





Endometriosis is an estrogen-dependent disease. P38 and C-jun NH2-terminal kinase (JNK) inhibitors may have a therapeutic effect on endometriosis through regulation of prostaglandin E2 (PGE2)-induced estrogen metabolism.
The objective of this study was to determine whether the activated MAPKs signaling pathway observed in human ectopic endometrial stromal cells (ESCs) from ovarian endometriomas influences levels of aromatase and estrogen receptor β (ERβ) protein regulated by PGE2. In turn, the effects of inhibiting MAPKs in the presence of PGE2 on estrogen production were investigated in vitro and in vivo.
Expression of aromatase and ERβ regulated by PGE2 were much higher in ESCs than eutopic ESCs from the same person. Activation of p38, JNK, ERK 1/2 and ERK 5 MAPKs by PGE2 were observed in ESCs, where PGE2-stimulated aromatase and ERβ expression mainly through p38 and JNK pathway. P38 and JNK inhibition or small interfering RNA knockdown blocked PGE2-induced aromatase and ERβ expression. PGE2 enhanced binding of downstream p38 and JNK transcription factors activating transcription factor-2 and c-Jun to aromatase and ERB promoter regions in ESCs. Moreover, treatment of endometriosis xenografts with inhibitors of p38 and JNK abrogated PGE2-amplified estradiol synthesis and xenograft growth.
PGE2 activates p38 and JNK signaling pathways, further stimulating c-Jun and activating transcription factor-2 binding to aromatase and ERB promoter regions with elevated estradiol production. Inhibition of JNK and P38 may be a potential method of treating human endometriosis.
Endometriosis is an estrogen-dependent disease (1). Hormonal treatment or conservative surgery achieves significant pain relief, but recurrence of symptoms is high. Thus, there is a clear need to develop novel and effective therapies for endometriosis (2, 3). Estradiol is a key hormone for the growth and persistence of endometriotic tissue (4). Endometriotic lesions show high estradiol biosynthesis compared with normal endometrium (5).
A growing body of evidence shows strong association between prostaglandin E2 (PGE2) and the synthesis of estradiol in human ectopic endometrial stromal cells (ESCs) (6–8). Increased PGE2 concentrations in the peritoneal fluid of women with endometriosis and ovarian endometriomas have been reported (9).
P450 aromatase is a key enzyme for estrogen biosynthesis (10). PGE2 appears to be the most potent stimulator of aromatase in ESCs (7). Although PGE2-stimulated expression of aromatase is observed in endometriosis, signaling events downstream of protein kinase A leading to aromatase production are still unknown.
Estrogen action is mediated primarily via nuclear estrogen receptors (ERs) (11–13). Previous studies suggest that an ERβ-selective compound might be therapeutic in a rodent endometriosis model, and ERs are believed to play a key role in endometrial and endometriosis growth regulation (14). Currently, the biologic roles of ERβ and the relationship between PGE2 and ERβ in endometriosis are still not well understood.
MAPKs' pathways are activated in response to a variety of extracellular and intracellular stimuli (15–17). Stimulation of MAPKs requires dual phosphorylation, which occurs from sequential activation (18). Four distinct MAPK pathways have been described: ERK1/2, ERK5, p38 MAPK, and C-jun NH2-terminal kinase (JNK) (19).
The aim of this study was to investigate the molecular and cellular mechanism by which PGE2 regulates aromatase and ERβ expression in human ESCs and xenograft. We propose that PGE2 induced aromatase and ERβ expression by interacting with activating transcription factor-2 (ATF2) and c-Jun via the MAPK signaling pathway, thereby influencing estrogen production in human ESCs.
Materials and Methods
Full description of the materials and methods used in this work is provided in the Supplemental Material. The experimental procedures were approved by the Institutional Review Board of Peking University First Hospital (No. 2014[789] and No. 2014[790]). Animal studies were approved and performed in accordance with Peking University Animal Care and Use Committee (J201403). Briefly, human ESCs derived from ovarian endometriomas (n = 18) and human eutopic endometrial stromal cells (EMs) from the same person (n = 12) were primary cultured with PGE2. Aromatase, ERβ, and cyclooxygenase 2 (COX2) mRNA levels were measured by real-time quantitative PCR (RT-qPCR). PGE2 and estrogen production were measured by enzyme-linked immunosorbent assay (ELISA) kits and aromatase activity was measured by [3H]water release assay (20). ERβ, ERK 1/2, ERK5, p38, JNK, ATF2, and c-Jun protein expression were measured by Western blot assay. Association between c-Jun and ATF2 was measured by coimmunoprecipitation assay and binding of ATF2 and c-Jun to the aromatase and ERB promoter region were detected by chromatin immunoprecipitation (ChIP) (21). A xenograft mouse model was adapted from the model previously described by Bruner-Tran et al (22). Tissue sections from ovarian endometrioma, eutopic endometrial tissues, and animal xenografts were processed for immunohistochemistry. Immunohistochemical staining for c-Jun and ATF2 were performed. All experiments were done at least three times. Statistical analysis for comparison of two treatment groups was performed by Student's t test. One-way ANOVA followed by Tukey's multiple-comparison test was used for comparison of more than two groups. A P value < .05 was considered significant. All values are given as the mean, with the bars showing mean ± SE.
