





The 22q11.2 deletion syndrome (DS) is a common multiple anomaly syndrome in which typical features include congenital heart defects, facial dysmorphism, and palatal anomalies. Hypocalcemia due to hypoparathyroidism is a common endocrine manifestation resulting from variable parathyroid hypoplasia, but hypercalcemia has not previously been reported in 22q11.2 DS.
Our patient is a 16-year-old adolescent male with dysmorphic facial features and delayed motor and speech development. At 2 years of age, 22q11.2 DS was confirmed by fluorescence in situ hybridization. In contrast to hypoparathyroidism that is usually seen in 22q11.2 DS, this patient had early childhood-onset hypercalcemia with inappropriately high PTH levels and hypocalciuria. Genomic DNA was obtained from the proband and screened for calcium-sensing receptor (CASR) mutations with negative results. No parathyroid tissue could be localized by imaging or surgical exploration. As a result of symptomatic hypercalcemia, the patient was treated with a calcimimetic (cinacalcet). During the treatment, plasma calcium normalized with mild symptoms of hypocalcemia. After discontinuation of cinacalcet, calcium returned to high pretreatment levels. Further DNA analysis of adaptor protein-2 σ subunit (AP2S1) showed a heterozygous missense mutation c.44 G>T, resulting in a p.R15L substitution; the mutation was absent in the healthy parents and two siblings.
Hypercalcemia in our patient with 22q11.2 DS could be explained by the de novo mutation in AP2S1. Identification of a genetic cause for hypercalcemia is helpful in guiding management and avoiding unnecessary treatment.
The 22q11.2 deletion syndrome (22q11.2 DS), also known as velocardiofacial syndrome or DiGeorge syndrome, is a common multiple anomaly syndrome caused by a microdeletion on chromosome 22 at q11.2. This syndrome is inherited as an autosomal dominant trait with 100% penetrance and a highly variable phenotype. Most cases of 22q11.2 DS are sporadic. In about 70% of patients, congenital heart disease is present with a high percentage of conotruncal anomalies. Palatal anomalies and facial dysmorphism are general features. Several other structural anomalies have been associated with 22q11.2 DS. Developmental, behavioral, and psychiatric disorders emerge frequently (1). The most common endocrine manifestation of 22q11.2 DS is hypoparathyroidism, which may range from severe neonatal hypocalcemia to latent hypoparathyroidism (2). Parathyroid dysfunction has been reported in approximately 50% of patients with 22q11.2 DS (2, 3). We describe an adolescent male with 22q11.2 DS and hypercalcemia from infancy. Fifteen years later, the etiology of hypercalcemia was resolved.
Subjects and Methods
Subjects
The index case, his parents, and two siblings were included in the study. All subjects underwent plasma calcium determinations and genetic evaluation. Written informed consent was obtained from all participants.
Laboratory methods and genetic testing
Plasma calcium, phosphate, alkaline phosphatase, and creatinine were measured by standard methods, and plasma fasting PTH was measured by an immunometric assay (Immulite 2000; Diagnostic Products Corporation). Serum 25-hydroxyvitamin D (25OHD) was assessed by liquid chromatography and 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] by RIA. Genomic DNA was isolated from the peripheral blood and screened for calcium-sensing receptor (CASR) mutations of the affected adolescent and adaptor protein-2 σ subunit (AP2S1) gene mutations of the affected adolescent, his siblings and parents. Exon 2 of the AP2S1 gene was PCR amplified and Sanger sequenced.
Case Description
This 16-year-old adolescent was born to healthy parents after an uncomplicated pregnancy with appropriate birth measures (weight 3890 g, +1.4 SD score; length 50 cm, +0.2 SD score). He had delayed motor and speech development. At the age of 1 year, he was admitted to the hospital because of pneumonia. His dysmorphic features (small, dysmorphic, and low-set ears, large mouth with a thin upper lip, small, short nose, mild micrognathia, excessive postaxial finger rudiments, umbilical and inguinal hernias, and growth retardation [length, −2.6 SD scores]) prompted further evaluation. On ultrasound examinations, no cardiac malformations were observed, but the left kidney was not visualized, and kidney aplasia was verified by a dimercaptosuccinic acid renal scan. The anatomy of the right kidney was normal. The plasma creatinine level had been within the normal range, confirming normal kidney function. Biochemical studies showed constantly elevated plasma calcium concentrations, whereas the PTH levels were in the normal range (Table 1). At 2 years of age, a fluorescence in situ hybridization test for a 22q11.2 deletion verified the diagnosis of 22q11.2 DS.
