





Abstract
Microbial strategies for biomass deconstruction involve an incredible repertoire of enzymatic, structural, and regulatory proteins. From carbohydrate active enzymes to cellulosomes, bacteria, yeast, and filamentous fungi adapt their functional machinery to grow from alternative carbon sources such as lignocellulose and survive starvation. In that context, microbes must be able to sense, bind, degrade, and utilize lignin, cellulose, and hemicelluloses. Nature has developed specialized protein modules, RNA structures, and regulatory systems operating at a genomic, transcription, and translation level. This review briefly summarizes the main regulatory pathways involved in lignocellulose microbial degradation, including carbon catabolite repression; anti-sigma factors; regulatory RNA elements such as small RNAs, antisense RNA, RNA-binding proteins, and selective RNA processing and stabilization; and transcriptional regulators and unfolded protein response. Interplay with global regulators controlling pH response and nitrogen utilization is also revised.
This work will inspire the design of novel microbial systems for consolidated bioprocessing of lignocellulosic biomass into biofuel and high-value chemicals, allowing the raising of new biorefineries for a greener, sustainable, and circular economy.
Introduction
In the past decades, microbial lignocellulose degradation has been raising interest due to the current need of transforming biomass into energy. As microorganisms do, we sense a decrease in availability of carbon sources easily transformed into energy and seek for alternatives such as biofuels. In consequence, biomass, which was formerly considered as waste material, is gaining more value for global economy. Today, we count for bioengineered microorganisms that efficiently transform simple sugars obtained from biomass into biofuel. However, the transformation of biopolymers into simple sugars is a bottleneck in this process. Chemical and physical methods are available to disrupt such material, although they are expensive and negative for the environment, and may interfere with processes downstream. Microbial lignocellulose degradation is a greener alternative for this challenge. It may be performed by microbial enzymes, whole microorganisms, or microbial consortia. In a consolidated bioprocessing (CBP), all steps from lignocellulose deconstruction to biofuel occur in the same bioreactor with the aid of living microorganisms (Lynd et al. 2005). However, natural microbial processes must be improved since they occur at time rates that are prohibitory for the industry and are governed by underexplored environmental factors. Additionally, the yield of lytic enzymes biosynthesized by the natural microbial producer is often low and does not meet industrial requirements (Liu and Qu 2021). Two approaches for improving these microbial processes have been considered (Mazzoli 2012): first, the heterologous expression of lignocellulolytic systems by model microorganisms such as Escherichia coli and Saccharomyces cerevisiae (Huang et al. 2014) and second, the improvement of the natural cellulolytic strain by different approaches (Lynd et al. 2005, Chaves et al. 2019). For both purposes, engineering the microbial catabolic pathways for lignocellulose deconstruction requires a deeper exploration of the regulatory pathways involved (Chaves et al. 2019, Ashokkumar et al. 2022). Several strategies involving regulatory elements in the host such as choice of promoter, modulation of messenger ribonucleic acid (mRNA) stability, translation efficiency, and posttranslational modifications have been developed for the improvement of expression and secretion of heterologous cellulolytic proteins (Mazzoli et al. 2012). Regulatory engineering successfully improved the cellulase activity in models of industrial strains when grown in cellulosic substrates (Table 1), which further encourages the development of the CBP model. To note, concerted engineering of different regulatory elements belonging to the same regulatory network leads to remarkable results. Regarding natural cellulolytic microorganisms, efforts have been made to understand the natural pathways in which a microbe interacts with lignocellulose and utilizes it as a carbon source. However, the precise signals and following regulatory mechanisms that control these processes in each microorganism are far from being understood (Ghosh and Das 2020). Several microorganisms that naturally degrade cellulose or lignin such as Clostridium cellulolyticum and Clostridium cellulovorans (Xin et al. 2019), Clostridiumthermocellum (Mazzoli and Olson 2020) and many other thermophilic microorganisms (Blumer-Schuette et al. 2014), and Rhodococcus opacus (Anthony et al. 2019) have been proposed as natural chassis to enhance the production of biofuel and high-value products from biomass. Engineering of regulatory elements of cellulolytic pathways in microorganisms has shown to be a successful approach, not only for enhancing the cellulolytic activity of the strains (Sukumaran et al. 2021) but also for improving biofuel production (Liu and Qu 2021). Promoter insertion in cellulolytic operons increased cellulose hydrolysis in C. cellulolyticum, while insertion of successful promoters into a mutant defective in lactate production improved ethanol titer by 65% (Tao et al. 2020). In Trichoderma harzianum, the constitutive expression of the positive regulator XYR1 increased cellulase, xylanase, and β-glucosidase activities that further caused an enhancement of 25% in the saccharification of sugarcane bagasse when compared with wild type (da Silva Delabona et al. 2017).
