when making the recommendation; in some instances, panelists offer technical suggestions for testing conditions, dosing, and monitoring. These technical comments reflect the best available evidence applied to a typical person being treated. Often this evidence comes from the panelists' values, preferences, and unsystematic observations; therefore, these remarks should be considered as suggestions. To make the guideline comprehensive, the task force included several overarching statements to emphasize some accepted clinical principles that are not supported by clear direct evidence. These statements are explicitly marked as ungraded best practice statements.
The Endocrine Society maintains a rigorous conflict-of-interest review process for the development of clinical practice guidelines. All Task Force members must declare any potential conflicts of interest. These are reviewed before members are approved to serve on the Task Force and reviewed periodically during the development of the guideline. The Clinical Guidelines Subcommittee vets the conflict-of-interest forms before the members are approved by the Society's Council to participate on the guideline Task Force. Most participants who help develop the guideline must have no conflict of interest related to the matter under study.
Those participants who do have conflicts of interest must disclose all conflicts. The Clinical Guidelines Subcommittee and the Task Force have reviewed all disclosures for this guideline and resolved or managed all identified conflicts of interest. Most members of the Task Force have relationships with companies that make pharmaceuticals for the treatment of CS. The Task Force explicitly discussed the possibility of a perception of conflict and agreed that the sections on individual pharmaceuticals would be written by an individual without such a conflict; subsequent changes were approved by the entire group.
Conflicts of interest are defined as receiving any compensation from commercial interest(s) in the form of grants; research support; consulting fees; salary; ownership interest (eg, stocks, stock options, or ownership interest excluding diversified mutual funds); honoraria or other payments for participation in speakers' bureaus, advisory boards, or boards of directors; or other financial benefits. Completed forms are available through the Endocrine Society office.
The Endocrine Society was the only funding source for this guideline; the Task Force received no funding or remuneration from commercial or other entities.
Commissioned systematic reviews
The three reviews summarized data from 29 case series of radiotherapy in CD, 21 case series of radiosurgery in CD, and 87 case series of treatment-naive CD patients who received first-line transsphenoidal surgery. The outcomes of interest were biochemical remission and biochemical recurrence rates. In each review, we tested several patient, surgeon, and procedure variables to determine whether they were predictors of remission and recurrence rates.
Overall, analyses were underpowered to determine important independent predictors of the outcomes of interest. The quality of the evidence for recurrence and remission outcomes was low due to high risk of bias, heterogeneity, and imprecision.
1. Treatment goals for Cushing's syndrome
1.1 In patients with overt CS, we recommend normalizing cortisol levels or action at its receptors to eliminate the signs and symptoms of CS and treating comorbidities associated with hypercortisolism. (1|⊕⊕⊕○)
1.2 We recommend against treatment to reduce cortisol levels or action if there is not an established diagnosis of CS. (1|⊕○○○)
1.3 We suggest against treatments designed to normalize cortisol or its action when there is only borderline biochemical abnormality of the hypothalamic-pituitary-adrenal (HPA) axis without any specific signs of CS. The benefit of treating to normalize cortisol is not established in this setting. (2|⊕○○○)
Evidence
CS is a condition of pathological hypercortisolism that includes demonstrable clinical features. The goals of treating CS are to eliminate its primary cause and achieve remission so as to eliminate the associated signs, symptoms, and comorbidities and to improve quality of life (QOL). In 1952, before effective treatment was available, patients with CS had a median survival of 4.6 years (3, 4). Sixty years later, despite available treatments for comorbidities, patients with active CS continue to have an increased standardized mortality rate that is 1.7 to 4.8-fold greater than the general population (5–9). In contrast, when CS and its associated comorbidities are successfully treated, the standardized mortality rate improves. Whether it is similar or not to the general population or remains significantly increased remains debatable (5–9). As expected, patients with persistent or recurrent hypercortisolism continue to have higher than expected mortality (Hazard Ratio, 2.8–16) (6, 9–11).
Cardiovascular disease, venous thrombosis, and infections are the primary causes of the excess mortality rate we see in CS. Research indicates that the risk of infection is lower in patients with mild to moderate vs severe hypercortisolism, which indirectly supports intervention (12). Altogether, these data suggest that prompt treatment of CS and its comorbidities is important to reduce mortality.
Restoring eucortisolism leads to clinical and biochemical improvements regarding obesity, arterial hypertension, insulin resistance, glucose tolerance, dyslipidemia, bone mineral density (BMD), linear growth in children, cognition, psychiatric disorders, and health-related QOL (HRQOL) (13). However, these complications persist in many patients and should be addressed therapeutically before and after remission from CS (14, 15) (see below).
Although there are no controlled studies in pediatric CS, growth and body composition generally improve after treatment. Most patients reach an adult height within their predicted parental target range, although they may need GH therapy to achieve this (16).
Because all treatments carry risk, clinicians should establish a diagnosis of CS before administering them (17). However, in life-threatening situations, a clinical diagnosis with minimal available biochemical data may justify prompt treatment. Similarly, the consequences of mild or cyclic hypercortisolism are not clear, so that treatment guidelines cannot be generalized to those patients. However, because CS tends to progress to severe hypercortisolism, it is possible that early recognition and treatment of mild or cyclic disease (values < 1.5-fold upper reference range) would reduce the risk of residual morbidity. Unfortunately, few data address this assumption. If the clinician is uncertain of the clinical diagnosis (regardless of the magnitude of biochemical perturbations), further testing over time is always the best approach. The treatment of subclinical CS in the context of the evaluation of an adrenal incidentaloma is outside the scope of this guideline on clinical CS.
2. Optimal adjunctive management
2.1 We recommend providing education to patients and their family/caretaker(s) about their disease, treatment options, and what to expect after remission. (Ungraded best practice statement)
2.2 We recommend that all patients receive monitoring and adjunctive treatment for cortisol-dependent comorbidities (psychiatric disorders, diabetes, hypertension, hypokalemia, infections, dyslipidemia, osteoporosis, and poor physical fitness). (Ungraded best practice statement)
2.3 We recommend that a multidisciplinary team, including an experienced endocrinologist, takes patient values and preferences into consideration and provides education about the treatment options to the patient (Ungraded best practice statement)
2.4 We suggest evaluating CS patients for risk factors of venous thrombosis. (2|⊕⊕○○)
2.5 In patients with CS undergoing surgery, we suggest perioperative prophylaxis for venous thromboembolism. (2|⊕⊕○○)
2.6 We recommend that clinicians discuss and offer age-appropriate vaccinations to CS patients—particularly influenza, Herpes zoster, and pneumococcal vaccinations—due to an increased risk of infection. (Ungraded best practice statement)
Evidence
Severe hypercortisolism impairs immunity and predisposes to severe, systemic infection and/or sepsis due to bacterial, fungal, and opportunistic pathogens (12, 18–20). Based on this, we suggest immunization against influenza, shingles, and pneumonia. Hypercortisolism alters coagulation-factor profiles for up to 1 year after a surgical cure (21) and carries an increased risk of venous thrombosis, especially in the 4 weeks after surgery (21–24). Clinicians caring for patients with CS should be aware of this increased risk, should evaluate their patients for thrombosis and bleeding (25, 26), and should consider anticoagulation treatments. Clinicians should treat all other comorbidities aggressively and seek appropriate consultations (including rehabilitation medicine). We suggest that patients receive written information regarding their disorder.
3. First-line treatment options
3.1 We recommend initial resection of primary lesion(s) underlying CD, ectopic and adrenal (cancer, adenoma, and bilateral disease) etiologies, unless surgery is not possible or unlikely to significantly reduce glucocorticoid excess. (1|⊕⊕⊕⊕)
Evidence
Complete surgical resection of the causal tumor(s) (or adrenal hyperplasia) is the optimal treatment of CS because it alleviates hypercortisolism while potentially leaving the normal HPA axis intact (except for bilateral adrenal disorders). The common causes of CS include intrinsic adrenal gland abnormalities and ACTH secretion from a corticotrope tumor (CD) or from an ectopic tumor (EAS). Thus, differential diagnostic testing and tumor localization studies are critical to a successful outcome (27).
3.1a We recommend unilateral resection by an experienced adrenal surgeon for all cases of benign unilateral disease. (1|⊕⊕⊕○)
Evidence
In experienced hands, unilateral adrenalectomy is curative in nearly 100% of adults and children with cortisol-producing adrenal adenomas; the complication rate is higher when performed by surgeons with less experience (28). Adrenalectomy is generally performed via either trans or retroperitoneal laparoscopy unless other factors preclude this. The role of adrenalectomy (vs intensive medical treatment of comorbidities) in patients with adrenal incidentaloma and subtle cortisol dysregulation (so-called subclinical CS) is not clear. However, retrospective studies suggest that patients with metabolic abnormalities are likely to benefit (29).
Patients with ACC have a poor prognosis, and clinicians should attempt definitive treatment via complete resection. In children, complete resection was associated with survival rates of 80%, whereas the outlook for unresectable disease was very poor (30). Surgeons increasingly perform transperitoneal laparoscopic adrenalectomy, particularly for small tumors, but this is controversial because many recommend open surgery to assess stage and achieve en bloc resection (31, 32). Because hypercortisolism is associated with increased mortality, medical treatment may be needed to achieve eucortisolism (33, 34).
