





Abstract
Uropathogenic Escherichia coli (UPEC) are a major cause of urinary tract infection (UTI), one of the most common infectious diseases in humans. UPEC are increasingly associated with resistance to multiple antibiotics. This includes resistance to third-generation cephalosporins, a common class of antibiotics frequently used to treat UTI.
We employed a high-throughput genome-wide screen using saturated transposon mutagenesis and transposon directed insertion-site sequencing (TraDIS) together with phenotypic resistance assessment to identify key genes required for survival of the MDR UPEC ST131 strain EC958 in the presence of the third-generation cephalosporin cefotaxime.
We showed that blaCMY-23 is the major ESBL gene in EC958 responsible for mediating resistance to cefotaxime. Our screen also revealed that mutation of genes involved in cell division and the twin-arginine translocation pathway sensitized EC958 to cefotaxime. The role of these cell-division and protein-secretion genes in cefotaxime resistance was confirmed through the construction of mutants and phenotypic testing. Mutation of these genes also sensitized EC958 to other cephalosporins.
This work provides an exemplar for the application of TraDIS to define molecular mechanisms of resistance to antibiotics. The identification of mutants that sensitize UPEC to cefotaxime, despite the presence of a cephalosporinase, provides a framework for the development of new approaches to treat infections caused by MDR pathogens.
Introduction
Uropathogenic Escherichia coli (UPEC) are a major cause of urinary tract infection (UTI), a disease of major significance to global human health.1 UPEC (and other UTI pathogens) are increasingly associated with resistance to multiple antibiotics, with strains that produce ESBLs conferring resistance to third-generation cephalosporins such as cefotaxime, ceftriaxone and ceftazidime, as well as the oxyimino-monobactam aztreonam, being frequently isolated. This has led to an increased rate of UTI treatment failure where conventional antibiotics have been used and has increased our dependence on last-line therapies such as carbapenems. With few new antibiotics in development, identifying new antibacterial targets and novel agents to treat infections caused by these pathogens is an urgent priority.
Many MDR UPEC strains belong to specific clones that are frequently isolated from urinary tract and bloodstream infections. E. coli ST131 is the most globally dominant MDR UPEC clone and causes infections associated with limited treatment options. ST131 was originally identified in 2008 as a major clone linked to the spread of the CTX-M-15 ESBL gene,2 encoding the most widespread CTX-M ESBL enzyme worldwide. ST131 strains have now been identified in both hospital and community settings from all parts of the globe. The largest subclonal lineage of E. coli ST131 is resistant to fluoroquinolones and contains the type 1 fimbriae fimH30 (H30) allele.2 The temporal and spatial relationships of a global collection of ST131 strains were recently investigated and highlighted the important role of mobile genetic elements and recombination in the evolution of this pathogenic lineage.3–7 The emergence of ST131 strains resistant to last-line carbapenems, due to acquisition of the blaNDM-1 MBL gene, has also been described,8,9 highlighting the alarming scenario of the spread of a highly successful UPEC clone that is resistant to all current effective therapy.
EC958 represents one of the most well-characterized ST131 strains in the literature. EC958 is a phylogenetic group B2, CTX-M-15-positive, fluoroquinolone-resistant, H30 ST131 strain isolated from the urine of an 8-year-old girl presenting in the community in March 2005 in the UK.10 The strain belongs to the PFGE-defined UK epidemic strain A and has an O25b:H4 serotype. EC958 contains multiple genes associated with UPEC virulence, including genes encoding adhesins, autotransporter proteins and siderophore receptors. EC958 expresses type 1 fimbriae and this is required for adherence to and invasion of human bladder cells, as well as colonization of the mouse bladder.10 In mice, EC958 causes acute and chronic UTI,11 as well as impairment of ureter contractility.12 EC958 is also resistant to the bactericidal action of human serum and we previously used transposon directed insertion-site sequencing (TraDIS) to characterize the complement of genes that define this phenotype.13
Cephalosporins are bactericidal β-lactam antibiotics. There are currently five generations of cephalosporins, with each generation indicated for different clinical uses. The third-generation cephalosporins are recommended for treatment of infections caused by Haemophilus influenzae and Enterobacteriaceae including E. coli, Klebsiella pneumoniae and Proteus mirabilis that do not produce ESBLs or the AmpC β-lactamase. Cefotaxime, a third-generation cephalosporin, is a bactericidal antibiotic that acts by inhibiting bacterial cell wall synthesis, targeting PBP3 (also known as FtsI).14,15 It has excellent activity against many Gram-negatives and an extremely low toxicity profile.14 Cefotaxime was used to treat acute UTIs caused by E. coli,16 but its effectiveness is now limited due to the increasing prevalence of CTX-M-15-positive ST131.