Results
Expression of aromatase and ERβ and regulation of aromatase and ERβ expression by PGE2 in ESCs and EMs
RT-qPCR, aromatase activity assays, and Western blotting results demonstrated differential expression of aromatase and ER in ESCs (n = 8) and in EMs from the same woman. Aromatase (n = 8; P < .01; t test; Figure 1A, upper) and ERβ (n = 8; P < .05; Figure 1A, lower) expression was significantly higher in ESCs than in EMs. ESCs (n = 12) incubated in the presence (1 μmol/L) or absence of PGE2 for different durations displayed elevated levels of aromatase and ERB mRNAs in a time-dependent way, peaking at 24 hours and 48 hours, respectively (P < .01; Figure 1B). In ESCs, PGE2 profoundly induced aromatase mRNA levels (5.9-fold; P < .01) and activity (11.3-fold; P < .01; Figure 1C, upper). PGE2 also significantly stimulated ERB mRNA levels and protein expression (3.9-fold; P < .01, Figure 1C, lower). EMs from the same women (n = 12) contained low levels of aromatase and ERB mRNAs, which could not be stimulated by PGE2 Supplemental Figure 1A, P > .05). Levels of estradiol were higher in PGE2-treated ESCs compared with vehicle control (Figure 1D; P < .01).
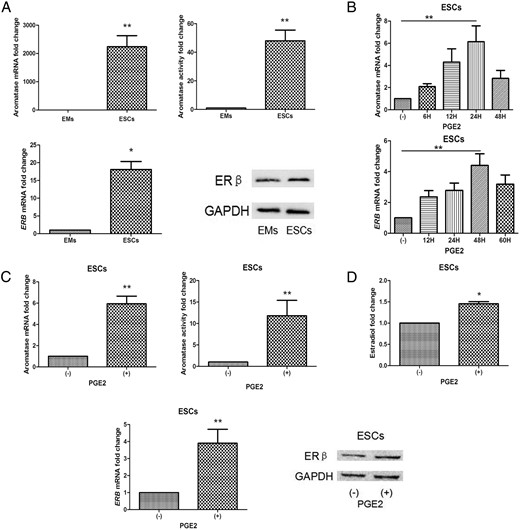
Expression and regulation of aromatase, ERβ, and PGE2 in ESCs and EMs.
Cells were incubated in the presence (or absence) of PGE2, dibutyryl cAMP, or phorbol diacetate for 24 (aromatase) and 48 hours (ERβ). Western blotting, aromatase activity assays, and quantification of aromatase mRNA levels were performed as described. Values are means ± SEM. A, Aromatase mRNA and enzyme activity in ESCs were higher than in EMs (n = 8; **t test, P < .01; upper). ERB mRNA and ERβ protein levels in ESCs were higher than in EMs (n = 8; *, P < .05; lower). B, Time-dependent PGE2-induced aromatase and ERβ expression peaked at 24 and 48 hours, respectively (n = 5; P < .01). C, PGE2 augmented aromatase mRNA and enzyme activity in ESCs, but not in EMs (n = 12; **, P < .01, upper). PGE2 elevated ERB mRNA and ERβ protein expression in ESCs, but not in EMs (n = 12; **, P < .01, lower). D, Effects of PGE2 on production of estradiol (n = 8; *, P < .05). GADPH, glyceraldehyde-3-phosphate dehydrogenase.
PGE2 receptors (EPs) 1–4 were each detected in ESCs and EMs by RT-qPCR. EP1 and EP3 were of lower abundance in ESCs and in EMs in comparison to EP2 and EP4 (n = 6; Supplemental Figure 1B). The cAMP analog dibutyryl cAMP (0.5 mmol/L) strongly induced aromatase and ERB mRNA levels (n = 8; 3.8-fold, 2.7-fold, respectively; P < .01), aromatase activity, and ERβ protein levels (10.0-fold; P < .01), whereas phorbol diacetate showed no significant effect (P > .05; Supplemental Figure 1C).
These findings indicate that aromatase and ERB appear to be key PGE2-induced genes in ESCs. Additionally, PGE2 activated aromatase and ERB expression in a cAMP-dependent manner.