Engineered regulatory pathways in model strains to increase cellulase production in the presence of cellulosic substrates.
| Wild-type strain | Target regulatory genes | Regulatory elements | Increase in cellulase activity | Ref. |
|---|---|---|---|---|
| N. crassa OR8-1a | gna-3,gnb-1 | Transporter involved in CCR | >3-fold | Collier et al. (2020) |
| N. crassa 74-OR23-1V | clr-2 | Transcription factor | 10-folda | Matsu-ura et al. (2018) |
| P. oxalicum 114-2 (CGMCC 5302) | clr-B, creA | Transcription factors | 11- to 58-foldb | Li et al. (2015) |
| P. oxalicum 114-2 (CGMCC 5302) | bgl2,creA,clrB | Transcription factors | 10-fold | Yao et al. (2015) |
| Wild-type strain | Target regulatory genes | Regulatory elements | Increase in cellulase activity | Ref. |
|---|---|---|---|---|
| N. crassa OR8-1a | gna-3,gnb-1 | Transporter involved in CCR | >3-fold | Collier et al. (2020) |
| N. crassa 74-OR23-1V | clr-2 | Transcription factor | 10-folda | Matsu-ura et al. (2018) |
| P. oxalicum 114-2 (CGMCC 5302) | clr-B, creA | Transcription factors | 11- to 58-foldb | Li et al. (2015) |
| P. oxalicum 114-2 (CGMCC 5302) | bgl2,creA,clrB | Transcription factors | 10-fold | Yao et al. (2015) |
Cellulase overproduction. bDepending on enzyme.
Engineered regulatory pathways in model strains to increase cellulase production in the presence of cellulosic substrates.
| Wild-type strain | Target regulatory genes | Regulatory elements | Increase in cellulase activity | Ref. |
|---|---|---|---|---|
| N. crassa OR8-1a | gna-3,gnb-1 | Transporter involved in CCR | >3-fold | Collier et al. (2020) |
| N. crassa 74-OR23-1V | clr-2 | Transcription factor | 10-folda | Matsu-ura et al. (2018) |
| P. oxalicum 114-2 (CGMCC 5302) | clr-B, creA | Transcription factors | 11- to 58-foldb | Li et al. (2015) |
| P. oxalicum 114-2 (CGMCC 5302) | bgl2,creA,clrB | Transcription factors | 10-fold | Yao et al. (2015) |
| Wild-type strain | Target regulatory genes | Regulatory elements | Increase in cellulase activity | Ref. |
|---|---|---|---|---|
| N. crassa OR8-1a | gna-3,gnb-1 | Transporter involved in CCR | >3-fold | Collier et al. (2020) |
| N. crassa 74-OR23-1V | clr-2 | Transcription factor | 10-folda | Matsu-ura et al. (2018) |
| P. oxalicum 114-2 (CGMCC 5302) | clr-B, creA | Transcription factors | 11- to 58-foldb | Li et al. (2015) |
| P. oxalicum 114-2 (CGMCC 5302) | bgl2,creA,clrB | Transcription factors | 10-fold | Yao et al. (2015) |
Cellulase overproduction. bDepending on enzyme.
We present here a unique study summarizing all the regulatory pathways of microbial lignocellulose deconstruction described to date with focus on genetic and epigenetic systems prone to engineering. We first introduce lignocellulose as substrate, as well as the enzymatic machinery and particularly efficient and specialized structures involved in its degradation. Once the effectors are presented, we move to the regulatory elements that govern the synthesis of these enzymes and structural proteins, focusing on processes occurring at membranes and inside the cell: sensing of the substrate, activation/inhibition of intracellular mechanisms, interaction with DNA or RNA sequences by proteins or RNA, induction/inhibition of genes encoding catalytic enzymes and structural proteins assisting deconstruction of lignocellulose. Starting with carbon catabolite repression (CCR) in yeast, filamentous fungi, and different genera of bacteria, we explore posttranscription and posttranslational regulatory mechanisms. Finally, but not less important, we summarize the main global regulators controlling cellulolytic genes in fungi and bacteria.
Lignocellulose-degrading enzymes
Cellulose is the most abundant polysaccharide in nature and the major constituent of plant cell wall. It consists of β-1,4 linked d-glucose units that form linear polymeric chains of about 8000–12 000 units (Fig. 1). In crystalline cellulose, these polymeric chains are packed together by hydrogen bonds forming highly insoluble structures. The complete degradation of cellulose into glucose involves a concerted action of several enzymes including cellobiohydrolases, endoglucanases, and β-glucosidases (Østby et al. 2020). Endoglucanases cut the amorphous regions of the cellulose chains, allowing cellobiohydrolases to act at the chain ends. Finally, β-glucosidases hydrolyze cellobiose to glucose.
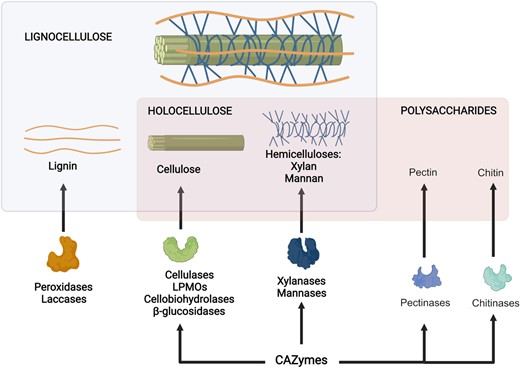
Lignocellulose consists of lignin, a polyphenolic polymer, cellulose, and hemicellulose. Cellulose is a polymer consisting solely in glucose, while hemicelluloses are heteropolymers formed by different monosaccharides. Holocellulose refers to the polysaccharide of lignocellulose, the remaining portion when lignin is not considered. Carbohydrate active enzymes (CAZymes) are enzymes involved in the catabolism of polysaccharides. Cellulose and hemicelluloses are substrates for these enzymes, together with other polysaccharides found in nature, such as chitin and pectin. Different enzymes depolymerize lignin. Laccases and peroxidases degrade not only lignin but also the corresponding monomers and other phenolic substrates. Created with BioRender.com.