Follow-up, optimally by an adrenal cancer-specific multidisciplinary team, may include cytotoxic chemotherapy and adjuvant radiotherapy or mitotane treatments (35). Further discussion is beyond the scope of these guidelines.
3.1b We recommend localizing and resecting ectopic ACTH-secreting tumors with node dissection as appropriate. (1|⊕⊕⊕⊕)
Evidence
In the absence of overt metastatic disease, ectopic ACTH-producing tumor resection cured 76% of patients, as demonstrated by observational studies (36).
3.1c We recommend TSS by an experienced pituitary surgeon as the optimal treatment for CD in pediatric and adult patients. (1|⊕⊕⊕⊕)
Evidence
Many centers have replaced the transnasal route with an endoscopic endonasal approach (37), but both methods can be effective (38). Successful resection is most likely when performed by an experienced neurosurgeon who has a high volume of these cases.
These are typically benign microadenomas (<1 cm diameter) and are evident on pituitary MRI in approximately 60% of adults (39) and approximately 55% of children (40, 41). Some, but not all, reports demonstrate a greater success when clinicians identify the tumor by MRI before surgery (42–44). However, because 10% of healthy adults have pituitary lesions ≤ 6 mm that are visible on MRI (45), the presence of a lesion does not definitively confirm the diagnosis of CD or identify the causal tumor. In addition, there is a 12% rate of abnormal pituitary MRI scans in patients with EAS (18) and a 12% rate of false localization by MRI in patients with surgically proven CD (46). When feasible, all patients with ACTH-dependent CS and no obvious causal neoplasm (>6 mm) should be promptly referred to an experienced center that can safely and reliably perform inferior petrosal sinus sampling to distinguish a pituitary from a nonpituitary (ectopic) cause.
Inferior petrosal sinus sampling does not reliably identify the tumor site because a side-to-side gradient of > 1.4 correctly predicted location in only 56–69% of adults (46–48) and 59–81% of children (40, 49). Hemihypophysectomy, based on inferior petrosal sinus sampling lateralization, cured only 50% of patients in one study, an outcome no better than chance (47).
3.1ci We recommend measuring serum sodium several times during the first 5–14 days after transsphenoidal surgery. (1|⊕⊕○○)
3.1cii We recommend assessing free T4 and prolactin within 1–2 weeks of surgery, to evaluate for overt hypopituitarism. (1|⊕⊕○○)
3.1ciii We recommend obtaining a postoperative pituitary MRI scan within 1–3 months of successful TSS. (Ungraded best practice statement)
Evidence
As with any TSS, potential complications include electrolyte disturbances, hemorrhage, and meningitis (50). Hyponatremia occurs in 5–10% of patients, usually between postoperative days 5 and 10 (51). This complication is more common after extensive gland exploration in menstruating women. Diabetes insipidus is relatively common in the first few postoperative days but is usually transient. We recommend measuring serum sodium several times during the first 5–14 days after surgery to address both possibilities, either daily or guided by the patient's intake and output. It is helpful to provide patients with education regarding post-transsphenoidal water balance disorders, including when to seek emergency care for the accompanying symptoms. It is advisable to provide the patient with information about how to reach an endocrinologist in this circumstance because some emergency rooms are not familiar with this cause of hyponatremia.
Because of the 5- to 7-day half-life of T4, a decrease of free or total T4 within 1 week of surgery may identify significant hypothyroidism (when compared to preoperative values). Acquired prolactin deficiency is a marker for hypopituitarism that may occur immediately after TSS (52). Hormonal deficits may be secondary to hypercortisolism and also transient; therefore, we recommend re-evaluating the need for replacement therapy (see below). Permanent hypopituitarism is more common after surgery for a microadenoma secreting ACTH than for those secreting GH. This probably reflects a tendency to more aggressive surgery but is not fully explained.
There are few data about the timing and need for postoperative imaging in patients with surgical remission, but typical practice may include obtaining a postoperative scan 1–3 months after surgery to serve as a new baseline in case of future recurrence.
3.1d We recommend surgical resection of bilateral adrenal disorders (1|⊕⊕○○) and suggest medical therapy to block aberrant hormone receptors for bilateral macronodular adrenal hyperplasia (BMAH) (2|⊕⊕○○).
Bilateral macronodular adrenal hyperplasia
The terminology in this field is changing. In primary BMAH, the disease eventually affects both adrenals, although it may present initially as an asymmetric unilateral nodule (53).
Recent reports demonstrate an inherited mutation in the armadillo repeat-containing 5 (ARMC5) gene in patients with BMAH and family members with unsuspected BMAH (54, 55) (and in some with meningioma) (56). These findings do not provide new treatment(s). However, screening family members (with dexamethasone 1 mg) is indicated until genetic tests are available.
BMAH can be treated by an appropriate antagonist if aberrant receptors are demonstrated that clearly couple to cortisol release, but the detailed workup of such patients is a research undertaking beyond the scope of these guidelines (57). Bilateral adrenalectomy, generally via the laparoscopic route, is the surgical treatment of choice, although some advocate for selective removal of the larger adrenal in older patients in whom nonfunctional tumors are a possibility, especially if there is only a single nodule on each side (58).
In children, McCune-Albright syndrome may present as BMAH, with a variable prognosis. It may be aggressive and life threatening, with severe clinical features and hypertension (59), in which case bilateral adrenalectomy is indicated. Alternatively, research has reported spontaneous remission after medical therapy to control hypercortisolemia (60). Therefore, in milder cases, clinicians may consider medical therapy.
Primary pigmented nodular adrenal disease
Laparoscopic bilateral adrenalectomy is the definitive treatment of choice and is curative in most cases of primary pigmented nodular adrenal disease, regardless of age (61, 62). However, in some pediatric patients, unilateral adrenalectomy leads to significant clinical and biochemical improvement (63), although studies have reported a subsequent relapse requiring the removal of the second gland (58, 64). It is important to screen patients with suspected primary pigmented nodular adrenal disease at intervals for features of Carney complex, particularly for atrial myxoma (62). If clinicians detect Carney complex, they should also test family members.
Evidence
The advantage of bilateral adrenalectomy is the swift and definitive control of hypercortisolism; its disadvantages include the need for lifelong glucocorticoid and mineralocorticoid replacement therapy.
A review (65) of 23 studies reporting 739 patients undergoing bilateral adrenalectomy (about 70% laparoscopic) showed a surgical morbidity of 18% and a median mortality of 3%. In patients with CD, the surgical mortality was < 1%. In 3–34% of cases, there was residual cortisol secretion due to accessory adrenal tissue or adrenal remnants, but < 2% had a relapse of CS. Adrenal crises occurred in 9.3 cases per 100 patient-years. Overall mortality was 17% during a 41-month follow-up (range, 14–294 mo). However, 46% of the deaths occurred within the first year after surgery, suggesting that careful management of cortisol-dependent comorbidities is needed. The 10-year mortality after bilateral adrenalectomy for CD, bilateral adrenal hyperplasia, and EAS is 3, 10, and 44%, respectively (65).
4. Remission and recurrence after surgical tumor resection
4.1 We suggest an individualized management approach based on whether the postoperative serum cortisol values categorize the patient's condition as hypocortisolism, hypercortisolism, or eucortisolism. (Ungraded best practice statement)
4.2 We recommend additional treatments in patients with persistent overt hypercortisolism. (1|⊕⊕⊕⊕)
4.3 We recommend measuring late-night salivary or serum cortisol in patients with eucortisolism after TSS, including those cases where eucortisolism was established by medical treatment before surgery. (1|⊕⊕○○)
4.4 We recommend using tests to screen for hypercortisolism to assess for recurrence in patients with ACTH-dependent CS. (1|⊕⊕⊕○)
Evidence
Postoperative initial remission
There is no consensus on the criteria for remission after resecting an ACTH-producing tumor. (In CD, because of the significant recurrence rate, the term “remission” is preferable to “cure.”) Normal corticotropes are suppressed by sustained hypercortisolism; therefore, ACTH and cortisol levels are low after resecting the ACTH-producing tumor. Remission is generally defined as morning serum cortisol values < 5 μg/dL (<138 nmol/L) or UFC < 28–56 nmol/d (<10–20 μg/d) within 7 days of selective tumor resection. Although clinicians have used glucocorticoid dependence to characterize surgical response, we advocate measuring serum cortisol levels as a preferable and quantifiable alternative.
Adult series report a remission rate of 73–76% for selectively resected microadenomas, but a lower remission rate for macroadenomas (∼43%) (66–68).
In two pediatric series using a strict criterion for remission (post-TSS serum cortisol of < 1 μg/dL [28 nmol/L] [41] or < 1.8 μg/dL [50 nmol/L] [40]), remission rates were 100 and 69%, respectively. Follow-up data suggest that hypercortisolemia recurrence was uncommon (40, 70). Other series reported remission rates of 70–98% (71, 72).
A recent report of 200 cases of pediatric CD (72) associated initial postoperative remission with adenoma identification at surgery. The study also associated long-term remission with younger age, smaller adenoma, and morning serum cortisol of < 1 μg/dL after surgery (72). In one study of adults, the initial remission rate for microadenomas was greater than for macroadenomas (72.8 vs 42.9%) (66). Technical aspects, surgeon experience, tumor size, and the lack of dural invasion likely contribute most to successful outcome in patients of any age (213).