Biochemical Parameters and BMD Measurements Before, During, and After Cinacalcet Treatment
| P-Ca, mmol/L | P-Ca ion, mmol/L | P-Pi, mmol/L | P-PTH, ng/L | P-ALP, U/L | 25OHD, nmol/L | 1,25(OH)2D3, pmol/L | P-Cr, μmol/L | U-Ca/Cr | BMD Z-Score | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS | FH | FN | WB | ||||||||||
| Reference range | 2.05–2.70 | 1.16–1.30 | 1.10–1.80 | 8–73 | 115–460 | >40 | 63–228 | 10–76 | <0.6 | ||||
| Before treatment | |||||||||||||
| 1 y, 7 mo | 2.99 | 1.39 | 22 | 386 | 44 | ||||||||
| 6 y, 3 mo | 3.02 | 1.43 | 0.72 | 51 | 361 | 32 | 96 | 28 | 0.04 | ||||
| 8 y, 2 mo | 2.81 | 1.46 | 1.10 | 72 | 182 | 20 | 110 | 30 | <0.17 | ||||
| 9 y, 6 mo | 2.79 | 1.51 | 1.04 | 70 | 186 | 44 | 116 | 32 | <0.13 | −3.0 | −0.3 | −1.0 | −0.3 |
| Treatment with cinacalcet (starting at 10 y) | |||||||||||||
| 10 y, 3 mo | 2.66 | 1.40 | 1.14 | 43 | 38 | 0.07 | |||||||
| 10 y, 6 mo | 2.33 | 1.23 | 1.39 | 34 | 0.09 | ||||||||
| 10 y, 10 mo | 2.17 | 1.17 | 1.32 | 16 | 189 | 41 | 105 | <0.15 | −2.3 | −0.5 | −1.0 | −1.3 | |
| 12 y, 4 mo | 1.22 | 1.16 | 69 | 266 | 58 | 45 | 0.13 | −2.5 | −1.0 | −1.3 | −2.0 | ||
| 13 y, 5 mo | 1.38 | 1.10 | 64 | 290 | 53 | 58 | <0.09 | −2.0 | −1.4 | −1.7 | −1.8 | ||
| After treatment | |||||||||||||
| 13 y, 10 mo | 1.45 | 0.99 | 75 | 262 | 198 | 0.02 | |||||||
| 14 y, 4 mo | 1.42 | 0.83 | 56 | 183 | 43 | 221 | <0.07 | ||||||
| 15 y, 4 mo | 2.87 | 1.47 | 1.06 | 187 | 46 | 207 | 0.06 | ||||||
| P-Ca, mmol/L | P-Ca ion, mmol/L | P-Pi, mmol/L | P-PTH, ng/L | P-ALP, U/L | 25OHD, nmol/L | 1,25(OH)2D3, pmol/L | P-Cr, μmol/L | U-Ca/Cr | BMD Z-Score | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS | FH | FN | WB | ||||||||||
| Reference range | 2.05–2.70 | 1.16–1.30 | 1.10–1.80 | 8–73 | 115–460 | >40 | 63–228 | 10–76 | <0.6 | ||||
| Before treatment | |||||||||||||
| 1 y, 7 mo | 2.99 | 1.39 | 22 | 386 | 44 | ||||||||
| 6 y, 3 mo | 3.02 | 1.43 | 0.72 | 51 | 361 | 32 | 96 | 28 | 0.04 | ||||
| 8 y, 2 mo | 2.81 | 1.46 | 1.10 | 72 | 182 | 20 | 110 | 30 | <0.17 | ||||
| 9 y, 6 mo | 2.79 | 1.51 | 1.04 | 70 | 186 | 44 | 116 | 32 | <0.13 | −3.0 | −0.3 | −1.0 | −0.3 |
| Treatment with cinacalcet (starting at 10 y) | |||||||||||||
| 10 y, 3 mo | 2.66 | 1.40 | 1.14 | 43 | 38 | 0.07 | |||||||
| 10 y, 6 mo | 2.33 | 1.23 | 1.39 | 34 | 0.09 | ||||||||
| 10 y, 10 mo | 2.17 | 1.17 | 1.32 | 16 | 189 | 41 | 105 | <0.15 | −2.3 | −0.5 | −1.0 | −1.3 | |
| 12 y, 4 mo | 1.22 | 1.16 | 69 | 266 | 58 | 45 | 0.13 | −2.5 | −1.0 | −1.3 | −2.0 | ||
| 13 y, 5 mo | 1.38 | 1.10 | 64 | 290 | 53 | 58 | <0.09 | −2.0 | −1.4 | −1.7 | −1.8 | ||
| After treatment | |||||||||||||
| 13 y, 10 mo | 1.45 | 0.99 | 75 | 262 | 198 | 0.02 | |||||||
| 14 y, 4 mo | 1.42 | 0.83 | 56 | 183 | 43 | 221 | <0.07 | ||||||
| 15 y, 4 mo | 2.87 | 1.47 | 1.06 | 187 | 46 | 207 | 0.06 | ||||||
Abbreviations: FH, femoral head; FN, femoral neck; LS, lumbar spine; 25OHD, serum concentration of 25OHD; 1,25(OH)2D3, serum concentration of 1,25(OH)2D3; P-ALP, plasma alkaline phosphatase; P-Ca, plasma calcium; P-Cr, plasma creatinine; P-Pi, plasma inorganic phosphate; U-Ca/Cr, urine calcium/creatinine ratio; WB, whole body.