Hemicelluloses (Fig. 1) are the second most abundant polysaccharides in nature. With a heterogeneous composition of various sugar units, hemicelluloses are classified according to the main sugar residues. Among the most abundant hemicelluloses, xylan structure comprises backbones of β-1,4 linked d-xylose, while glucomannan consists of chains of alternated β-1,4 linked d-mannose and d-glucose units. The less abundant arabinan consists of α-1,5 linked l-arabinose, while galactan chains consist of β-1,3 linked d-galactose units. These main chains can be linked to lateral ramifications of 4-O-methylglucuronic acid, arabinose, galactose, and acetylated xylose. Hemicellulose hydrolysis is a result of the concerted action of endo-enzymes that internally cleave the backbone, exo-enzymes that release monomeric sugars and associated enzymes that cut side chains of polymers or oligosaccharides to finally release mono- or disaccharides. For example, endo-1,4-β-d-xylanase and β-xylosidase act on the main sugar chain of xylan, while α-glucuronidase and acetyl xylan esterase cleave the side chains (Emami et al. 2009).
Cellulose can be found in a crystalline state, where intermolecular forces form an ordered structure that confers special mechanical and physicochemical properties to the polymer (de Souza 2013). Crystalline cellulose and other recalcitrant polysaccharides such as chitin are degraded by lytic polysaccharide monooxygenases (LPMOs) found in both bacteria and fungi (Agostoni et al. 2017, Sagarika et al. 2022).
Since all these enzymes have polysaccharides as substrates, they qualify as carbohydrate active enzymes (CAZymes). This huge group of enzymes acts on glycosidic bonds or linkages of moieties attached to saccharides by hydrolysis and other mechanisms. They are classified into families according to their genetic sequences, and function is predicted based on the activities described for other members of the same family.
Lignocellulose is also composed of a nonpolysaccharide polymer. Lignin is an insoluble complex branched phenolic polymer consisting of an extensive cross-linked network of substituted phenylpropane linked by carbon–carbon and ether linkages. Lignin degradation, a prerequisite for hydrolysis of cellulose and hemicelluloses, is a critical step due to the hydrophobicity and complex irregular structure of this phenolic polymer. Contrarily to hydrolytic cellulases and hemicellulases, ligninases catalyze oxidative, nonspecific reactions and exhibit broad substrate specificity (Janusz et al. 2017). Fungal ligninolytic enzymes are mainly manganese peroxidases (MnP), lignin peroxidases (LiP) catalyzing a variety of oxidative reactions that are dependent on H2O2, and laccases that are multicopper phenol oxidases that oxidize various phenolic compounds and aromatic amines reducing molecular oxygen to water (Weng et al. 2021). Although the most efficient lignin degraders in nature are white-root fungi (Kamimura et al. 2019), there are some ascomycetes like Aspergillussydowii that grow on lignin as sole carbon source with the aid of LiP, MnP, and laccases (Cong et al. 2017). Available information on bacterial lytic enzymes acting directly on the polymer is restricted to specific bacterial genera, and associate pathways are not fully understood (Silva et al. 2021). DyP-type peroxidases and laccases have been identified in lignin-degrading bacterial genera such as Streptomyces, Pseudomonas, Rhodococcus, Bacillus, Enterobacter, and others (Guan et al. 2018; Lee et al. 2019). The regulatory mechanisms of lignin degradation are scarcely documented in filamentous fungi and bacteria. There is evidence that lignin deconstruction is also assisted by arrangements of structural proteins. Packaging of ligninolytic enzymes into outer-membrane vesicles has been described in Pseudomonas putida (Salvachúa et al. 2020). While validation of the role of bacterial enzymes in lignin depolymerization is evolving (Silva et al. 2021), regulatory mechanisms controlling microbial ligninases and associated genes are still not clear (Park et al. 2022). Therefore, in this review, we further describe those regulatory pathways involving cellulases and hemicellulases, i.e. enzymes acting on holocellulose.
Cellulosomes
Cellulosomes are multienzyme complexes where enzymes are attached to structural proteins called scaffoldins (Artzi et al. 2017, Wang et al. 2019b). The dockerin module of the enzymes connects with the cohesin module of scaffoldins (Fig. 2). These structures display different levels of complexity: scaffoldins can also contain a dockerin module for binding to other scaffoldins and a carbohydrate-binding module (CBM) for targeting the complex and its enzymes to appropriate sites on the plant cell wall substrate.
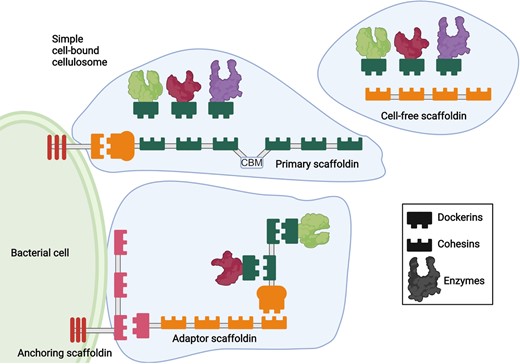
Anaerobic cellulolytic bacteria and filamentous fungi degrade cellulases and hemicellulases with highly specialized structures called cellulosomes. Degrading enzymes are attached to structural proteins called scaffoldins by complementary modules: dockerins and cohesins. Some species display cellulosomes attached to the membrane by anchoring scaffoldins, while others possess free cellulosomes. Created with BioRender.com.