Patients with mild or cyclic CS and those rendered eucortisolemic by medical treatment before surgery may not have suppressed corticotropes. Their postoperative UFC and morning cortisol may be normal. In these patients, clinicians must measure late-night serum or salivary cortisol. If there is a normal diurnal rhythm, ie, an appropriately low late-night serum or salivary cortisol level, then it is likely that the patient is in remission. Conversely, the lack of a diurnal rhythm suggests persistent disease. Patients who are eucortisolemic after resection, especially those with moderate preoperative hypercortisolism, may have residual tumors and are more prone to recurrence than patients with prolonged postoperative hypocortisolism (67).
After bilateral adrenalectomy, patients have undetectable serum cortisol values, and morning plasma ACTH levels are often between 200 and 500 pg/mL. Morning cortisol values are generally < 1.8 μg/dL (50 nmol/L) after resecting an adrenal adenoma.
Recurrence
Recurrence rates of hypercortisolemia in pediatric CD are very low (40, 70), especially when postoperative plasma ACTH and cortisol levels are undetectable. Recurrence is a more significant problem in adults. In one study, after initial remission, 23% of adults with microadenomas and 33% with macroadenomas had recurrence. Recurrence rates vary from 15–66% within 5–10 years of initially successful surgery (68, 73, 74).
Clinicians should evaluate patients for possible CD recurrence when the HPA axis recovers, and then annually, or sooner if they have clinical symptoms. Early recovery (within 6 mo) of HPA axis function may indicate an increased risk of recurrence (66). Two longitudinal studies demonstrated that elevated late-night serum/salivary cortisol is one of the earliest biochemically detectable signs of recurrence and almost always precedes elevated urine cortisol (75, 76). Although many such patients evolve to significant hypercortisolism, it is uncertain whether there is any benefit to treating patients with a mild early recurrence if they are asymptomatic.
Although studies have reported that a number of tests (eg, CRH, desmopressin) predict recurrence risk, diagnostic accuracy is not high enough to provide certainty regarding long-term outcomes. Thus, clinicians must monitor every patient in remission from CD for the possibility of recurrence (73, 77).
Rarely, residual adrenocortical tissue (usually in the surgical bed, but sometimes in the gonads) regrows as a result of prolonged ACTH hypersecretion. If patients become cushingoid after bilateral adrenalectomy, clinicians should test endogenous cortisol secretory capacity by withholding glucocorticoid for 24 hours and checking serum/salivary cortisol levels.
Recurrence after ectopic ACTH-secreting tumor resection generally reflects metastasis.
5. Glucocorticoid replacement and discontinuation, and resolution of other hormonal deficiencies
5.1 We recommend that hypocortisolemic patients receive glucocorticoid replacement and education about adrenal insufficiency after surgical remission. (1|⊕⊕⊕⊕)
5.2 We recommend follow-up morning cortisol and/or ACTH stimulation tests or insulin-induced hypoglycemia to assess the recovery of the HPA axis in patients with at least one intact adrenal gland, assuming there are no contraindications. We also recommend discontinuing glucocorticoid when the response to these test(s) is normal. (1|⊕⊕⊕○)
5.3 We recommend re-evaluating the need for treatment of other pituitary hormone deficiencies in the postoperative period. (1|⊕⊕⊕○)
Evidence
After successful surgery, glucocorticoid replacement is required until the HPA axis recovers, which in adults occurs about 6–12 months after resecting ACTH-producing tumors and about 18 months after unilateral adrenalectomy (78, 79). In one large pediatric series, recovery occurred at a mean of 12.6 ± 3.3 months after surgery (80). (Obviously, bilateral adrenalectomy results in a need for lifelong glucocorticoid and mineralocorticoid replacement.)
Despite the use of physiological glucocorticoid replacement, many patients suffer from glucocorticoid withdrawal. Patients should be warned that this is common and expected (81). Symptoms include anorexia; nausea; weight loss; and other nonspecific symptoms such as fatigue, flu-like myalgias and arthralgias, lethargy, and skin desquamation. Accordingly, patients usually feel worse within a few days or weeks after successful surgery. Adults may experience persistent or new-onset atypical depressive disorders, anxiety, or panic symptoms (82). Recovery from the glucocorticoid withdrawal syndrome may take 1 year or longer.
The syndrome may persist even after the HPA axis has recovered and may even occur in patients who do not develop secondary adrenal insufficiency after TSS for CD (83). The pathophysiology of the steroid withdrawal syndrome is not known. Patients may improve with a temporary increase in the glucocorticoid dose, but it is important to reduce the dose as soon as possible to avoid iatrogenic CS. Administering serotonin-specific reuptake inhibitors may help, but this has not been systematically studied. Generally, the most important intervention is frequent support and reassurance by the medical team. Family, friends, and patient support groups also may be helpful.
We recommend glucocorticoid replacement with hydrocortisone, 10–12 mg/m2/d in divided doses, either twice or thrice daily, with the first dose taken as soon as possible after waking (84). Although this dose is somewhat higher than recently reported cortisol production rates (85), it works well clinically, probably because of interindividual differences in hepatic and adipose metabolism and glucocorticoid receptor polymorphisms. Hydrocortisone is preferred because more potent synthetic glucocorticoids with a longer half-life may prolong HPA axis suppression.
When hydrocortisone is not available, or patients request once-daily dosing, clinicians can administer other glucocorticoids at the lowest possible replacement dose. Written instructions about stress dosing for intercurrent illnesses, injectable emergency steroids, and the need to obtain and wear a medical alert tag indicating adrenal insufficiency/glucocorticoid replacement are essential (84).
Although some practitioners prescribe supraphysiological doses (eg, hydrocortisone 20 mg two to three times daily) in the immediate postoperative period, there are no controlled studies that address whether this (or a slower taper) minimizes the glucocorticoid withdrawal syndrome. Other clinicians use only physiological replacement doses to avoid continued excessive glucocorticoid exposure.
There are a variety of tapering and discontinuation strategies, none of which has been systematically studied; the following are general comments. Some centers reduce the hydrocortisone dose as weight decreases and discontinue abruptly when the HPA axis is recovered; others taper the dose at fixed intervals. Clinicians can assess HPA axis recovery with a morning cortisol level obtained (before that day's glucocorticoid dose) every 3 months, followed by an ACTH stimulation test starting when the level is 7.4 μg/dL (200 nmol/L) or more. The axis has recovered if the baseline or stimulated level is approximately 18 μg/dL (500 nmol/L) or greater. Patients with cortisol levels below 5 μg/dL (138 nmol/L) should remain on glucocorticoids until retested in 3–6 months. Stimulation testing may be helpful with intermediate values. However, various studies propose different cutoffs, and assays differ, so clinical judgment should be used. It is rare that the HPA axis does not eventually recover.
Any etiology of hypercortisolism can cause preoperative functional central hypothyroidism and central hypogonadism. Although these may resolve after 6 postoperative months (86), patients may need continued replacement therapy. Clinicians should repeat testing to establish when and if the patient has recovered. GH deficiency, frequently present during hypercortisolism, persisted in over 50% of children during the first 12 months after cure (87) and to a lesser extent into adult life (88). It might not be possible for patients to attain normal linear growth after surgically cured CD, with patients often not reaching their genetic target (89). For this reason, some centers test GH stimulation at 3 months after surgery and initiate human GH replacement, combined with GnRH agonist therapy, in pubertal subjects to maximize growth potential in children with abnormal responses (16). The use of GH therapy in adults after remission should follow previously published guidelines (90). Because hypercortisolemia can affect the GH axis, some experts advise waiting at least 12 months after remission to test for deficiency of this hormone in adults (91).
6. Second-line therapeutic options
6.1 In patients with ACTH-dependent Cushing's syndrome who underwent a noncurative surgery or for whom surgery was not possible, we suggest a shared decision-making approach because there are several available second-line therapies (eg, repeat transsphenoidal surgery, radiotherapy, medical therapy, or bilateral adrenalectomy). (2|⊕⊕○○)
Evidence
When surgery is not possible or is noncurative, the choice of second-line therapy must take into account patient preferences; treatment goals; biochemical control; the size and location of residual tumors; the urgency to treat; other medications (drug-drug interactions); the patient's personal history; the method of delivery, side effects, and cost of medication; gender; age; and the availability of medical therapies.
After unsuccessful transsphenoidal surgery, if cortisol levels remain consistently elevated, the prompt titration of medical therapies (or bilateral adrenalectomy) is needed. Even if cortisol levels are normal, careful observation is needed because cortisol levels may fall over subsequent weeks (67).
6.1a We suggest bilateral adrenalectomy for occult or metastatic EAS or as a life-preserving emergency treatment in patients with very severe ACTH-dependent disease who cannot be promptly controlled by medical therapy. (2|⊕⊕⊕○)
6.1b We recommend regularly evaluating for corticotrope tumor progression using pituitary MRIs and ACTH levels in patients with known CD who undergo bilateral adrenalectomy and in patients who undergo this procedure for presumed occult EAS (because some of the latter have a pituitary and not ectopic tumor). (1|⊕⊕⊕○)
Evidence
Medical therapy may be an initial approach in any patient with ACTH-dependent CS who has failed surgery, who has persistent or metastatic disease, or who has an occult tumor. It is also useful in severely ill patients (92).