PGE2 activate p38, JNK, ERK1/2, and ERK5 MAPKs in ESCs
To identify the cAMP downstream signaling events involved in PGE2-induced aromatase and ERB expression, we examined the abundance and phosphorylation status of important agents within MAPK pathways, including p38, JNK, ERK1/2, and ERK5 in endometriotic cells. p38, JNK1, ERK2, and ERK5 were highly expressed in ESCs and EMs from the same patients. No significant differences were observed between basal levels of p38, JNK1, ERK2, and ERK5 mRNAs (n = 6; P > .05; t test; Figure 2A, upper) and no marked changes in mRNA levels were observed after PGE2 treatment (n = 6; Figure 2A, lower). Protein phosphorylation of p38, JNK1/2, ERK1/2, and ERK5 were rapidly induced by PGE2 stimulation (Figure 2B) and maximal by 5–15 minutes, declining to basal level within 1–2 hours in ESCs. Hence, PGE2 may activate all four MAPK signaling pathways in endometriosis.
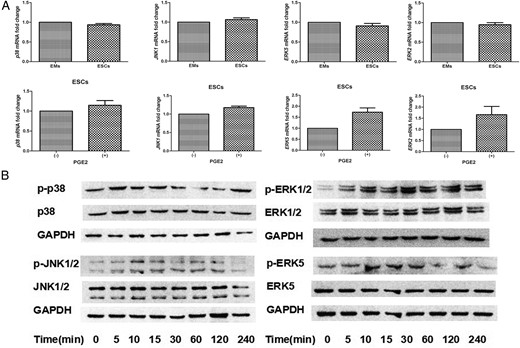
PGE2 treatment stimulates the p38, JNK1/2, ERK1/2, and ERK5 MAPK pathways in ESCs.
A, SYBR Green-based RT-qPCR analysis showed that P38, JNK1, ERK2, and ERK5 mRNAs were highly expressed in both ESCs and EMs, with no differences between transcript levels in either cell type (n = 6; t test, P > .05; upper). After starvation overnight, cells were treated with 0 or 1 μmol/L PGE2 (P) for 24 hours. Neither treatment altered the basal mRNA levels of p38, JNK1, ERK2, or ERK5 (n = 6; P > .05; lower). B, ESCs were serum-starved overnight and treated with 1 μmol/L PGE2 for the indicated times. Whole-cell lysates were prepared and subjected to SDS-PAGE and analyzed by Western blot with the indicated antibodies (n = 6). GADPH, glyceraldehyde-3-phosphate dehydrogenase.
PGE2-stimulated aromatase and ERβ expression via MAPK pathways in ESCs
To determine whether p38, JNK, ERK1/2, and ERK5 activation by PGE2 were required for PGE2-stimulated aromatase expression and aromatase activity in ESCs, we used inhibitor compounds to block phosphorylation/activation of p38, JNK, ERK1/2, and ERK5. We measured aromatase activity and mRNA levels in the presence or absence of the p38 inhibitor SB202190 (SB), JNK inhibitor AS601245 (AS), ERK1/2 inhibitor PD98059 (PD), or ERK5 inhibitor BIX02189 (BIX) following PGE2 treatment for 24 hours (n = 10). PGE2 induced a 5.9-fold increase in total aromatase mRNA, which was reduced to basal levels in the presence of 1 μmol/L SB or AS. Addition of either SB2or AS markedly reduced this induction by 87% and 80%, respectively (P < .01, ANOVA). When PD or BIX were used to block ERK1/2 or ERK5 phosphorylation/activation, PGE2-induced total aromatase mRNA levels were not significantly altered (P > .05). Hence, increased aromatase activity with PGE2 treatment results from elevated aromatase gene expression. PGE2 treatment resulted in an 11.3-fold increase in aromatase activity, whereas this increased induction was significantly reduced by 82% and 75% with either SB or AS addition, respectively (P < .01, Figure 3A).
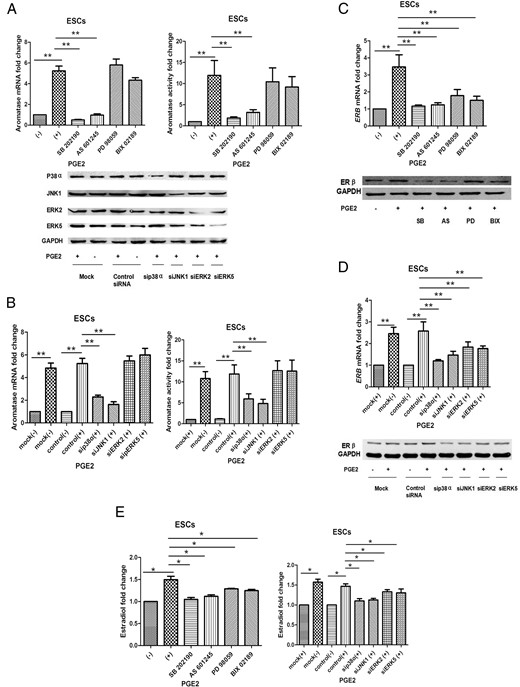
PGE2 stimulated aromatase and ERβ expression via MAPK pathways in ESCs.