To date, fungal and bacterial cellulosomes were exclusively found in anaerobic microorganisms (Gilmore et al. 2020) typically inhabiting the rumen. Cellulosome-producing bacteria can be thermophilic or mesophilic. Most of them belong to the genus Clostridium or Bacillus. Cellulosomes can be attached to the bacterial cell surface or can be released as cell-free cellulosomes (Fig. 2). Simple cellulosome systems common in mesophiles consist of a single scaffoldin that incorporates up to nine enzymatic subunits and usually contains a cellulose-binding module CBM that targets the substrate. Cellulosomes not only hydrolyze carbohydrates but also facilitate access to polysaccharide surfaces. It has been shown that the composition of the cellulosome depends on the available carbon source (Artzi et al. 2017). Accordingly, cellulosomes may include not only cellulases but also hemicellulases, ligninases, pectinases, mannanases, and chitinases (Wang et al. 2019b).
Similar structures involved in polysaccharide sensing and degradation have been found in marine bacteria (Dürwald et al. 2021). As well, other systems have been described in rumen bacteria such as Fibrobacter succinogenes S85. In this system, the cellulolytic machinery is not released, nor arranged into cellulosomes, but localized on the cell envelope (Raut et al. 2019). Cellulosomal genes are absent in F. succinogenes S85 and Tetratrico Peptide Repeat (TPR) domain-containing proteins would play the role of cohesins and dockerins. Extracellular vesicles containing cellulases, small RNAs, and heat shock proteins have been also described in the fungus Trichoderma reesei when grown in cellulose (de Paula et al. 2019).
Regulation triggered by simple sugars
Carbon catabolite repression (CCR)
For most microorganisms, glucose is the preferred carbon source. Its catabolism produces the greatest energetic yield, and its preferential consumption is coordinated with the sensing of intra- or extracellular carbon sources. To shift the metabolism toward glucose or preferred carbon sources, CCR prevents the utilization of alternative carbon sources by inhibiting the transcription of catalytic enzymes.
Fungal CCR
Mechanisms involved in fungal CCR have been mainly clarified in S. cerevisiae. When glucose is present, utilization of alternative carbon sources such as ethanol and glycerol is repressed. The increase of phosphorylated glucose (G6P) and sensing of glucose via G-protein coupled receptors (GPCR) produces a burst of cAMP and consequent activation of PKA (Fig. 3). Both G6P and PKA promote Snf1 inhibition in the phosphorylated state. In turn, phosphorylated Snf1 prevents the translocation of the repressor Mig1 to the cytoplasm (Milanesi et al. 2021). Genes required for the utilization of the alternative carbon source are thus repressed, being repression regulated by both nuclear localization of Mig1p and nucleosome positioning (Brown et al. 2014).
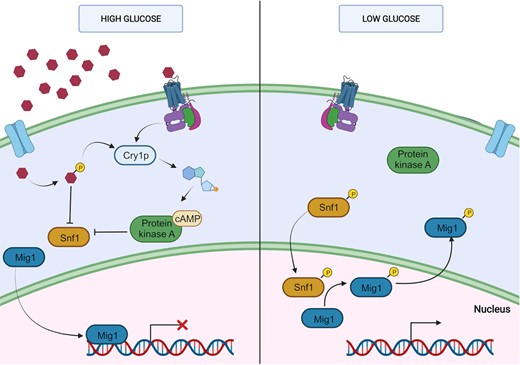
Simplified CCR mechanism described in S. cerevisiae. Filamentous fungi share this general mechanism with the participation repressors such as CRE1/cre1/CreA. When the preferred carbon source is available (left), it enters the cell by hexose transporters or G-protein receptors (GPCR). Phosphorylated glucose and GPCR activate the adenylyl cyclase Cry1p. The increase in cAMP activates the protein kinase A (PKA) that inhibits the protein Snf1 (sucrose nonfermenting 1). Glucose phosphate also inhibits this protein. In this state, the repressor Mig1 is in the nucleus impeding the transcription of genes necessary for the catabolism of alternative carbon sources. When the concentration of glucose is low. There, Mig1 is phosphorylated and translocated to the cytoplasm allowing transcription of catabolic genes. Created with BioRender.com.
Aspergillus nidulans, Neurospora crassa, and T. reesei are considered as model filamentous fungi in the study of CCR mechanisms (Brown et al. 2014). Trichoderma reesei and A. nidulans detect glucose by the cAMP-dependent PKA pathway. In A. nidulans, the protein GprH (a GPCR subtype) plays an important role in carbon starvation among other activities related to fungal growth. Genes homologous to GprH have been found in many filamentous fungi suggesting that this is a putative common glucose sensor in many other fungal species (Martín et al. 2019).
As described for filamentous fungi, a Cys2-His2 type DNA-binding zinc finger repressor protein named Mig1p controls the transcription of alternative carbon usage genes during CCR repression in S. cerevisiae. In filamentous fungi, this function is exerted by the Cys2-His2 type DNA-binding zinc finger repressor protein named Cre1 and homologs CRE1 and CreA. For most filamentous fungi, these repressor proteins bind to upstream regulatory elements inhibiting the expression of the transcription factors and metabolic enzymes required for alternative carbon usage (Brown et al. 2014). The nuclear localization of CreA/CRE1/Cre1 depends on carbon availability. In A. nidulans, CreA regulates xylanase genes and the expression of the xylanase transcriptional regulator, xlnR. The same was observed for CRE1 in N. crassa, A. nidulans, and T. reesei. In A. nidulans, N. crassa, and T. reesei, CreA/Cre1/CRE1 derepression only occurs if complex carbohydrates such as lignocellulose are present, whereas high concentrations of alternative, simple carbon sources mainly cause CreA/Cre1 nuclear localization (Daly et al. 2015). In organisms capable of consuming a wide variety of carbon sources, such as saprophytic filamentous fungi, CCR involves a range of preferred carbon sources and can be activated not only by exogenously added sugars, but also by cell wall degradation breakdown products (Huberman et al. 2016). Elicitation of fungal CCR by simple sugars may be dose dependent. For example, it was observed that xylanase was induced by little amounts of xylose but repressed at higher concentrations of the monosaccharide (Daly et al. 2015).