In patients with CD, the primary corticotroph adenoma remains in situ after adrenalectomy. Therefore, both children and adults have a significant risk of developing macroscopic (>1 cm) enlargement of the tumor and hyperpigmentation—the so-called Nelson's syndrome.
After bilateral adrenalectomy for CD, 21% of adults developed Nelson's syndrome (65), whereas corticotroph tumor progression without all of the features described by Nelson was observed on MRI in 50% of cases (93). Nelson's syndrome was less likely in patients without visible tumor at the time of adrenalectomy (93). It is not known conclusively whether RT mitigates this risk.
The risk of corticotroph tumor progression is associated with increased plasma ACTH, but other predictors are not understood. Thus, lifelong follow-up is important and should include clinical examinations for hyperpigmentation, ACTH measurements, and MRI scans. These should be performed initially and at annual intervals with a decrease in frequency based on previous findings.
Patients with presumed occult EAS may in fact have CD and thus should be regularly examined for the emergence of a pituitary tumor (94).
6.2 Repeat transsphenoidal surgery
6.2 We suggest repeat transsphenoidal surgery, particularly in patients with evidence of incomplete resection, or a pituitary lesion on imaging. (2|⊕⊕○○)
In 12 of 17 patients with persistent hypercortisolism after TSS, early repeat transsphenoidal surgery induced hypocortisolism. All patients had tumors that were found during the initial procedure (95). Repeat surgery for recurrent hypercortisolism led to hypocortisolism in 22 of 31 patients with previous surgical remission. Evidence of previous incomplete resection, the presence of a pituitary lesion on imaging, and intraoperative tumor detection predicted remission (96). Repeat surgery carries an increased risk of hypopituitarism compared to initial surgery. Although remission is less likely than after the first surgery, it can be achieved rapidly compared to some other second-line treatments and is important to consider, particularly when there is access to an expert pituitary surgeon.
6.3 Radiation therapy/radiosurgery for Cushing's disease
6.3 We recommend confirming that medical therapy is effective in normalizing cortisol before administering RT/radiosurgery for this goal because this will be needed while awaiting the effect of radiation. (1|⊕○○○)
6.3a We suggest RT/radiosurgery in patients who have failed TSS or have recurrent CD. (2|⊕⊕○○)
6.3b We recommend using RT where there are concerns about the mass effects or invasion associated with corticotroph adenomas. (1|⊕⊕⊕○)
6.3c We recommend measuring serum cortisol or UFC off-medication at 6- to 12-month intervals to assess the effect of RT and also if patients develop new adrenal insufficiency symptoms while on stable medical therapy. (1|⊕⊕⊕○)
Evidence
Although pituitary radiation may serve as a primary treatment for CD in individuals who cannot undergo surgery or when the tumor is invasive or unresectable, it is most often used as a second-line treatment when surgery fails. The effects of radiation occur over months to years, so it is important to medically manage cortisol excess until cortisol is controlled off-medication. If pituitary RT is planned, one should first demonstrate that adequate biochemical control is possible by medical means to ensure control while waiting for radiation effects to occur. RT of some type may be indicated for an invasive, expansive, atypical corticotroph tumor, in which case it is possible that cortisol may be controlled in a different way (eg, adrenalectomy) or may not be elevated.
Several different forms of radiation are available. The term “conventional radiation” typically refers to fractionated photon beam RT. Many centers traditionally administered RT from a linear accelerator, via a three-field technique (two lateral, one frontal) to deliver a total dose of 45 Gy over 6 weeks (97). However, intensity-modulated RT is increasingly used to modulate the dose to accommodate tumor contours and spare nearby critical structures.
Conventional RT results in remission in up to 83% of adult patients, from 6–60 months after treatment, but often within 2 years. In children, conventional RT without adjunctive medical treatment was effective in less time than in adults (98). In pediatric series, 18 of 23 (78%) children were cured in 9–18 months after RT alone (99, 100).
Stereotactic radiation uses a frame to position the patient accurately and includes computer-assisted planning combined with MRI to deliver radiation to the tumor through many ports so as to minimize radiation to surrounding structures. Using single-dose stereotactic radiation is often termed “radiosurgery”; several millimeters of clearance between the tumor and the neurovisual apparatus are required to avoid damaging the optic nerves. Gamma knife, linear accelerator, and proton beam are administered in this way; there are no direct comparisons of effectiveness and safety, but for CD, results appear to be similar. There are no reported series of stereotactic radiosurgery in pediatric CD.
Radiosurgery increases patient convenience by allowing a single treatment day, rather than 6 weeks of therapy (101). It may provide more rapid biochemical control of cortisol excess than conventional radiation and confer less risk of radiation damage to surrounding brain structures, but these anecdotal findings have not been definitively established.
Some experts recommend radiosurgery only when there is a clear target on MRI, bearing in mind that this may not be an ACTH-secreting tumor, whereas others advise using radiosurgery in all patients with adequate clearance of the neurovisual apparatus, targeting the whole sella and a few millimeters beyond if no lesion is visible.
Reports of radiation effectiveness in CD include various methods, definitions of biochemical and tumor control, and lengths of follow-up, making comparison across studies difficult. Among publications over the last 15 years with at least 20 subjects, cortisol excess was biochemically controlled in 28–86% of patients (102–104). Recurrences may develop, so patients need long-term monitoring. It has been suggested that recurrence may be more frequent after radiosurgery than after conventional RT, but this point is not firmly established. Tumor size was better controlled (83–100% success rate) than cortisol (102–104); this treatment option is particularly valuable for large tumors.
A normal diurnal rhythm is not necessarily achieved after RT, so increased late-night cortisol levels should not be a criterion for remission. The increased nocturnal value may represent the persistence of mild residual hypercortisolism in patients with normalized UFC, but this possibility has not been investigated.
Clinicians use medications to normalize cortisol until radiation takes effect. We recommend assessing serum cortisol or UFC off-medication at 6- to 12-month intervals and if patients develop new adrenal insufficiency symptoms while on stable medical therapy.
Hypopituitarism is a risk with all forms of radiation. Up to two-thirds of patients develop anterior pituitary hormone deficiency after RT (101, 105). All patients should undergo a careful assessment of anterior pituitary function post-therapy annually (at least) or sooner if hormone deficiency symptoms develop. Additional radiation risks include optic neuropathy (1–2%) and other cranial neuropathies (2–4%) as well as a small risk of secondary neoplasia within the radiation field (most commonly meningiomas) (101).
In children, GH deficiency and hypogonadism were common within 1 year of RT; thyroid hormone deficiency was not reported (106). Administering human GH for GH deficiency is indicated if linear growth potential exists. A reassessment of GH secretion at growth completion is advised because only one of six patients in one study showed unequivocally normal GH secretion (88). Changes in cognitive function have not been studied after RT for pediatric or adult CD.
Values
Patients should consider the cost, accessibility, and convenience of radiosurgery vs RT when choosing between the two.
6.4 Medical treatment
6.4 We recommend steroidogenesis inhibitors under the following conditions: as second-line treatment after TSS in patients with CD, either with or without RT/radiosurgery; as primary treatment of EAS in patients with occult or metastatic EAS; and as adjunctive treatment to reduce cortisol levels in ACC. (1|⊕⊕⊕○)
6.4a We suggest pituitary-directed medical treatments in patients with CD who are not surgical candidates or who have persistent disease after TSS. (2|⊕⊕⊕○)
6.4b We suggest administering a glucocorticoid antagonist in patients with diabetes or glucose intolerance who are not surgical candidates or who have persistent disease after TSS. (2|⊕⊕⊕○)
6.4c We suggest targeted therapies to treat ectopic ACTH syndrome. (2|⊕○○○)
Values
The choice of medical therapy should be guided by efficacy, individual patient factors, and cost. The goal is clinical normalization using cortisol levels as a proxy endpoint (except for mifepristone, see below). This can be achieved either with a “block and replace” strategy in which circulating cortisol is reduced to minimally detectable levels and glucocorticoid replacement is added (avoiding supraphysiological doses) (107, 108) or with a “normalization” strategy aimed to achieve eucortisolism (109, 110). If there is evidence of significant cyclicity, block and replace may be preferable, but it carries additional risk if higher doses and multiple medications are needed.
Remarks
Hypoadrenalism may occur when treating with mifepristone or any of the steroidogenesis inhibitors, due to overtreatment, the inability to mount a cortisol response to intercurrent infection, or cyclical or variable hypercortisolism. The gastrointestinal symptoms of hypoadrenalism overlap in many cases with side effects of the drugs. Thus, the possibility of adrenal insufficiency must be addressed (see below) (Table 1).