A, Inhibition of p38 or JNK, not ERK1/2 or ERK5, blocks PGE2-stimulated induction of aromatase mRNA and enzyme activity (n = 10; **ANOVA, P < .01). Following overnight serum-starvation, ESCs were pretreated with DMSO (-), 5 μmol/L SB202190, 5 μmol/L AS601245, or 10 μmol/L PD98059 and 5 μmol/L BIX 02189 for 1 hour, then subsequently treated with 1 μmol/L PGE2 for 24 hours. B, SiRNA-mediated knockdown of p38a or JNK1, but not ERK reduced PGE2-stimulated aromatase expression in ESCs (n = 10; **, P < .01). ESCs were mock-transfected or transfected with the indicated siRNAs, serum-starved overnight, and treated with 0 or 1 μmol/L PGE2 for 24 hours. Cells were harvested for immunoblotting with anti-p38, anti-JNK, anti-erk1/2, anti-ERK5, and anti-GAPDH antibodies for verification of siRNA knockout efficiency. Cells were harvested for TaqMan-based RT-qPCR and aromatase activity assays. C, Inhibition of p38, JNK, ERK1/2, and ERK5 blocks PGE2-stimulated induction of ERB mRNA and ERβ protein levels (n = 10; **, P < .01). D, SiRNA-mediated knockdown of p38a, JNK1, ERK2, or ERK5 reduced PGE2-stimulated ERB expression in ESCs (n = 10; **, P < .01). Cells were harvested for TaqMan-based RT-qPCR and Western blotting. E, PGE2 elevated estradiol production via MAPK pathways, especially p38 and JNK (n = 8; *, P < .05). Conditioned media were collected and assayed by ELISA. Basal aromatase and ERβ expression with DMSO pretreatment or mock-transfection were normalized to 1. Error bars indicate ± SEM. DMSO, dimethyl sulfoxide; GADPH, glyceraldehyde-3-phosphate dehydrogenase.
To further validate the roles of p38, JNK, ERK1/2, and ERK5 in PGE2 regulation of aromatase, we used small interfering RNAs (siRNAs) to knockdown p38a, JNK1, ERK2, and ERK5. The efficacy of siRNAs knockdown was determined by Western blotting. In mock or negative control siRNA-transfected ESCs, PGE2 treatment resulted in a marked increase in aromatase total mRNA levels and aromatase activity. ESCs transfected with sip38a exhibited profoundly diminished p38 protein levels and a significant reduction in PGE2-induced total aromatase mRNA levels of 60% (n = 10, P < .01). Likewise, transfection of siJNK1, which abolished expression of JNK1, had a similar effect (63%, n = 10, P < .01). Transfection of sip38a or siJNK1 suppressed PGE2-stimulated aromatase activity by 53% and 72%, respectively (n = 10, P < .01). However, transfection of siERK2 or siERK5 did not significantly alter PGE2 induction of aromatase activity (P > .05, Figure 3B).
To examine the effect of p38, JNK, ERK1/2, and ERK5 phosphorylation on ERB gene expression, we cotreated ESCs (n = 10) with or without PGE2 and the inhibitors. Addition of the four inhibitors all markedly reduced PGE2-induced ERB mRNA expression by 90, 86, 32, and 47% (P < .01), respectively. ERβ protein levels were also significantly suppressed by the four inhibitors (Figure 3C). Transfection of sip38a, siJNK1, siERK2, or siERK5 suppressed PGE2-stimulated ERB mRNA expression by 83, 72 (P < .01), 39, and 44% (P < .05), respectively, with equivalent effects on ERβ protein levels (Figure 3D). Taken together, these results indicate that activation of p38 and JNK, but not ERK1/2 or ERK5, is necessary for PGE2 induction of aromatase expression. Although PGE2 induced ERβ expression via p38, JNK, ERK1/2, and ERK5 in ESCs, P38 and JNK appeared to play a more important role. Effects of p38, JNK, ERK1/2, and ERK5 on PGE2 induction of estradiol production are presented in Figure 3E (P < .05, n = 10).