Bacterial CCR
CCR via transcriptional regulators follows distinct pathways in Gram-negative and Gram-positive bacteria (Fig. 4), since different, unrelated transcriptional regulators CRP and CcpA, respectively, are used (Galinier and Deutscher 2017). However, the phosphoenolpyruvate (PEP):carbohydrate phosphotransferase system (PTS) plays a major role in both mechanisms (Deutscher et al. 2006).
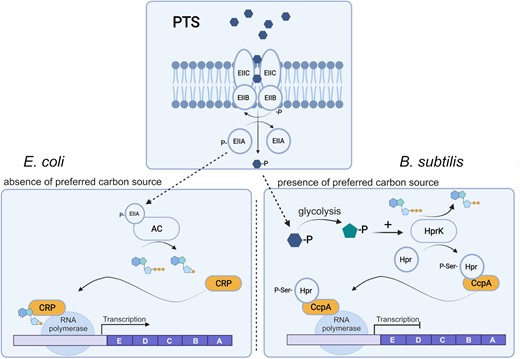
CCR in E. coli and Bacillus subtilis as representatives of Gram-negative and Gram-positive bacteria, respectively. Both mechanisms depend on the PTS. When glucose, the preferred carbon source for most bacteria, is available, it is transported into the cell by EIIA, B, and C. In this process, glucose is phosphorylated and EIIA stays unphosphorylated. At low glucose concentration, phosphorylated EIIA prevails and activates adenylyl cyclase. cAMP binds CRP protein inducing the transcription of catabolic genes by interacting with RNA polymerase. CCR in B. subtilis is triggered by the increase in intracellular glucose phosphate. As a result of glycolysis, fructose 1,6 phosphate also increases and stimulates the Hpr kinase, which phosphorylates the Hpr protein in a serine residue. Phosphorylated Hpr binds CcpA, and together prevent RNA polymerase from initializing transcription of genes involved in lignocellulose catabolism. Created with BioRender.com.
In Gram-negative bacteria such as E. coli, the phosphorylation state of the glucose-specific EIIAGlc protein regulates the activity of adenylyl cyclase and, consequently, the level of cAMP in the cell. When a rapidly metabolizable carbohydrate such as glucose is available, EIIAGlc is mostly in the unphosphorylated form. The contrary occurs at low concentrations of glucose. In the phosphorylated form, EIIAGlc stimulates adenylyl cyclase, and the intracellular cAMP concentration increases. The catabolite activator protein (CAP also known as cAMP receptor protein CRP), which needs to interact with cAMP to bind to DNA, recognizes its target DNA sequences and expression of CCR-sensitive operons is induced. At sufficiently high levels, cAMP binds to the transcriptional regulator CRP, which induces binding to specific DNA sequences in the promoter region of the target genes, where it activates the initiation of transcription through interaction with the polymerase (Kremling et al. 2015). In the Gram-negative F. succinogenes S85, the intracellular concentration of cyclic-di-GMP increases when cells are grown in cellulose, suggesting that this mediator is involved in the regulation of cellulose degradation by CCR (Raut et al. 2019).
In Gram-positive mesophilic bacteria such as B. subtilis, the signaling intermediate is HPr and not EIIAGlc (Warner and Lolkema 2003, Lin and Xu 2013). HPr(Ser-P) binds to the transcriptional regulator CcpA, inducing the recognition of the CRE sites (catabolite responsive element) in the genome. The binding of this complex to target sequences prevents transcription of genes.
Inducers and regulators of transcription of cellulolytic genes
While glucose is a repressor of fungal cellulolytic enzymes, cellobiose, sophorose, lactose, and oligosaccharides may act as inducers in filamentous fungi. However, transglycosylation of glucose to another sugar could lead to induction instead of repression (Behera et al. 2017). The disaccharide sophorose, probably produced from cellobiose by the transglycosylation activity of β-glucosidase, is one of the most powerful inducers in T. reesei. Induction and repression may depend on the concentration of the sugar, as shown by xylose in Aspergillus niger and T. reesei. Preferred carbon sources vary among fungal species, as well as their catalytic efficiency toward the same substrate. For example, A. nidulans is less effective in degrading crystalline cellulose than N. crassa and is specifically more proficient at inducing xylanases, ß-mannanases, and pectate lyases (Coradetti et al. 2013). Major transcriptional regulators of cellulose- and hemicellulose-degrading enzymes in ascomycetes include the homologs XlnR/XYR1/XLN1, homologs CLR-1/ClrA, and homologs CLR-2/ClrB (Benocci et al. 2017). XlnR/XYR1/XLN1 may control xylanases, cellulases, or both depending on the species. Some regulators are species specific, such as ACEII (activator of cellulase expression) found in T. reesei or use different regulatory strategies depending on the strain (Rosolen et al. 2022). Additionally, an interplay between regulators has been reported (Mattam et al. 2022). The ClbR (cellobiose response regulator) of Aspergillus aculeatus regulates CAZyme encoding genes in response to cellulose and cellobiose both in an XlnR-dependent and -independent manner (Coradetti et al. 2013).