Medical Treatment of CS
| Drug | Pros | Cons | Dosea |
|---|---|---|---|
| Steroidogenesis inhibitors | |||
| Ketoconazoleb | Quick onset of action | Adverse effects: GI, hepatic dyscrasia (death), male hypogonadism; requires acid for biological activity; DDIs | 400–1600 mg/d; every 6–8 h dosing |
| Metyraponeb | Quick onset of action | Adverse effects: GI, hirsutism, HT, hypokalemia; accessibility variable across countries | 500 mg/d to 6 g/d; every 6–8 h dosing |
| Mitotanec | Adrenolytic, approved for adrenal cancer | Slow onset of action; lipophilic/long half-life, teratogenic; adverse effects: GI, CNS, gynecomastia, low WBC and T4, ↑ LFTs; ↑ CBG, DDIs | Starting dose, 250 mg; 500 mg/d to 8 g/d |
| Etomidate | Intravenous, quick onset of action | Requires monitoring in ICU | Bolus and titrate |
| Pituitary-directed | |||
| Cabergoline | Adverse effects: asthenia, GI, dizziness | 1–7 mg/wk | |
| Pasireotided | Most successful when UFC <2-fold normal; sc administration; adverse effects: diarrhea, nausea, cholelithiasis, hyperglycemia, transient ↑ LFTs; ↑QTc | 600–900 μg twice daily | |
| Glucocorticoid receptor-directed | |||
| Mifepristonee | Difficult to titrate (no biomarker); abortifacient; adverse effects: fatigue, nausea, vomiting, arthralgias, headache, hypertension, hypokalemia, edema, endometrial thickening | 300–1200 mg/d |
| Drug | Pros | Cons | Dosea |
|---|---|---|---|
| Steroidogenesis inhibitors | |||
| Ketoconazoleb | Quick onset of action | Adverse effects: GI, hepatic dyscrasia (death), male hypogonadism; requires acid for biological activity; DDIs | 400–1600 mg/d; every 6–8 h dosing |
| Metyraponeb | Quick onset of action | Adverse effects: GI, hirsutism, HT, hypokalemia; accessibility variable across countries | 500 mg/d to 6 g/d; every 6–8 h dosing |
| Mitotanec | Adrenolytic, approved for adrenal cancer | Slow onset of action; lipophilic/long half-life, teratogenic; adverse effects: GI, CNS, gynecomastia, low WBC and T4, ↑ LFTs; ↑ CBG, DDIs | Starting dose, 250 mg; 500 mg/d to 8 g/d |
| Etomidate | Intravenous, quick onset of action | Requires monitoring in ICU | Bolus and titrate |
| Pituitary-directed | |||
| Cabergoline | Adverse effects: asthenia, GI, dizziness | 1–7 mg/wk | |
| Pasireotided | Most successful when UFC <2-fold normal; sc administration; adverse effects: diarrhea, nausea, cholelithiasis, hyperglycemia, transient ↑ LFTs; ↑QTc | 600–900 μg twice daily | |
| Glucocorticoid receptor-directed | |||
| Mifepristonee | Difficult to titrate (no biomarker); abortifacient; adverse effects: fatigue, nausea, vomiting, arthralgias, headache, hypertension, hypokalemia, edema, endometrial thickening | 300–1200 mg/d |
Abbreviations: GI, gastrointestinal; DDI, drug-drug interactions; HT, hypertension; CNS, central nervous system; WBC, white blood cell count; LFTs, liver function tests; CBG, corticosteroid binding globulin; ICU, intensive care unit; QTc, corrected QT interval.
Except as noted, the lowest dose may be used initially, unless the patient has severe hypercortisolism (UFC more than five times normal), in which case the starting dose may be doubled.
Ketoconazole and metyrapone are approved by the European Medicines Agency for the treatment of CS.
Mitotane has FDA approval for treatment of adrenal cancer.
Pasireotide has FDA approval for treatment of patients with CD who are not surgical candidates or have failed surgery. The agent is also approved in Europe.
Mifepristone has FDA approval for treatment of patients with CS and diabetes or glucose intolerance who are not surgical candidates or have failed surgery.
The use of other agents listed here represents an off-label use for the treatment of CS.
Medical Treatment of CS
| Drug | Pros | Cons | Dosea |
|---|---|---|---|
| Steroidogenesis inhibitors | |||
| Ketoconazoleb | Quick onset of action | Adverse effects: GI, hepatic dyscrasia (death), male hypogonadism; requires acid for biological activity; DDIs | 400–1600 mg/d; every 6–8 h dosing |
| Metyraponeb | Quick onset of action | Adverse effects: GI, hirsutism, HT, hypokalemia; accessibility variable across countries | 500 mg/d to 6 g/d; every 6–8 h dosing |
| Mitotanec | Adrenolytic, approved for adrenal cancer | Slow onset of action; lipophilic/long half-life, teratogenic; adverse effects: GI, CNS, gynecomastia, low WBC and T4, ↑ LFTs; ↑ CBG, DDIs | Starting dose, 250 mg; 500 mg/d to 8 g/d |
| Etomidate | Intravenous, quick onset of action | Requires monitoring in ICU | Bolus and titrate |
| Pituitary-directed | |||
| Cabergoline | Adverse effects: asthenia, GI, dizziness | 1–7 mg/wk | |
| Pasireotided | Most successful when UFC <2-fold normal; sc administration; adverse effects: diarrhea, nausea, cholelithiasis, hyperglycemia, transient ↑ LFTs; ↑QTc | 600–900 μg twice daily | |
| Glucocorticoid receptor-directed | |||
| Mifepristonee | Difficult to titrate (no biomarker); abortifacient; adverse effects: fatigue, nausea, vomiting, arthralgias, headache, hypertension, hypokalemia, edema, endometrial thickening | 300–1200 mg/d |
| Drug | Pros | Cons | Dosea |
|---|---|---|---|
| Steroidogenesis inhibitors | |||
| Ketoconazoleb | Quick onset of action | Adverse effects: GI, hepatic dyscrasia (death), male hypogonadism; requires acid for biological activity; DDIs | 400–1600 mg/d; every 6–8 h dosing |
| Metyraponeb | Quick onset of action | Adverse effects: GI, hirsutism, HT, hypokalemia; accessibility variable across countries | 500 mg/d to 6 g/d; every 6–8 h dosing |
| Mitotanec | Adrenolytic, approved for adrenal cancer | Slow onset of action; lipophilic/long half-life, teratogenic; adverse effects: GI, CNS, gynecomastia, low WBC and T4, ↑ LFTs; ↑ CBG, DDIs | Starting dose, 250 mg; 500 mg/d to 8 g/d |
| Etomidate | Intravenous, quick onset of action | Requires monitoring in ICU | Bolus and titrate |
| Pituitary-directed | |||
| Cabergoline | Adverse effects: asthenia, GI, dizziness | 1–7 mg/wk | |
| Pasireotided | Most successful when UFC <2-fold normal; sc administration; adverse effects: diarrhea, nausea, cholelithiasis, hyperglycemia, transient ↑ LFTs; ↑QTc | 600–900 μg twice daily | |
| Glucocorticoid receptor-directed | |||
| Mifepristonee | Difficult to titrate (no biomarker); abortifacient; adverse effects: fatigue, nausea, vomiting, arthralgias, headache, hypertension, hypokalemia, edema, endometrial thickening | 300–1200 mg/d |
Abbreviations: GI, gastrointestinal; DDI, drug-drug interactions; HT, hypertension; CNS, central nervous system; WBC, white blood cell count; LFTs, liver function tests; CBG, corticosteroid binding globulin; ICU, intensive care unit; QTc, corrected QT interval.
Except as noted, the lowest dose may be used initially, unless the patient has severe hypercortisolism (UFC more than five times normal), in which case the starting dose may be doubled.
Ketoconazole and metyrapone are approved by the European Medicines Agency for the treatment of CS.
Mitotane has FDA approval for treatment of adrenal cancer.
Pasireotide has FDA approval for treatment of patients with CD who are not surgical candidates or have failed surgery. The agent is also approved in Europe.
Mifepristone has FDA approval for treatment of patients with CS and diabetes or glucose intolerance who are not surgical candidates or have failed surgery.
The use of other agents listed here represents an off-label use for the treatment of CS.
Monitoring includes assessing clinical response and the biochemical evaluation (24-h UFC, morning serum cortisol, or serum cortisol day curves [except for mifepristone; see below]) to evaluate for hypercortisolism control. Assessing adrenal insufficiency in patients on medical treatment is mainly done clinically; dose interruption or reduction should be considered when adrenal insufficiency is suspected.
Except as noted, none of the medical treatments discussed have U.S. Food and Drug Administration (FDA) approval for the treatment of hypercortisolism.
Many of these agents stimulate or inhibit CYP3A4, which may lead to significant drug-drug interactions (111). Thus, review of all other medications is important when initiating therapy and when adding other medications. These agents also may increase the QT interval (Table 1).
Evidence
Ketoconazole
Ketoconazole, an imidazole derivative with antifungal activity, impairs adrenal and gonadal steroidogenesis by inhibiting side-chain cleavage, 17,20-lyase, and 11-β hydroxylase enzymes (112).
Taking all case series together, ketoconazole monotherapy (at daily doses of 400–1200 mg) normalized UFC in 57 of 82 patients with presumed CD (25–93% rate in individual studies); normalization was not dependent on the dose or duration of treatment (113–120). Recent data confirm these findings with a greater than 50% drop in UFC in 75% of 200 patients, and with clinical improvements in diabetes, hypertension, and hypokalemia (121). Efficacy in the ectopic ACTH syndrome is lower: of nine patients, only four (44%) achieved eucortisolism (113–115, 122).