PGE2 enhances binding of ATF2 and c-Jun to the aromatase and ERB promoter region in ESCs
Previous studies indicate that ATF2 and c-Jun are transcription factors downstream p38 and JNK pathways (15, 23). Given the dependence of PGE2-induced aromatase and ERβ expression on JNK and p38, their substrates c-Jun and/or ATF2 may regulate aromatase and ERB transcription by interacting directly or as a complex with the aromatase and ERB promoter regions in endometriosis. First, we demonstrated that ATF2 and c-Jun were highly expressed in ovarian endometriomas and EMs from the same patients (n = 10; Figure 4A, upper). Meanwhile, there was no difference between the mRNA basal levels of ATF2 and c-Jun in EMs and ESCs (n = 6; Figure 4A, lower) and no marked change in the expression level of either mRNA after PGE2 treatment (n = 6; Figure 4B, upper). PGE2 treatment activated ATF2 and c-Jun in ESCs. Phosphorylation of ATF2 and c-Jun were rapidly induced by PGE2 stimulation, reaching a peak by 5–30 minutes, and declining to basal level by 1–2 hours (n = 6; Figure 4B, lower). To further determine the interaction between ATF2 and c-Jun, we conducted coimmunoprecipitation and observed an increase in complex formation between ATF2 and c-Jun with PGE2 treatment for 30 minutes (n = 5, P < .05, t test; Figure 4C). To confirm the direct interaction between c-Jun and the aromatase and ERB promoter region in vivo, we performed chromatin immunoprecipitation experiments targeting aromatase and ERB promoter region in ESCs. First, we treated ESCs with PGE2 for 30 minutes. ChIP experiments were carried out with an anti-c-Jun antibody followed by aromatase promoter region II (PII)-specific semiquantitative PCR. In the absence of PGE2, a low level of c-Jun binding to aromatase PII was detected. Following PGE2 treatment, c-Jun binding to aromatase PII was manifestly enhanced. Likewise, we conducted ChIP experiments with an anti-ATF2 antibody and found that treatment with PGE2 for 30 minutes resulted in enhanced recruitment of ATF2 to aromatase PII (n = 8, Figure 4D). Then, we conducted SYBR Green qPCR analysis and observed PGE2 stimulation of c-Jun binding to aromatase PII by 4.8-fold and ATF2 binding to aromatase PII by 9.2-fold (n = 8, P < .05; Figure 4D). Similar results were observed for the ERB promoter region by semiquantitative PCR and qPCR analysis in which PGE2 stimulated c-Jun binding to the ERB promoter region by 2.3-fold and ATF2 binding to the ERB promoter region by 2.6-fold (n = 8, P < .05; Figure 4E).
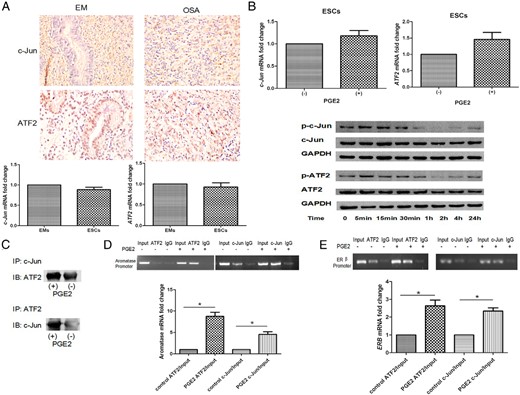
PGE2 enhances binding of ATF2 and c-Jun to the aromatase and ERβ promoter region in ESCs.
A, Immunohistochemical analysis of human ovarian endometriomas and eutopic endometrial tissues showed that ATF2 and c-Jun were highly expressed in patients with endometriosis (n = 10; upper). Original magnification, ×40. SYBR Green-based RT-qPCR analysis showed that there was no difference between ATF2 and c-Jun mRNA expression in EMs and ESCs (n = 6; t test, P > .05; lower). B, After serum-starvation overnight, ESCs were treated with 0 or 1 μmol/L PGE2 (P). Neither treatment altered basal mRNA levels of ATF2 or c-JUN at 24 hours (n = 6; P > .05; upper). PGE2 magnified ATF2 and c-Jun phosphorylation in a time-dependent manner (n = 6; lower). C, ESCs were treated (or not) with 1 μmol/L PGE2 for 30 minutes, and cell lysates were collected and immunoprecipitated with anti-c-Jun or anti-ATF2 antibody and analyzed by Western blotting with anti-ATF2 or anti-c-Jun antibodies. Association between c-Jun and ATF2 was increased by PGE2 treatment (n = 5, P < .05). D, ESCs were treated (or not) with 1 μmol/L PGE2 for 30 minutes, then subjected to chromatin immunoprecipitation with anti-ATF2 and anti-c-Jun antibodies or control IgG followed by aromatase PII-specific semiquantitative PCR (upper) and SYBR Green-based qPCR analysis (lower). PGE2 enhanced binding of ATF2 and c-Jun to aromatase PII (n = 8; *, P < .05). E, PGE2 enhanced the binding of ATF2 and c-Jun to the ERB promoter region (n = 8; *, P < .05). Basal aromatase and ERB promoter specific mRNA levels with no PGE2 treatment were normalized to 1. -, control; IB, immunoblot; IP, immunoprecipitation.
Taken together, these findings indicate that PGE2 specifically enhances c-Jun and ATF2 binding to aromatase and the ERB promoter region, resulting in transcriptional activation of aromatase and ERB gene expression.