Biochemical Parameters and BMD Measurements Before, During, and After Cinacalcet Treatment
| P-Ca, mmol/L | P-Ca ion, mmol/L | P-Pi, mmol/L | P-PTH, ng/L | P-ALP, U/L | 25OHD, nmol/L | 1,25(OH)2D3, pmol/L | P-Cr, μmol/L | U-Ca/Cr | BMD Z-Score | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS | FH | FN | WB | ||||||||||
| Reference range | 2.05–2.70 | 1.16–1.30 | 1.10–1.80 | 8–73 | 115–460 | >40 | 63–228 | 10–76 | <0.6 | ||||
| Before treatment | |||||||||||||
| 1 y, 7 mo | 2.99 | 1.39 | 22 | 386 | 44 | ||||||||
| 6 y, 3 mo | 3.02 | 1.43 | 0.72 | 51 | 361 | 32 | 96 | 28 | 0.04 | ||||
| 8 y, 2 mo | 2.81 | 1.46 | 1.10 | 72 | 182 | 20 | 110 | 30 | <0.17 | ||||
| 9 y, 6 mo | 2.79 | 1.51 | 1.04 | 70 | 186 | 44 | 116 | 32 | <0.13 | −3.0 | −0.3 | −1.0 | −0.3 |
| Treatment with cinacalcet (starting at 10 y) | |||||||||||||
| 10 y, 3 mo | 2.66 | 1.40 | 1.14 | 43 | 38 | 0.07 | |||||||
| 10 y, 6 mo | 2.33 | 1.23 | 1.39 | 34 | 0.09 | ||||||||
| 10 y, 10 mo | 2.17 | 1.17 | 1.32 | 16 | 189 | 41 | 105 | <0.15 | −2.3 | −0.5 | −1.0 | −1.3 | |
| 12 y, 4 mo | 1.22 | 1.16 | 69 | 266 | 58 | 45 | 0.13 | −2.5 | −1.0 | −1.3 | −2.0 | ||
| 13 y, 5 mo | 1.38 | 1.10 | 64 | 290 | 53 | 58 | <0.09 | −2.0 | −1.4 | −1.7 | −1.8 | ||
| After treatment | |||||||||||||
| 13 y, 10 mo | 1.45 | 0.99 | 75 | 262 | 198 | 0.02 | |||||||
| 14 y, 4 mo | 1.42 | 0.83 | 56 | 183 | 43 | 221 | <0.07 | ||||||
| 15 y, 4 mo | 2.87 | 1.47 | 1.06 | 187 | 46 | 207 | 0.06 | ||||||
| P-Ca, mmol/L | P-Ca ion, mmol/L | P-Pi, mmol/L | P-PTH, ng/L | P-ALP, U/L | 25OHD, nmol/L | 1,25(OH)2D3, pmol/L | P-Cr, μmol/L | U-Ca/Cr | BMD Z-Score | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS | FH | FN | WB | ||||||||||
| Reference range | 2.05–2.70 | 1.16–1.30 | 1.10–1.80 | 8–73 | 115–460 | >40 | 63–228 | 10–76 | <0.6 | ||||
| Before treatment | |||||||||||||
| 1 y, 7 mo | 2.99 | 1.39 | 22 | 386 | 44 | ||||||||
| 6 y, 3 mo | 3.02 | 1.43 | 0.72 | 51 | 361 | 32 | 96 | 28 | 0.04 | ||||
| 8 y, 2 mo | 2.81 | 1.46 | 1.10 | 72 | 182 | 20 | 110 | 30 | <0.17 | ||||
| 9 y, 6 mo | 2.79 | 1.51 | 1.04 | 70 | 186 | 44 | 116 | 32 | <0.13 | −3.0 | −0.3 | −1.0 | −0.3 |
| Treatment with cinacalcet (starting at 10 y) | |||||||||||||
| 10 y, 3 mo | 2.66 | 1.40 | 1.14 | 43 | 38 | 0.07 | |||||||
| 10 y, 6 mo | 2.33 | 1.23 | 1.39 | 34 | 0.09 | ||||||||
| 10 y, 10 mo | 2.17 | 1.17 | 1.32 | 16 | 189 | 41 | 105 | <0.15 | −2.3 | −0.5 | −1.0 | −1.3 | |
| 12 y, 4 mo | 1.22 | 1.16 | 69 | 266 | 58 | 45 | 0.13 | −2.5 | −1.0 | −1.3 | −2.0 | ||
| 13 y, 5 mo | 1.38 | 1.10 | 64 | 290 | 53 | 58 | <0.09 | −2.0 | −1.4 | −1.7 | −1.8 | ||
| After treatment | |||||||||||||
| 13 y, 10 mo | 1.45 | 0.99 | 75 | 262 | 198 | 0.02 | |||||||
| 14 y, 4 mo | 1.42 | 0.83 | 56 | 183 | 43 | 221 | <0.07 | ||||||
| 15 y, 4 mo | 2.87 | 1.47 | 1.06 | 187 | 46 | 207 | 0.06 | ||||||