Ketoconazole's side-effect profile (Table 1) is relatively benign, except for idiosyncratic severe hepatic dyscrasia, which is estimated to occur in one in 15 000 exposed individuals (115, 122–124). The FDA issued a black box warning for this in 2013, and the European Medicines Agency has restricted access to the agent to physicians specialized in treating CS (125). Among 33 cases of potential ketoconazole-induced liver injury submitted to the FDA from the time of initial marketing in 1980, 18 patients had an 8-fold elevation in transaminases, five had cholestatic injury, and nine had a mixture of the two (126). Thus, monitoring liver function is necessary. Mild asymptomatic elevation in serum transaminases occurs in approximately 10–15% of cases (121, 126), usually when therapy starts or when doses increase, so close monitoring is needed at these times. Values typically return to normal within 2–4 weeks after stopping therapy or reducing doses. If liver enzyme elevations remain less than three times the upper limit of normal, most clinicians will continue therapy. However, when enzyme levels are higher, discontinuation or dose reduction is advised.
Metyrapone
Metyrapone inhibits 11-β hydroxylase, which catalyzes the conversion of 11-deoxycortisol to cortisol. Its 2-hour half-life necessitates three to four doses daily. Phenytoin and phenobarbital accelerate metyrapone metabolism, and estrogens reduce it; metyrapone is excreted in breast milk.
Metyrapone controls hypercortisolemia in 50–75% of patients with CS. In the largest published single center series (91 patients), chronic therapy controlled hypercortisolemia in CD despite a rise in serum ACTH (127). These findings were confirmed recently in 195 patients (128). Although no medication for CS is approved for use during pregnancy, metyrapone has been given occasionally in pregnant women with CS with no apparent adverse effects to mother or offspring (129–132).
Adverse effects of metyrapone are most common when therapy starts or doses increase, and they mainly consist of gastrointestinal disturbances (in the absence of hypoadrenalism). However, this reaction is uncommon when the medication is taken with food or milk. With chronic therapy, hirsutism and acne may worsen due to the accumulation of androgenic precursors secondary to the blockade of cortisol synthesis; the accumulation of mineralocorticoid precursors requires monitoring for hypokalemia, edema, and hypertension.
Remarks
Metyrapone causes a gross elevation of circulating levels of the cortisol precursor 11-deoxycortisol, which can cross-react in many immunoassays for serum and urinary cortisol. This artificially elevates apparent cortisol values, potentially masking biochemical hypoadrenalism. Liquid chromatography-tandem mass spectrometry assays for cortisol circumvent this issue (133). With no cross-reactivity issues, the treatment target is either UFC in the normal range or mean serum cortisol levels that are between 5.4 and 10.8 μg/dL (150–300 nmol/L) throughout the day (134).
Combination therapy
Metyrapone and ketoconazole may be combined to enhance the control of severe hypercortisolemia (92, 135).
Mitotane
Mitotane is primarily used to treat adrenal carcinoma. It inhibits CYP11A1 (P450 side-chain cleavage) and has a direct cytotoxic action on the adrenal cortex (136). Mitotane has a long half-life and sustained effects because of its storage in adipose tissue. Hence, the dose may be increased at weekly intervals, and UFC normalization takes almost 6 months (107). The dose and mitotane plasma levels required to control hypercortisolism in CD were lower than those needed to achieve ACC antineoplastic activity, with medians around 2.7 g of mitotane equivalent per day and 8.5 mg/L, respectively (107).
Mitotane as monotherapy does not cure CD. It is an effective adjunctive therapy in patients with CD as a first- or second-line treatment (after unsuccessful TSS) while awaiting the effects of pituitary RT or when surgery is not possible. In three studies of patients with CD receiving mitotane for these indications, 72–82% had sustained hypercortisolism remission (107, 108, 110).
Because mitotane increases the production of cortisol-binding globulin, plasma total cortisol levels increase proportionally (137). Biochemical monitoring therefore relies on UFC or salivary cortisol measurements. In patients who develop adrenal insufficiency, the usual hydrocortisone replacement dose should be increased because mitotane strongly activates CYP3A4 and increases hydrocortisone clearance (138). A safe approach is to increase the initial hydrocortisone daily dose by one-third. Dexamethasone, a strong CYP3A4 inducer, should be avoided (139). At the doses used for CD, mitotane has less adrenolytic effects on the zona glomerulosa; mineralocorticoid replacement may not be required (110).
Side effects lead to discontinuation of the drug in up to 28% of patients (107). Mitotane is a teratogen; pregnancy should be avoided for years after stopping the drug because measurable plasma levels may persist for months. Measuring plasma levels may help guide this decision. Potential drug interactions are numerous (140) (Table 1).
Glucocorticoid receptor antagonist
In one study, Mifepristone, a glucocorticoid receptor antagonist and antiprogestin, led to an improvement in hypertension and/or diabetes in 40 and 60%, respectively, of 34 patients (141). At least one other clinical parameter (weight, depression, cognition, clinical appearance, or QOL) improved in 87%. Mifepristone is approved in the United States for the control of diabetes or glucose intolerance secondary to hypercortisolism in patients who failed surgery or are not surgical candidates. Mean ACTH levels increased by more than 2-fold in 31 of 43 patients with CD followed for a median of 11.3 months. Three macroadenomas increased in size, whereas one regressed (142).
Cortisol levels remain unchanged or may increase during mifepristone treatment, and therefore practitioners cannot use hormonal measurements to guide efficacy or to diagnose adrenal insufficiency. Because practitioners must use clinical cortisol-dependent variables for these purposes, it is difficult to estimate the correct dose. For this reason, clinicians should start mifepristone at 300 mg/d, titrate it slowly, and base dose adjustment on clinical parameters, primarily glucose, and weight reduction. Adverse events include symptoms of cortisol insufficiency (fatigue, nausea, vomiting, arthralgias, and headache), evidence of increased mineralocorticoid action (hypertension, hypokalemia, edema), and antiprogestin effects (endometrial thickening) (Table 1). One study treated suspected adrenal insufficiency with drug discontinuation or 2–8 mg of dexamethasone daily (141).
Etomidate
Etomidate is the only medical treatment available for severe hypercortisolism in seriously ill patients of any age who are not immediate surgical candidates and who cannot take oral medications. It is also useful in an emergency setting with acute unmanageable symptoms such as respiratory failure or severe psychosis (143, 144) and can be an effective bridge to other medical or surgical therapies (145). Etomidate is an imidazole derivative (like ketoconazole) that is often used for anesthesia induction. Subhypnotic doses rapidly decrease steroidogenesis within 12–24 hours by inhibiting 11β-hydroxylase and cholesterol side-chain cleavage (146). Due to the need for iv infusion, these patients should be managed in an intensive care unit. It may be prudent to administer etomidate preparations containing propylene glycol (which may cause thrombophlebitis and pain on injection) through a central venous line. Measuring cortisol levels every 4–6 hours is required, and clinicians can titrate the infusion rate to achieve a stable serum cortisol level between 10 and 20 μg/dL (280–560 nmol/L) or they can use a block and replace strategy. A loading dose of 3–5 mg is followed by a continuous infusion of 0.03–0.10 mg/kg/h (2.5–3.0 mg/h). Studies have not reported sedation at these doses; however, patients may need a dose reduction if renal failure occurs, due to a resulting increase in free etomidate concentrations.
Medical pituitary-directed treatments
Cabergoline and pasireotide act directly on corticotroph tumors to inhibit ACTH production. They are generally not effective in adrenal causes of CS, and their role in the treatment of ectopic ACTH production remains to be determined.
Cabergoline
Cabergoline is a dopamine agonist with high affinity for the dopamine receptor subtype 2, which is expressed by most corticotroph adenomas (147, 148).
In small studies, 30–40% of patients responded and continued to have normal UFC levels after 2–3 years of cabergoline treatment (149, 150). However, the cortisol-lowering effect did not last in 29% of initial responders. The serum prolactin concentration did not predict long-term UFC response. The doses patients received were up to 7 mg orally per week, with a median dose of 3.5 mg/wk in one study and a mean dose of 2.1 mg/wk in the other, which is higher than typical doses for hyperprolactinemia (149, 150).
Systolic and diastolic blood pressure, fasting glucose, and insulin improved. Tumor volume decreased or remained stable in the small number of reported patients with visible adenomas (149, 150).
Side effects were typical of dopamine agonist use, such as nausea, dizziness, and asthenia (Table 1), which were not reported as adrenal insufficiency (150). In one study, a single patient progressed from mild to moderate tricuspid regurgitation on echocardiogram after 2 years of treatment; in the other, no patients who had echocardiograms showed any significant valvulopathy (149, 150).
Cabergoline has been combined with other medications to treat CD (151, 152). In 12 patients with persistent hypercortisolism after TSS, cabergoline monotherapy (up to 3 mg/wk) normalized UFC in three patients at 6 months. Adding ketoconazole (up to 200 mg twice daily) normalized UFC in six of the nine nonresponders (152). In another study, adding cabergoline (up to 6 mg/wk) to pasireotide normalized UFC in four patients who were not controlled on pasireotide alone (151). We need larger trials to establish the role of combination therapies.