P38 and JNK inhibition in vivo abrogates PGE2-induced estradiol synthesis and growth of endometriosis grafts
To test the effects of SB202190 and AS601245 on human endometriotic tissues in vivo, tissue fragments from ovarian endometriomas were injected sc into nude ovariectomized mice. Each mouse received two injections of tissue, one in each flank. The 60 mice were randomly assigned to one of four treatment arms: E2 (E), E + PGE2, E + PGE2 + SB, and E + PGE2 + AS (n = 15 each group; Figure 5A, upper). E2 was included in all treatment arms for establishment of endometriotic lesions, as previously described (24). Two weeks later, mice were sacrificed, grafts and blood were obtained, and pathology diagnosed (Figure 5A, lower). Tumor volume was not significantly increased in the PGE2 treatment group, compared with controls (33.5 mm3 vs 32.8 mm3, P > .05, ANOVA). However, trends toward decreased tumor volume were noted with both SB (22.36 mm3, P < .05) and AS (23.87 mm3, P < .05). In all four treatment arms, mice maintained body weight, and there were no signs of heart or liver toxicity upon histopathological evaluation. Mouse body weights among the four arms did not vary significantly (Figure 5B).
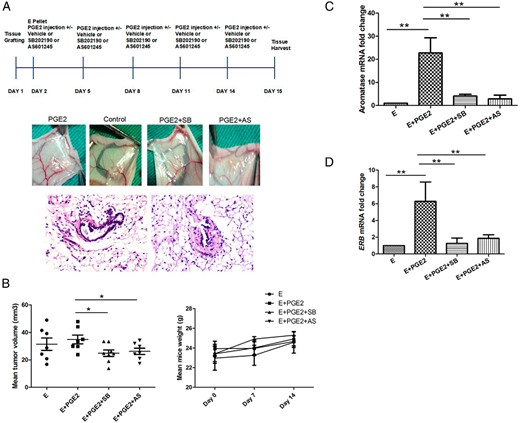
P38 and JNK inhibition in vivo abrogates PGE2-induced estradiol production and growth of endometriosis grafts.
A, Endometriosis tissue fragments were injected sc into both flanks of the mice. On the next day, estradiol hormone pellets were implanted and PGE2 (30 ng/g/d) injected sc in nude mice dosed orally with 1 mg/kg/d SB202190, AS601245, or vehicle. PGE2 injection, SB202190, and AS601245 were delivered orally every 3 days for 2 weeks. After 2 weeks, mice were sacrificed, xenografts and blood were obtained (n = 40, upper), and immunohistochemical staining (n = 10, lower) was performed on graft tissue. Original magnification, ×40. B, Xenograft volumes were measured and independent trends toward decreased mean tumor volume were noted with SB202190 and AS601245 treatments (*ANOVA, P < .05). Data are expressed as mean ± SEM from 78 lesions in 60 mice. SYBR Green-based RT-qPCR quantification of C, aromatase and D, ERB mRNAs was performed. Aromatase and ERB mRNA levels were each significantly lower in the SB202190- and AS601245-treated groups than in the E + PGE2 group (n = 8 each group; **, P < .01).
Treatment of SB or AS (1 mg/kg/d p.o. every 3 days) commenced 1 day after injection of the endometrial fragments and continued for total 15 days. SYBR Green-based qPCR analysis of the tissue grafts showed that aromatase mRNA levels were significantly lower in the SB and AS groups than in the E + PGE2 group (P < .01; Figure 5C). The levels of ERB mRNA exhibited the same trend as for aromatase. ERB mRNA levels were highest with E + PGE2, whereas treatment with SB or AS decreased levels of ERB expression (P < .01; Figure 5D). Together, these data suggest that PGE2 induced the growth of endometriosis grafts and in vivo estradiol synthesis, at least in part, through p38 and JNK signaling pathways. Thus, we propose the use of p38 and JNK pathway inhibitors in endometriosis treatment.
P38 and JNK inhibition suppresses E2-induced PGE2 production in ESCs
To further explore the relationship between E2 and PGE2 (25) and dissect the roles of p38 and JNK in E2 regulation of PGE2 production, we used drug inhibitors and siRNAs to depress function of p38 and JNK. ESCs incubated in the presence or absence of E2 for varying durations and at different doses revealed that COX2 mRNA levels increased in a time- and dose-dependent fashion, peaking at 24 hours and 10–7 M, respectively (n = 5, Figure 6A). E2 (10–7 M) exposure resulted in a 3.2-fold increase in COX2 mRNA levels at 24 hours (P < .05, ANOVA), which was reduced to approximately basal levels in the presence of 1 μmol/L SB or AS. Addition of either SB or AS markedly reduced this induction by 76 and 79%, respectively (n = 8; P < .01; Figure 6B, upper). Addition of either SB or AS also reduced PGE2 accumulation by 30% and 33% (n = 8; P < .05; Figure 6B, lower). In mock or negative control siRNA transfected ESCs, PGE2 treatment resulted in a marked increase in COX2 mRNA levels. ESCs transfected with sip38a or siJNK1 displayed a significant reduction in E2-induced COX2 mRNA levels of 38% and 45% (n = 8; P < .01; Figure 6C, upper). Likewise, the results of PGE2 ELISA showed that transfection of sip38a or siJNK1 had a similar effect in reducing PGE2 production by 21 and 24% (n = 8; P < .05; Figure 6C, lower). Taken together, PGE2 increased aromatase and ERβ expression via activation of p38 and JNK, which further activated downstream transcription factors c-Jun and ATF2 binding to the aromatase and ERB promoter regions, augmenting aromatase and ERB gene transcription and inducing E2 production. Increased E2 production in turn stimulated PGE2 expression, which could be suppressed by the inhibitory effects of p38 and JNK (Figure 6D).