In cellulolytic anaerobes, CCR of cellulosomes is induced by glucose and cellobiose among others (Koeck et al. 2014), and as seen for xylose in filamentous fungi, it seems to be dose dependent in certain cellulolytic species (Xu et al. 2013, Li et al. 2017). Bacterial repressors of cellulolytic genes are XylR found in Bacillus (Rodionov et al. 2001), and TmcR in Myxobacter sp. (Ramírez-Ramírez et al. 2009). Additionally, two-component systems (TCSs) have been implicated in regulating both cellulosomal (xyl-doc cluster) and noncellulosomal CAZymes and associated transporters in C. cellulolyticum (Xu et al. 2013).
In Streptomycesspp., CCR is not under strict control of the PTS, but other regulatory systems including glucose kinase and transcription regulators such as CebR, DasR, and ArgR (Romero-Rodríguez et al. 2017). The CebR system causes repression in the presence of glucose and induction in the presence of cellulose oligosaccharides in Streptomyces spp. (Anderson et al. 2012, Book et al. 2016). The operon regulated by CebR in Streptomyces reticuli consists of an ABC transporter Ceb, integral membrane proteins CebF and CebG, and the lipoprotein CebE that binds specifically cellobiose and cellotriose. CebR binds to the operator in the presence of glucose and is released in the presence of cellopentaose. A limited number of Streptomyces spp. encode this operon (Book et al. 2016). Additionally, CebR binding specificity for cellulose oligomers may be different among species (Schrempf 2017). The expression of cellulolytic PMOs is also regulated by CebR in the presence of cellulose. A similar mechanism involving the regulator DasR regulates the expression of chitinolytic PMOs (Agostoni et al. 2017). However, PMOs have not been found to be part of operons.
Regulatory elements binding polysaccharides in cellulolytic bacteria: anti-sigma factors
Cellulosomal genes are expressed at higher levels in the presence of complex natural polymers than in the presence of simple oligosaccharides, and, at high growth rate, cellulosomal genes are downregulated. This requires substrate sensing and the coordinated expression of relevant cellulosomal genes. In C. thermocellum, some cellulosomal genes are regulated by several anti-sigma factors and alternative sigma factors (Kahel-Raifer et al. 2010, Nataf et al. 2010). In the absence of substrate, the transmembrane anti-sigma factor is attached to an alternative sigma factor in the cell. Each anti–sigma factor also has an exocellular CBM-like component that binds the polysaccharide substrate in the external medium. Binding of an appropriate substrate to this CBM results in a conformational change of the anti–sigma factor that releases the alternative sigma factor, which then interacts with RNA polymerase, thereby initiating transcription of cellulosomal genes. Ortiz de Ora et al. (2018) showed how a collection of five alternative sigma factors, namely σI1, σI2, σI3, σI4, and σI6, regulates the expression of 17 genes encoding different cellulosomal components in this species. More recently, it was shown that the cellulolytic bacterium Cytophaga hutchinsonii senses cellulose by the sigma factor σcel1 and regulates cellulose utilization by a unique mechanism that has not been fully elucidated yet (Wang et al. 2019a).
Microbial posttranscriptional regulatory mechanisms
Antisense RNAs are single-stranded RNA molecules complementary to coding mRNA transcripts. These antisense molecules impede translation when binding its complementary transcript. Cis natural antisense transcripts (cis-NATs) regulate the catabolic response in anaerobic lignocellulolytic fungi (Solomon et al. 2018). As part of the CCR in T. reesei, long noncoding RNAs interact with proteins such as the transcriptional factor Xyr1 involved in xylanase expression (Till et al. 2020).
In bacterial genomes, functionally related genes are frequently organized as an operon. They are usually transcribed in the same polycistronic mRNA molecule. Regulatory mechanisms such as the selective RNA processing and stabilization (SRPS) are based on the modulation of the stability of mRNA transcripts to either prevent or enable its degradation, thus increasing or decreasing expression, respectively (Fig. 5a). The cip-cel operon and the xyl-cel operon encoding genes of the cellulosomes of C. cellulolyticum are an example of regulation by SRPS (Xu et al. 2015a). In these operons, a secondary structure of the transcript called stem-loop confers stability to mRNAs. Small loops favor the degradation of the transcript and decrease the expression of the encoded gene. In this manner, those transcripts forming proper loops will allow the highest expression. Additionally, different sorts of RNA regulatory elements have been found taking part in CCR of cellulolytic anaerobes. In Clostridium papyrosolvens, the 5′-UTR (untranslated region) of the cellulosomal xyl-doc cluster blocks transcription. Since some substrates such as corn stover can release the repression of 5′-UTR, it is speculated that 5′-UTR of xyl-doc is a putative riboswitch regulating the expression of downstream cellulosomal genes (Zou et al. 2018).