Abbreviations: FH, femoral head; FN, femoral neck; LS, lumbar spine; 25OHD, serum concentration of 25OHD; 1,25(OH)2D3, serum concentration of 1,25(OH)2D3; P-ALP, plasma alkaline phosphatase; P-Ca, plasma calcium; P-Cr, plasma creatinine; P-Pi, plasma inorganic phosphate; U-Ca/Cr, urine calcium/creatinine ratio; WB, whole body.
During follow-up, plasma PTH concentrations remained inappropriately high (for hypercalcemia) but within the reference range, and the urinary calcium excretion (calcium/creatinine ratio) was low in random samples (Table 1). In addition, the calcium/creatinine clearance ratios were low (<0.01). The patient had mild hypophosphatemia and low levels of 25OHD, whereas the 1,25(OH)2D3 concentrations were normal (Table 1). His parents and siblings were asymptomatic and normocalcemic. CASR mutation analysis for familial hypocalciuric hypercalcemia (FHH) type 1 showed no pathological changes. A parathyroid adenoma was excluded by a 99mTc-sestamibi scan and 99mTc-sestamibi/123I subtraction scanning followed by single-photon emission computed tomography/computed tomography of the neck and thorax. Furthermore, surgical exploration revealed no parathyroid tissue in the neck or thoracic region.
The patient's symptomatic hypercalcemia persisted, and his dual-energy X-ray absorptiometry–derived areal bone mineral density (BMD) at the lumbar spine showed low values, suggesting impaired bone mineralization (Table 1). Vitamin D supplementation (10 μg/d) was commenced at age 9 years based on low 25OHD levels (Table 1) to avoid the potential negative skeletal effects of vitamin D deficiency; this had no effect on serum calcium levels. At 10 years of age, cinacalcet (Mimpara), a calcimimetic agent, was started at a dose of 30 mg once daily and increased up to 30 mg twice daily to control hypercalcemia and inappropriately high PTH levels. Plasma calcium levels normalized, and PTH levels decreased slightly (Table 1). The patient reported occasional numbness and tingling in his legs. Since the benefits of cinacalcet remained uncertain and some potential adverse effects were observed, the drug was discontinued after 3 years of treatment. The biochemical parameters returned to the levels seen before cinacalcet treatment.
Consequently, because the clinical and biochemical findings in our patient appeared consistent with defective calcium sensing, we screened for mutations of AP2S1, the recently discovered gene underlying FHH3 (4). The patient was found to have a heterozygous missense mutation (c.44G>T; p.R15L) in the AP2S1 gene, suggesting the diagnosis of FHH3. Restriction enzyme analysis confirmed the mutation in the patient, whereas the parents and two siblings were negative for the mutation (Figure 1).