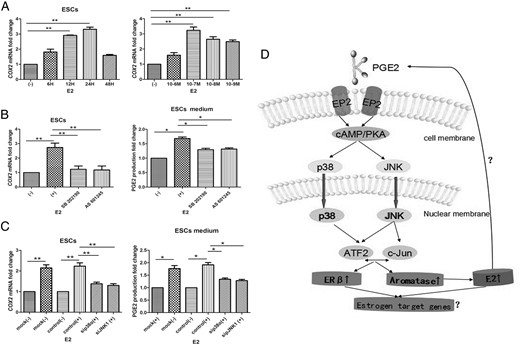
P38 and JNK inhibition suppresses E2-induced PGE2 production in ESCs.
A, Cells were serum-starved overnight, then incubated in the presence (or absence) of E2 for indicated times and dosages. Quantification of COX2 mRNA levels using SYBR Green-based RT-qPCR revealed E2-induced COX2 expression in a time- and dose-dependent fashion, peaking at 24 hours and 10–7 M, respectively (n = 5). B, Inhibition of p38 or JNK blocks E2-stimulated induction of COX2 mRNA levels (n = 8; **ANOVA, P < .01; upper) and PGE2 production (n = 8; *, P < .05; lower). Following overnight serum-starvation, ESCs were pretreated with DMSO (-), 5 μmol/L SB202190, or 5 μmol/L AS601245 for 1 hour. Cells were then treated with 10–7 M E2 for 24 hours. C, siRNA-mediated knockdown of P38a or JNK1 reduced E2-stimulated COX2 mRNA levels (n = 8; **, P < .01; upper) and PGE2 production (*, P < .05; lower). ESCs were mock-transfected or transfected with the indicated siRNAs, serum-starved overnight, and treated (or not) with 10–7 M E2 for 24 hours. Cells were harvested for detection of COX2 mRNA. Conditioned cell culture media were collected for PGE2 ELISA. Basal COX2 mRNA and PGE2 concentrations with DMSO pretreatment were normalized to 1. Values are means ± SEM. D, Schematic model of PGE2 regulation of aromatase and ERβ expression in endometriosis.
Discussion
In this study, we showed that inhibition of P38 or JNK can limit PGE2-induced aromatase and ERβ expression, which abrogates estrogen biosynthesis and action, resulting in decreased PGE2 levels in endometriosis.
Previous studies showed that although aromatase activity and mRNA expression were not detected in EMs of disease-free women, expression and activity were detected in ESCs and EMs of women with endometriosis, whereas detection in ESCs was much higher than that in EMs from the same individuals. Elevated PGE2 concentrations in the peritoneal fluid of women with endometriosis and ovarian endometriomas promote the development and severity of endometriosis. In this study, PGE2 increased cAMP, then activated p38 and JNK signaling pathways. This further stimulated binding of c-Jun and ATF2 to aromatase promoter regions and increased aromatase transcription. Increased aromatase activity promoted estrogen biosynthesis, leading to high levels of E2 accumulation. In turn, augmented E2 production stimulated COX2 expression and elevated PGE2 concentration.
ERβ is the only ER subtype expressed in primary placental villus endothelial cells and is essential for maintaining COX2 mRNA and protein levels in these cells (26). Previous studies have shown that epigenetic ERB promoter regulation can account for high ERβ levels, and ERβ is also responsible for low ERA expression in endometriotic stromal cells (27). Additionally, a positive role of ERβ on endometriotic stromal cell-cycle progression and proliferation has been proposed (28). Our study is the first to show that PGE2 stimulated binding of c-Jun and ATF2, not only to aromatase regulatory domains, but also to the ERB promoter, elevating ERB transcription and ERβ translation. Additionally, this may accentuate expression of estrogen-related genes, correlated with increased E2 production. Because PGE2 synthesis is also dramatically higher in ESCs, it is tempting to speculate that amplified levels of aromatase and ERβ are at least in part responsible for high PGE2 levels in endometriosis.