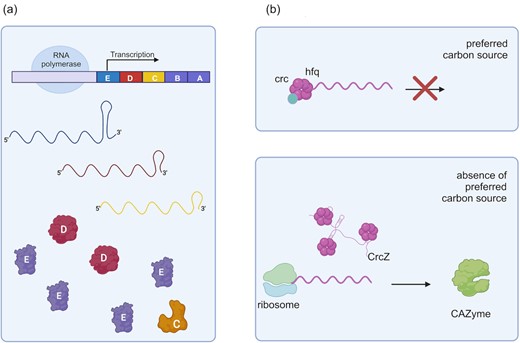
RNA bacterial regulatory pathways for cellulolytic genes. (a) The cip-cel operon of C. cellulolyticum is regulated by SRPS. Mature transcripts are stabilized by terminal stem-loops. Transcript degradation depends on the sequence and consequent secondary structure of the terminal mRNA stem-loop. Unstable mRNA (yellow) is more prone to degradation. Consequently, expression of the corresponding gene (C) will be decreased. (b) In Pseudomonas aeruginosa, CCR is mainly exerted by RNA regulatory pathways. When the preferred carbon source is available, transcripts of alternative carbon sources are inhibited by the chaperon Hfq associated or not with protein Crc. When the preferred carbon source is not available, the small regulatory RNA (sRNA) crcZ sequesters Hfq and releases transcripts allowing binding of ribosomes and further translation. Created with BioRender.com.
Resembling cis-NATs described for lignocellulolytic fungi, bacterial cis-encoded small regulatory RNAs (sRNAs) are transcribed from the opposite strand of their target genes and are complementary to their target transcripts. Polysaccharide utilization loci (PULs) encode genes dedicated to the uptake and utilization of a specific glycan or polysaccharide and include their own protein regulators for substrate-dependent induction of the locus (Grondin et al. 2017). Many of these PULs transcribe sRNAs from the opposite strand in Bacteroides spp. As an example, overexpression of DonS, an sRNA of Bacteroides fragilis, triggers the loss of the complementary PUL transcript, impeding the cell to utilize host glycans when dietary polysaccharides are available (Durica-Mitic et al. 2018).
Trans-encoded sRNAs regulate distantly encoded targets that can be either RNA or protein. Sgrs is an example of RNA-binding sRNA. High intracellular concentrations of phosphosugars induce the transcription of the sgrS gene in E. coli (Durica-Mitic et al. 2018). SgrS binds to the ptsG transcript, which encodes the glucose transporter gene ptsG, and facilitates its degradation, thus limiting glucose uptake and avoiding toxicity of phosphosugars in the PTS pathway of CCR (Fig. 4). Another example is the sRNA Spot 42, which binds transcripts required for utilization of alternative carbon sources by E. coli (Bækkedal and Haugen 2015). As previously mentioned, Hfq is a protein serving as a conserved RNA chaperon. Hfq can bind adenylate uridylate (AU-rich) sequences of target mRNA and facilitate the pairing interaction between mRNA and sRNA. Mutation of Hfq in Lysobacter enzymogenes almost abolished extracellular chitinase activity and significantly reduced the activity of both extracellular protease and cellulase while increasing the biosynthesis of secondary metabolites (Xu et al.2015b). In Gram-negative bacteria, trans-encoded sRNAs often require the assistance and protection of the Hfq protein. In Pseudomonas spp., CCR is regulated exclusively at the posttranscriptional level (Sonnleitner et al. 2009). Contrary to E. coli, Pseudomonas spp. do not prefer glucose. When the preferred carbon source is available, the protein Hfq binds the 5′-UTRs of mRNAs encoding transporters and catabolic enzymes for less-preferred carbon sources and directly blocks translation initiation (Fig. 5b). This complex is stabilized by the protein Crc. In the absence of the preferred carbon source, the expression of sRNA antagonists to Hfq is induced by a TCS, causing derepression of the cellulolytic transcripts (Pusic et al. 2021).
Posttranslational regulation in filamentous fungi
During the lignocellulolytic response in filamentous fungi, nascent cellulolytic enzymes are sent to the endoplasmic reticulum (ER) prior to secretion. The increased demand for protein folding and further posttranslational modifications may cause significant ER stress. Proteins incorrectly folded are prone to proteasomal degradation via the ER-associated degradation pathway. To avoid that, several regulatory feedback mechanisms are activated in the presence of cellulose or degrading products such as cellobiose, to reach the level of enzyme secretion required for plant cell wall degradation (Huberman et al. 2016; Xiong et al. 2021). In that manner, the activation of Ire1 and hac-1 upregulates a broad set of genes necessary to adapt the ER folding capacity to demand in the unfolded protein response. To note, this mechanism is also activated when S. cerevisiae expresses heterologous cellobiohydrolases (Ilmén et al. 2011). Besides, several components of the ER secretory pathway have been engineered in T. reesei to increase the production of cellulolytic enzymes (Shen et al. 2021).
Regulation by environmental factors
Carbon utilization depends mainly on environmental factors. Availability of carbon, nitrogen, and phosphate, as well as pH, osmolarity, and temperature, controls the life of most microorganisms. Therefore, it is expected that regulatory elements responsive to these factors display the highest hierarchy in the control of all cellular processes, including lignocellulose degradation.
Regulation of cellulose degradation by pH in filamentous fungi
In filamentous fungi, environmental pH is detected through a plasma membrane complex via a signaling cascade in which Pal proteins and the zinc finger transcription factor PacC or its homologs are involved (Tilburn et al. 1995, Peñalva et al. 2014). The Pal proteins sense alkaline pH in the environment and transmit a signal that leads to the activation of PacC. Activated PacC regulates the expression of several genes to adapt to the alkaline environment in filamentous fungi, including those related to xylan degradation (Peñalva and Arst 2002). This transcription factor regulates not only several CAZyme genes but also associated transcription factors in fungi (Wang et al. 2020). PacC is the pH response effector in the induction of cellulase and xylanase encoding genes in the ascomycete Humicola grisea, sharing regulatory pathways with CreA (Mello de Sousa et al. 2011). The same was observed in A. nidulans, where most genes under the control of ClrB were positively regulated by PacC (Kunitake et al. 2016). In T. reesei, only a few holocellulase genes that are differently expressed depending on pH were regulated by PACI, the ortholog of PacC. In this strain, an interplay with other transcription factors also seemed to be occurring, since the transcription of regulatory genes xyr1 and ace2 increased in the pac1 null mutant (He et al. 2014). In the same manner, the pH regulator PAC-3 in N. crassa was involved in the regulation of holocellulolytic genes and would be linked to the transcription of regulatory genes xlr-1,cre-1,clr-1, andclr-2 as well (Campos Antoniêto et al. 2017).