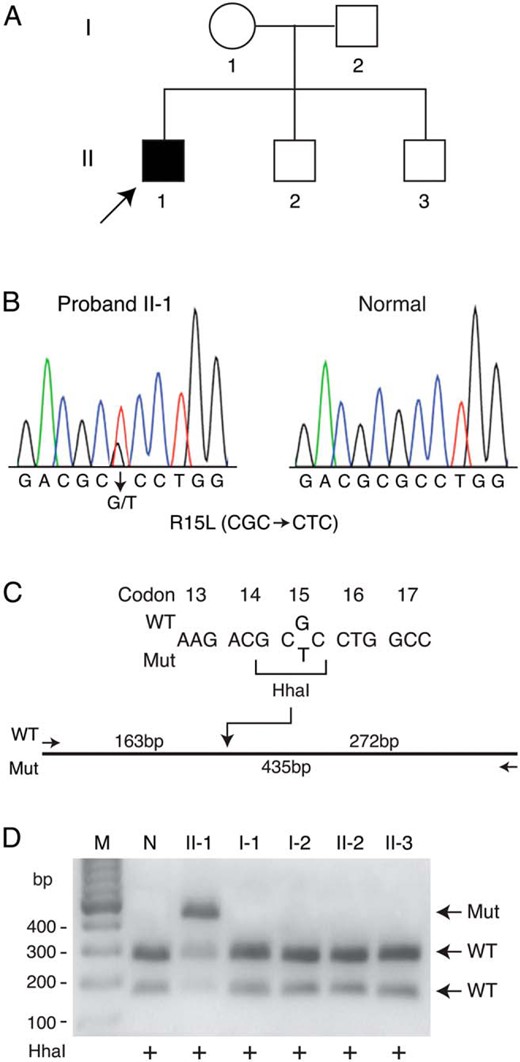
Detection of a mutation in the AP2S1 gene.
A, Pedigree of the family. Clinical status is indicated by the open symbols (unaffected) and solid symbol (affected). The proband is indicated by the arrow. B, Direct sequence analysis of the exon 2 genomic DNA amplicon of proband II-1 (left) revealed a heterozygous missense mutation not seen in an unrelated normal individual (right). A similar analysis of the DNA of family members, I-1, I-2, II-2, and II-3, revealed a normal sequence (data not shown). C, Wild-type (WT) and mutant (Mut) sequences of part of exon 2. The restriction enzyme HhaI recognition site destroyed by the mutation (CGC->CTC, codon 15) is bracketed. The HhaI site in the wild-type sequence is shown on the restriction map of the exon 2 amplicon. D, Gel electrophoretic separation of HhaI restriction digests of the exon 2 PCR product in an unrelated normal individual (N) and all family members (pedigree numbers as in panel A) confirmed the presence of the longer mutated restriction fragment (Mut) in proband II-1 and the absence in all other family members. Lane M, DNA markers with sizes to the left of the gel.
Discussion
FHH is an autosomal dominant disorder characterized by hypercalcemia, inappropriately high serum PTH (for hypercalcemia), and low urinary calcium excretion. About two-thirds of FHH cases are caused by heterozygous inactivating mutations in the CASR gene on chromosome 3q21.1, leading to FHH1 (5). The CASR is a guanine nucleotide-binding protein (G protein)–coupled receptor that signals through the G protein subunits α11 (Gα11) and αq (Gαq) (6). Heterozygous mutations in the GNA11 gene on chromosome 19p13.3 may lead to loss of function in Gα11 and underlie FHH2 (6). FHH1 and FHH2 are usually associated with normal circulating PTH concentrations and mild hypermagnesemia and are generally asymptomatic disorders (4).