The presence and activation by phosphorylation of three MAPK family members (P38, JNK, and ERK1/2) has been described in endometriosis (29). Here, we further demonstrate that ERK5 is also highly expressed and phosphorylated in ESCs. Drug-mediated inhibition or siRNA-modulated knockdown of p38 or JNK reduced PGE2-stimulated aromatase mRNA levels and activity in ESCs, indicating that activation of p38 and JNK, but not ERK1/2 or ERK5, is necessary for PGE2-induced aromatase expression. The discrepancy may in part be due to cellular differences or in the ligands employed; further investigations are required to resolve this disparity and reveal the specific mechanisms. Consistent with this, PGE2 elevated ERB expression largely through p38 and JNK signaling pathways, whereas ERK1/2 and ERK5 may similarly undergo activation. Moreover, although increased E2 production augmented COX2 expression and prompted elevated PGE2 levels, inhibition of p38 and JNK decreased E2-induced COX2 expression and PGE2 concentrations. In other words, this positive feedback between PGE2 and E2 in ESCs might be partly disrupted by p38 and JNK blockade.
The p38 and JNK downstream transcription factor ATF2 can form homodimers or heterodimers with members of the ATF and activator protein 1 (AP-1) families, including c-Jun, thereby directly interacting with gene promoter regions via CRE and/or AP-1 sites (30). The aromatase promoter region contains a CRE motif in the 211/199 base pair region. This CRE element has been identified as the c-Jun binding site in granulosa cells; however, c-Jun represses rather than promotes aromatase expression in such cells (31). Our chromatin immunoprecipitation assays showed that PGE2 enhances specific binding by c-Jun and ATF2 to the aromatase promoter, activating gene transcription. Genome-wide mapping of ERβ-binding motifs reveals extensive cross-talk with the transcription factor AP-1 in ERβ-overexpressing MCF7 breast cancer cells (32). In the present study, PGE2 directly activated c-Jun and ATF2 or formed a promoter region transcription complex to regulate ERB via an AP-1 site, induced binding of ATF2 and c-Jun to ERB promoter regions, resulted in enhanced ERB transcription in ESCs.
The role of MAPK signaling pathways has been investigated in many diseases (33, 34). Previous mouse studies show that SB203580, a p38 MAPK inhibitor, suppresses the development of endometriosis by downregulating proinflammatory cytokines and proteolytic factors (35). The role of JNK inhibitors is still not characterized in endometriosis. In this study, p38 and JNK inhibitors SB202190 and AS600125 were able to reduce PGE2-stimulated aromatase and ERB expression in human ESCs and in endometriosis xenografts. Thus, not only p38, but also JNK may represent new drug targets for tissue-specific ablation of aromatase and ERB expression in endometriosis. In the xenograft studies shown here, mouse body mass and histological examination of organs did not reveal any evidence of toxicity-related to the p38 and JNK inhibitors. Although we showed a potential way of reducing estradiol synthesis by inhibition of p38 or JNK in endometriosis, the potential side effects and teratogenicity of treatment need to be noticed because endometriosis is a disease that more or less only affects women of fertile age. Further studies are still needed.
In summary, our results indicate that PGE2 activates p38 and JNK signaling pathways, stimulating binding of c-Jun and ATF2 to aromatase and ERB promoter regions, resulting in upregulation of mRNA transcriptional activity and subsequent estrogen biosynthesis in pathogenic endometriosis. Inhibition of p38 or JNK could limit estrogen biosynthesis and action, thus decreasing PGE2 concentration in ESCs. Moreover, inhibition of p38 and/or JNK in vivo could impede or halt the growth of endometriomas. Thus, p38, JNK, and their downstream transcription factors, c-Jun and ATF2, may be potential new drug targets for tissue-specific ablation of aromatase and ERB expression in endometriosis.
Acknowledgments
We thank Professor Yu Qi and Professor Ding-Fang Bu for their generous advice regarding the study.
This work was supported by the National Natural Science Foundation of China (Grant 81270674) and the Beijing Municipal Natural Science Foundation (Grant 7132204).
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AP
activator protein
- AS
JNK inhibitor AS601245
- ATF
activating transcription factor
- BIX
ERK5 inhibitor BIX02189
- ChIP
chromatin immunoprecipitation
- COX
cyclooxygenase
- ELISA
enzyme-linked immunosorbent assay
- EM
eutopic ESC
- EP
PGE2 receptor
- ER
estrogen receptor
- ESC
ectopic endometrial stromal cell
- JNK
C-jun NH2-terminal kinase
- PD
ERK1/2 inhibitor PD98059
- PII
aromatase promoter region II
- PGE
prostaglandin
- RT-qPCR
real-time quantitative PCR
- SB
p38 inhibitor SB202190
- siRNA
small interfering RNA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}