Aminoacid starvation in filamentous fungi and yeast
Some global regulators were found in regulatory pathways of both nitrogen and carbon metabolism. The global regulator Gcn4 controlling the expression of aminoacid biosynthetic genes that are essential under aminoacid starvation is upregulated when the preferred carbon source is absent in S. cerevisiae. CPC-1 and CpcA are orthologs of Gcn4 in N. crassa and Aspergillusspp., respectively. This global regulator showed to be essential for cellulose utilization in filamentous fungi. The CpcA coding gene of the cellulase-producing fungus Penicillium oxalicum is required for rapid induction of cellulase synthesis during the early stage of cellulose utilization (Pan et al. 2020). A N. crassa Δcpc-1 mutant showed reduced ability to grow on cellulose and reduced cellulolytic activity (Tian et al. 2007, Schmoll et al. 2012). Another example of interplay between carbon and nitrogen regulatory pathways was observed in A. nidulans, where CpcA and CreA are both under the control of AreB, a regulator of genes involved in the metabolism of nitrogen (Chudzicka-Ormaniec et al.2019).
Bacterial global regulators
The carbon control protein CcpA, a pleiotropic regulator in C. cellulolyticum, represses xylose utilization genes when glucose is present (Ren et al. 2010), and is putatively involved in the regulation of the cip-cel operon (Abdou et al. 2008). In an evolutionary study, Warner and Lolkema (2003) suggested similarities between the CcpA-dependent CCR and the nitrogen regulation system (Ntr) in Proteobacteria. Although the link between regulation of carbon and nitrogen metabolism remains obscure, some common regulatory elements could be elucidated. To note, transcription of several genes involved in nitrogen assimilation, especially under nitrogen-limiting conditions, depends upon the sigma factor 54 encoded by the rpoN gene (Deutscher et al. 2006). Transcription initiation at sigma 54-dependent promoters requires the interaction with specific transcription activators such as NtrC or NifA, some of which can be activated by PTS-mediated phosphorylation via P-Ser-HPr, a regulatory element involved in bacterial CCR (Fig. 4).
In the actinobacterium Saccharopolyspora erythraea, the global regulators GlnR and PhoP bound the promoters of β-glucosidase encoding genes involved in cellulose digestion in response to nitrogen and phosphate availability (Xu and Ye 2018, Wei et al. 2019). PhoP is also a global regulator controlling transcription of cellulases in Xanthomonas spp. Moreover, genomic analyses showed that regulation of cellobiose utilization by GlnR and PhoP is likely conserved in actinobacteria. Additional bacterial global regulators involved in the control of cellulolytic genes are RpoS in the plant pathogenic Erwiniaspp. (Mukherjee et al. 1998), RbsR in Serratia (Lee et al. 2017), and HpaR1 and Clp in Xanthomonas (Liu et al. 2019).
Conclusions
Most of the current knowledge on microbial biomass degradation is based on model organisms such as the filamentous fungi A. nidulans, T. reesei, N. crassa, the yeast S. cerevisiae, and bacteria B. subtilis, E. coli, and C. cellulolyticum. This is a clearly small sample size if we consider the immense microbial diversity. Thus, we would not be far from truth if we state that most of regulatory mechanisms for biomass deconstruction are currently obscure and unknown. However, many regulatory transcriptional factors have been identified, especially in filamentous fungi, and their hierarchies in the control of cellular processes are beginning to be elucidated. The different forms of RNA as regulatory elements constitute an exciting and rapidly evolving field of research, not only for the novelty of such systems but also for the possibility of engineering those pathways in CBP platforms. While these pathways and regulatory elements begin to be discovered, there is exciting evidence of successfully engineered regulatory systems in industrial filamentous fungi for a higher yield of cellulolytic enzymes (Liu and Qu 2021). More work on this field will clarify which microorganisms provide the best chassis for engineering and which regulatory pathways are easily manipulated for a more efficient and faster biomass degradation, to finally assess a greener and sustainable production of biofuels and high-value chemicals.
Acknowledgments
This study was supported by grants PUE2017 CERZOS-CONICET and PGI 24/B294 SeGCyT-UNS (to M.S.V.G.). F.R.V. acknowledges CONICET for the doctoral degree scholarship and J.D. acknowledges the National Interuniversity Council of Argentina for the advanced student's scholarship.
Conflict of interest
No conflict of interest declared.
Author contributions
All authors made substantial contributions to conception and design or the acquisition and analysis of data, drafted or critically revised the manuscript, and approved the final submitted version. M.S.V.G. proposed the topic and designed the manuscript. F.R.V. and M.A.B. reviewed information on regulatory mechanisms for lignocellulose deconstruction in yeast, mushrooms, and filamentous fungi. M.S.V.G. and J.D. reviewed literature on bacterial regulatory pathways.
Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}