Pasireotide
Pasireotide is a somatostatin receptor (SST) agonist that binds to four of the five SST subtypes with substantially higher affinity for SST1 and SST5 than octreotide or lanreotide (153, 154). Corticotroph tumors have a high expression of SST5, and pasireotide decreased ACTH secretion and cell proliferation in cultured human corticotroph tumors (49, 155).
A phase 3 trial administered pasireotide 600 or 900 μg sc twice daily in 162 CD patients who had failed (or were not candidates for) surgery and had a mean baseline UFC level at least 1.5-fold above normal (156). After 6 months, 20% of the subjects attained a normal UFC. Systolic and diastolic blood pressure, triglycerides, low-density lipoprotein cholesterol, weight, and HRQOL improved. About 90% of patients who were not controlled by month 1 or 2 remained uncontrolled at 6 and/or 12 months; thus, a brief trial may predict the chance of biochemical control.
Mean tumor volume decreased by 44% in 75 patients with a lesion on MRI at the 900-μg dose (156). Most side effects were similar to those of other somatostatin analogs (predominantly gastrointestinal, including biliary sludge and gallstones) except for the important finding of hyperglycemia (73% of patients) (Table 1). Glucose and glycated hemoglobin increased soon after drug initiation in most patients, regardless of whether UFC was controlled; no patient developed diabetic ketoacidosis or hyperosmolar coma (156). Pasireotide was approved in 2012 in the European Union and the United States for the treatment of CD when surgery is not successful or cannot be performed.
Clinicians should correct hypokalemia and hypomagnesemia before initiating pasireotide. And they should administer tests for baseline liver function tests, thyroid function (including free thyroid hormone), IGF-1, fasting glucose/glycated hemoglobin, as well as gallbladder ultrasounds and electrocardiograms for corrected QT interval prolongation or bradycardia. Clinicians should evaluate changes in these parameters on pasireotide (hyperglycemia, prolonged quality corrected QT interval thyroid abnormalities, gallstones, and GH deficiency) based on clinical symptoms and signs. At a minimum, they should monitor these at 3–4 months after initiating treatment and after any dose increase. Because of hyperglycemia, clinicians should monitor postprandial glucose and also recommend that patients take the drug after (not before) meals and follow dietary recommendations for diabetes.
Targeted therapies for ectopic ACTH syndrome
ACTH-secreting tumors may express functional SST2 and Dopamine 2 receptors (157, 158). Drugs targeting these receptors may reduce ACTH secretion and, consequently, control hypercortisolism. Several case reports show that octreotide, a potent SST2 agonist, may control ACTH and cortisol secretion for a short- to midterm period in patients with recurrent or unresectable ectopic ACTH-secreting tumors (159, 160). However, octreotide treatment usually had little or no effect on tumor growth.
Studies have occasionally reported hormonal control with the dopamine-2 agonist receptor cabergoline given alone or in combination with an SST2 agonist (150, 161).
In three reports, the tyrosine kinase inhibitors vandetanib and sorafenib rapidly and fully controlled hypercortisolism caused by ACTH secretion from metastatic medullary thyroid carcinomas (162–164). The dissociation between the decrease in ACTH secretion and the lack of tumor reduction suggests a direct antisecretory effect.
7. Approach for long-term follow-up
7.1 We recommend treating the specific comorbidities associated with CS (eg, cardiovascular risk factors, osteoporosis and psychiatric symptoms) in all patients with CS throughout their lives until resolution. We also recommend testing for recurrence throughout life, except in patients who underwent resection of an adrenal adenoma with a computerized tomography (CT) density of < 10 Hounsfield units. (1|⊕⊕⊕○)
7.2 We recommend educating patients and families about the clinical features of remission. (Ungraded best practice statement)
7.3 In patients with adrenal adenoma, we suggest follow-up tests for the specific comorbidities associated with CS if the adenoma density on CT was < 10 Hounsfield units. (2|⊕⊕○○) For those with higher Hounsfield unit values or pathology consistent with possible carcinoma, we suggest evaluating for malignancy using imaging. (2|⊕○○○)
7.4 We recommend that patients with Carney complex have lifelong follow-up tests for cardiac myxoma and other associated disease (testicular tumors, acromegaly, thyroid lesions). (1|⊕⊕⊕⊕)
Evidence
Monitoring and treating Cushing's features and comorbidities
With CS, biochemical remission or a cure is usually associated with significant clinical improvement, but some comorbidities may not completely normalize. Evaluating and treating the long-term negative effects of chronic hypercortisolism may therefore be important to reduce morbidity, improve QOL, and reduce the long-term excess mortality associated with CS.
After surgical or medical remission of CS, a significant proportion of patients will either develop or have a recurrence of autoimmune or inflammatory diseases, such as hypothyroidism, psoriasis, celiac disease, ulcerative colitis, Crohn's disease, asthma, lupus, and rheumatoid arthritis after surgical or medical remission (165). There is no evidence that surgical or medical remission of CS increases the risk of the development recurrence of autoimmune or inflammatory diseases. It is likely that previous hypercortisolism suppressed the conditions.
Patients with Carney complex and primary pigmented nodular adrenocortical disease should be followed annually for the development of new or recurrent cardiac myxomas and other features of the syndrome (166).
Clinicians should tell patients that they may feel unwell for 6–9 months, that their mood will gradually improve, and that improvement may continue for more than 1 year. Generally, weight, bruising, and the physical appearance of the face change first. Patients should receive recommendations for physical therapy and nutrition to optimize the recovery of muscle strength and the normalization of weight. The recovery of comorbidities is variable, and clinicians should continue treating these conditions, tapering off or discontinuing treatment as goals are met (see below).
Metabolic syndrome and cardiovascular risk
Metabolic syndrome, including central obesity, arterial hypertension, insulin resistance, impaired glucose tolerance, and dyslipidemia, is present in at least two-thirds of patients with CS (167), and it is believed to contribute to increased cardiovascular morbidity.
After successful treatment, decreased body weight and fat mass, improved blood pressure, a reduction in antihypertensive treatment, or a reversal of hypertension usually occurs within weeks to a year. However, excess weight and hypertension may persist in up to 25% of patients (8, 14, 15, 168–176). Patients with a history of CD have increased blood pressure levels compared to age-, sex-, and body mass index (BMI)-matched patients (177). Persistent hypertension has been attributed to microvessel remodeling, abdominal obesity, and insulin resistance (15, 177). A longer duration of hypercortisolism is associated with persistent hypertension (15).
Similarly, glucose and lipid metabolism improve, and medications may be reduced and/or discontinued in many patients, but the prevalence of diabetes and dyslipidemia remains increased compared to BMI-matched controls several years after remission (8, 168, 171, 175, 177). Fasting blood glucose may underdiagnose diabetes mellitus compared to an oral glucose tolerance test (178). Clinicians should carefully monitor adult patients treated with GH replacement for diabetes mellitus (179). The persistence of central obesity plays a key role in continued insulin resistance (14, 171, 175, 177).
There is increased long-term risk of myocardial infarction (7, 8). Because hypertension and diabetes appear to be the main controllable determinants of cardiovascular events and mortality (8, 11, 174, 180), rigorous and repeated long-term follow-up evaluation and treatment of these conditions are mandatory. One study found that depression was associated with an increased risk of cardiovascular disease (180).
Mood and cognitive function
Psychiatric referral may benefit adult patients with CS. Many patients experience improvement in psychiatric symptoms, particularly depression and emotional lability. However, several years after CD is cured, patients have an increased prevalence of psychopathology, including anxiety, depression symptoms, and maladaptive personality compared to patients treated for other types of pituitary adenoma (173, 181–184). Although a few short-term studies indicate a significant improvement in psychopathology within the first year after successful surgical or medical remission (82, 182), little is known regarding long-term reversibility.
Cognitive impairment, particularly declarative short-term memory deficits, is a prominent feature of hypercortisolism. Deficits also occur in attention, visuospatial abilities, and working memory (185). These are likely related to the abnormal function of key central nervous system structures such as the hippocampus, amygdala, and frontal cortex that are peculiarly vulnerable to glucocorticoid excess. Several imaging studies identified atrophy in these specific brain areas including decreased hippocampal volume during active CS; this improves after remission in some but not all patients. Some, but not all, studies found a parallel improvement in cognitive function (186–190).
Pediatric patients show personality changes such as moodiness, irritability, compulsive behavior, and overachievement in school (191).
There are scarce data regarding the long-term recovery of cognitive impairments. Compared to matched controls, patients in long-term remission from CD or adrenal adenomas performed significantly worse in tests of executive function (185), memory (185, 190), and other cognitive tests (192). When evaluated, hypopituitarism and hydrocortisone dependency were associated with worse performance, but cognitive impairment was independent of coexistent chronic fatigue or affective disorders (192). The patients with memory scores below normative cutoff values showed decreased hippocampal volume on MRI.
Quality of life
Patients with active CS of any cause have impaired HRQOL compared to patients with other pituitary tumors (181, 193, 194). Because CS is associated with long-term physical, psychological, emotional, and cognitive after-effects despite cure, HRQOL is likely to be affected in the long term and is an important outcome measure to assess in patients with a previous history of CS.