The history of FHH3 started in the early 1990s when McMurtry et al (7) reported a large kindred with familial benign (hypocalciuric) hypercalcemia. In late 1990s, linkage studies with the same kindred mapped the FHH3 locus to chromosome 19q13 (8). With a second kindred, Nesbit et al (5) narrowed the gene localization on chromosome 19q13.3. Finally, the same group showed that missense mutations in the gene encoding AP2S1 cause FHH3 (4). Adaptor protein-2 is crucial for clathrin-mediated endocytosis and the recycling of various cell surface proteins, including G protein–coupled receptors, such as CASR (9). Thus, AP2S1 regulates CASR signaling. The prevalence of AP2S1 mutations has varied from 13% to 20% among the patients with FHH lacking CASR mutations (4, 10). Interestingly, all described AP2S1 mutations affect Arg15 and comprise Arg15Cys, Arg15His, and Arg15Leu mutations; the latter of these was found in our patient. The Arg15 residue is evolutionarily conserved and forms key contacts with dileucine-based motifs of clathrin-coated vesicles (4). The mutations described lead to decreased sensitivity of CASR-expressing cells to extracellular calcium and reduced CASR endocytosis (4).
Cinacalcet is a type II calcimimetic that binds to CASR, increasing its sensitivity to extracellular calcium and thus reducing PTH secretion by the parathyroid glands. By acting through CASR in renal tubules, cinacalcet also directly increases the urinary excretion of calcium. Cinacalcet (30 mg twice daily) has been reported to normalize serum calcium and PTH levels in a 6-year-old child with FHH1 without any adverse effects during the treatment (11). Recently, Howles et al (12) reported that cinacalcet reduced serum calcium concentrations by >20% and normalized PTH levels in 3 patients with FHH3 who had AP2S1 mutations. Similarly, the cinacalcet therapy proved to be effective in our patient, but it caused symptoms typical for hypocalcemia despite normocalcemia. In this case, the cinacalcet therapy would not have been necessary because of the benign course and favorable prognosis of FHH3.
Despite the negative findings of parathyroid imaging and surgery, parathyroid tissue was clearly present in this patient as shown by the circulating PTH within the normal or “high normal” range. The imaging techniques and surgery may not be sensitive enough to identify the reduced amount of parathyroid tissue (as it may be expected in 22q11.2 DS), or, alternatively, the parathyroid tissue may locate ectopically. Studies have reported parathyroid dysfunction in approximately 50% of patients with 22q11.2 DS; however, in the remaining patients, the parathyroid abnormality is not present at all or is relatively mild (3). Given the presence of parathyroid tissue in this patient, the de novo AP2S1 mutation behaves as in other reported patients with FHH3 and impairs CASR signaling in the parathyroid gland, leading to hypercalcemia (4, 10). Therefore, the combination of the phenotypic features of 22q11.2 DS with FHH resulted in the unusual presentation of this patient.
In conclusion, the unexpected combination of hypercalcemia and 22q11.2 DS in the absence of CASR mutations resulted in extensive examinations and treatment attempts. Our case indicates the importance of an accurate genetic diagnosis in patients with suspected defects in calcium sensing. The exact diagnosis enables health care professionals to offer the patient appropriate management, genetic counseling, and information about the disorder's course and prognosis.
Acknowledgments
This study was supported financially by grants from the Academy of Finland, the Swedish Research Council, the Swedish Childhood Cancer Foundation, the Folkhälsan Research Foundation, the Finnish Pediatric Research Foundation, the Sigrid Jusélius Foundation, the Helsinki University Hospital Research Funds (to O.M), and the Canadian Institutes of Health Research (to G.N.H.).
Authors' roles are as follows: study design: S.T., H.V., and O.M.; study conduct: S.T., G.N.H., H.V., D.E.C.C., and O.M.; data collection: S.T., G.N.H., H.V., L.C., B.S.P.L., B.Y.L.W., D.E.C.C., and O.M.; data analysis: S.T., G.N.H., H.V., L.C., B.S.P.L., B.Y.L.W., M.J.V., D.E.C.C., and O.M.; data interpretation: S.T., G.N.H., H.V., M.J.V., D.E.C.C., and O.M.; drafting the manuscript: S.T., H.V., and O.M.; revising the manuscript content: S.T., G.N.H., H.V., M.J.V., D.E.C.C., and O.M.; approving the final version of the manuscript: S.T., G.N.H., H.V., L.C., B.S.P.L., B.Y.L.W., M.J.V., D.E.C.C., and O.M.; S.T., G.N.H., D.E.C.C., and O.M. take responsibility for the integrity of the data analysis.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- BMD
bone mineral density
- 22q11.2 DS
22q11.2 deletion syndrome
- FHH
familial hypocalciuric hypercalcemia
- 25OHD
25-hydroxyvitamin D
- 1,25(OH)2D3
1,25-dihydroxyvitamin D3.

{kind=link}