There are few published cross-sectional studies of QOL during long-term remission. Most used generic QOL questionnaires. QOL improves in patients in remission compared to those with hypercortisolism, regardless of the cause of CS or treatment used (13, 194–196). Nevertheless, two large cross-sectional studies report long-term residual impairment in physical and social functioning as well as limitations in the ability to perform usual activities of daily living due to physical and emotional problems, pain, fatigue, sleep problems, reduced vitality, and feeling of health impairment (13, 197). Together with the residual psychological and cognitive impairments, chronic decreased perceived QOL has significant social and economic consequences, such as the inability to return to work (198).
The duration of remission did not affect QOL results in one study (197) but was associated with lower physical (but not psychological) component scores in another (13). Postoperative hypopituitarism may decrease QOL (197). However, it is unlikely to play a large role because GH-deficient females in remission from CD have a greater impairment in QOL than those with other causes of GH deficiency (199).
Osteoporosis
Osteoporosis results from direct effects of cortisol on bone cells and indirect events such as glucocorticoid-induced hypogonadism, secondary hyperparathyroidism, GH deficiency, and reduced bone strain due to myopathy (200). However, the prevalence of fractures is not known (201). Studies of adolescents and adults document major improvement and sometimes complete normalization of BMD after curing CS, although 4–5 years may be needed to achieve full recovery (202–205). Importantly, supraphysiological postoperative hydrocortisone doses may hamper bone recovery. The scant fracture data suggest that the increased symptomatic fracture risk disappears after curing CS, similar to that seen in exogenous glucocorticoid-induced osteoporosis (204, 206). It is not clear whether complete normalization occurs because the expected BMD for a given person cannot be predicted (8, 173, 200). We recommend a detailed fracture assessment, evaluating BMD at the spine and hip, obtaining lateral morphometric imaging of the spine, adequate calcium and vitamin D intake, avoidance of excessive glucocorticoid supplementation, and personalized long-term follow-up based on the results of bone evaluation. A recent consensus paper on evaluation and pharmacological intervention for (exogenous) glucocorticoid-induced osteoporosis provides useful guidance (207).
8. Special populations/considerations
8.1 We recommend urgent treatment (within 24–72 h) of hypercortisolism if life-threatening complications of CS such as infection, pulmonary thromboembolism, cardiovascular complications, and acute psychosis are present. (1|⊕⊕⊕○). The associated disorder(s) should be addressed as well (eg, anticoagulation, antibiotics).
Severe hypercortisolism may be a life-threatening condition that mandates immediate treatment, regardless of whether all diagnostic tests are completed. Most of these patients have ectopic ACTH syndrome associated with serious comorbidities including infections, pulmonary thromboembolism, and cardiovascular problems. However, sometimes relatively modest hypercortisolism due to CD can cause acute psychosis with a high suicide risk, so that prompt resolution of excess cortisol is imperative.
In addition to pulmonary thromboembolism and infections, patients may experience acute heart and respiratory failure, peritonitis due to gut perforation, pancreatitis, and untreatable psychosis (173, 208). Typical signs of bowel perforation, such as rebound, guarding, loss of bowel sounds, fever, and elevated white blood cell count may be lacking due to hypercortisolism.
Many experienced clinicians suggest specific treatments for each condition (eg, anticoagulation prophylaxis and prophylaxis for Pneumocystis jiroveci with trimethoprim-sulfamethoxazole [or dapsone in patients with sulfa allergies]), especially for bedridden or low-mobility patients or those with UFC > 5-fold normal. Immediate efforts to reduce cortisol may be lifesaving. Some patients who are seriously ill with comorbidities and have very high UFC levels need intensive care unit management with a multidisciplinary approach that includes experienced endocrinologists.
Bilateral adrenalectomy provides immediate hypercortisolism control and can be lifesaving. However, few data about bilateral adrenalectomy as a lifesaving therapy in critically ill patients are available, and a number of these studies treated patients with cortisol-lowering drugs before surgery. The risk of surgery must be balanced against the likelihood of medical control. If aggressive medical management does not control hypercortisolism, clinicians should consider bilateral adrenalectomy even in high-surgical-risk patients.
Steroidogenesis inhibitors such as ketoconazole and metyrapone can rapidly lower cortisol levels but often must be used together in severe hypercortisolism (209). In 11 critically ill patients, a high-dose regimen combining mitotane (3.0–5.0 g/d), metyrapone (3.0–4.5 g/d), and ketoconazole (400–1200 mg/d) decreased UFC to near normal levels within 24–48 hours with dramatic improvement in clinical condition and acceptable side effects (210). In more than 50% of patients, UFC remained low to normal, and metyrapone and ketoconazole could be discontinued after several months, whereas UFC remained controlled by mitotane monotherapy. In another series of 22 patients with severe CS due to ectopic ACTH syndrome and ACC, metyrapone and ketoconazole combination therapy was able to control hypercortisolism and dramatically improve clinical status within 1 month in 78% of patients (92).
The rapid onset of action of mifepristone is compatible with its use in life-threatening hypercortisolism (211), but data are lacking. Of note, it can ameliorate acute steroid psychosis within days (212).
As discussed above, etomidate may be helpful in patients with life-threatening hypercortisolemia who cannot take oral medications.
Future Directions and Recommended Research
We recommend the following research aims:
- 1.
Identify biological markers and tissue factors that explain the variable clinical effect of steroids among individuals and establish ideal tests so clinicians can:
- a.
Quantify the magnitude of glucocorticoid exposure to characterize the severity of illness and guide decision-making about when to initiate treatment.
- b.
Determine whether treatment has resulted in remission.
- c.
Accurately monitor patients for their response to medical therapy to guide dose optimization.
- a.
- 2.
Evaluate the clinical effects and benefits/risks of treating “subclinical” or mild CS.
- 3.
Evaluate new medical therapies and combinations of medical treatments with different mechanisms of action.
- 4.
Evaluate the utility of venous thromboembolism prophylaxis before and after remission.
- 5.
Compare RT and radiosurgery in terms of time to remission and risk of hypopituitarism and other side effects.
- 6.
Evaluate the effects of long-term hypopituitarism and other possible consequences of pediatric radiation.
- 7.
Assess long-term QOL and cognitive changes experienced in CS by adults and children and determine optimal treatment strategies.
- 8.
Ascertain the best follow-up strategy for early diagnosis of recurrence of CD after surgery.
- 9.
Conduct future studies that address the recurrence and remission rates of CD using the same biochemical tests that are required for initial diagnosis.
Financial Disclosures of the Task Force*
Lynnette K. Nieman, MD (chair)—Financial or Business/Organizational Interests: UpToDate; Significant Financial Interest or Leadership Position: HRA Pharma (Research Grant to Institution), UpToDate (Honoraria). Beverly M. K. Biller, MD—Financial or Business/Organizational Interests: American Association of Clinical Endocrinologist, Growth Hormone Research Society, American College of Physicians; Significant Financial Interest or Leadership Position: HRA Pharma (Consultant), Cortendo (Consultant, Research Grant to Institution), Novartis (Consultant, Research Grant to Institution), Novo Nordisk (Consultant, Research Grant to Institution), Pfizer (Consultant). James W. Findling, MD—Financial or Business/Organizational Interests: none declared; Significant Financial Interest or Leadership Position: Novartis (Consultant, Research Grant to Institution), Corcept (Consultant, Research Grant to Institution). Hassan M. Murad**, MD, MPH—Financial or Business/Organizational Interests: Mayo Clinic, Division of Preventive Medicine; Significant Financial Interest or Leadership Position: none declared. John Newell-Price, MD, FRCP, PhD—Financial or Business/Organizational Interests: Royal College of Physicians, Society of Endocrinology, The Pituitary Foundation; Significant Financial Interest or Leadership Position: Novartis (Consultant, Study Steering Committee), HRA Pharma (Consultant, Research Grants), Clinical Endocrinology (Senior Editor). Martin O. Savage, MD, FRCPCH—Financial or Business/Organizational Interests: none declared; Significant Financial Interest or Leadership Position: IPSEN (Consultant), Merke Serono (Consultant), Sandoz (Consultant), OPKO Health, Inc. (Consultant). Antoine Tabarin, MD—Financial or Business/Organizational Interests: Novartis, HRA Pharma; Significant Financial Interest or Leadership Position: Novartis (Board, Speaker, Honoraria, Research Grant to Institution), HRA Pharma (Speaker, Honoraria, Research Grant to Institution), IPSEN Biotech (Speaker, Honoraria).
* Financial, business, and organizational disclosures of the task force cover the year prior to publication. Disclosures prior to this time period are archived.
**Evidence-based reviews for this guideline were prepared under contract with the Endocrine Society.
Acknowledgments
Cosponsoring Association: European Society of Endocrinology.
The Intramural Research Program of the Eunice Kennedy Shriver Institute of Child Health and Human Development provided salary support to Dr Nieman for her work on this manuscript.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- ACC
adrenocortical carcinoma
- BMAH
bilateral macronodular adrenal hyperplasia
- BMD
bone mineral density
- CD
Cushing's disease
- CS
Cushing's syndrome
- CT
computerized tomography
- EAS
ectopic ACTH secretion
- HPA
hypothalamic-pituitary-adrenal
- HRQOL
health-related QOL
- MRI
magnetic resonance imaging
- QOL
quality of life
- RT
radiation therapy
- SST
somatostatin receptor
- TSS
transsphenoidal selective adenomectomy
- UFC
urine free cortisol.

{kind=link}