Here we employed a combination of saturated transposon mutagenesis and TraDIS together with a phenotypic resistance screen to discover genes whose inactivation sensitizes CTX-M-15-positive ST131 to the third-generation cephalosporin cefotaxime. Our analysis identified genes associated with cell division and the twin-arginine translocation (Tat) pathway, and their function was confirmed through the construction of mutants and phenotypic testing. Furthermore, we showed that these genes also contribute to resistance to other third-generation cephalosporins. Overall, our approach demonstrates the power of genome-wide screening as a tool for the discovery of novel genetic determinants that could be targeted for the development of new antimicrobials.
Materials and methods
Bacterial strains and growth conditions
The E. coli strain EC958 was isolated from the urine of a patient with community-acquired UTI in the UK.10 EC958 belongs to ST131 subclade C2 or H30Rx3,4 and is resistant to fluoroquinolones and third-generation cephalosporins.17 Bacterial strains were cultured in lysogeny broth (LB) or on LB agar with appropriate selection (chloramphenicol 30 mg/L) as required.
Transposon library screening with cefotaxime and multiplex TraDIS
Approximately 2 × 108 bacterial cells from the EC958 miniTn5-Cm library13 were inoculated into 100 mL of LB either with (test) or without (control) 4 mg/L cefotaxime. After 18 h of shaking incubation at 37°C, 1 mL of culture was removed for genomic DNA extraction using the Ultraclean Microbial DNA isolation kit (MoBio Laboratories). The screening assays were performed in duplicate. Genomic DNA from test and control samples was subjected to TraDIS analysis as described previously.18 In brief, the Illumina libraries were prepared using the Nextera DNA Sample Prep Kit (Illumina) following the manufacturer’s instructions, with modifications for Tn5-insertion-site sequencing, and sequenced with 100 bp single-end reads on the Illumina MiSeq platform at the IMB Sequencing Facility, University of Queensland.
Analysis of TraDIS data
Raw sequencing reads were filtered and trimmed to keep 30 bp immediately after the 12 bp Tn5-specific tag (5′-TATAAGAGACAG-3′) at their 5′ ends using FASTX-Toolkit version 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/index.html). These reads were aligned to the EC958 genome (HG941718) using Maq ‘easyrun’ (version 0.7.1).19 Subsequent analysis steps were carried out as previously described20 to calculate the number of sequence reads (raw read counts) and the number of different insertion sites for every gene. The read counts and insertion sites were visualized using Artemis version 13.0.21 Subsequent analysis steps were carried out in R (version 3.0.3) using the edgeR package (version 3.4.2)22 to estimate the log2 fold change (logFC) of read counts and false discovery rate (FDR) for each gene between test and control samples. To identify genes required for survival in the presence of cefotaxime, we used stringent criteria of logFC ≤ −2 and FDR ≤ 0.001. The full output from edgeR is included in Dataset S2 (available as Supplementary data at JAC Online), with the position and orientation of each insertion site listed in Dataset S3. The TraDIS reads have been deposited in the SRA under accession number PRJNA525739.
Molecular methods
Chromosomal DNA purification, PCR and DNA sequencing of PCR products were performed as previously described.18 Defined mutations were made using the λ-Red recombinase method with some modifications.23–25 The full list of primers used is outlined in Dataset S1.
Growth analyses
Bacterial growth kinetics were measured as changes in OD600 over time using the FLUOstar OPTIMA Microplate Reader (BMG Labtech). Briefly, overnight cultures were standardized to OD600 = 0.05 in LB or LB supplemented with 4 mg/L cefotaxime and loaded in triplicate onto a flat-bottom 96-well plate at 200 μL per well. The plate was then sealed using the Breathe-Easy sealing membrane (Diversified Biotech) and incubated at 37°C with shaking inside the microplate reader. Optical measurements were recorded every 15 min for 12 h; all experiments were performed in triplicate.
Antimicrobial susceptibility testing
MICs were determined by Etest (bioMérieux Australia) on Mueller–Hinton agar at 37°C. The procedure and interpretation of MICs were performed as recommended by the manufacturer using EUCAST breakpoints (http://www.eucast.org/clinical_breakpoints/). All MIC assays were performed in triplicate.
Live-cell microscopy
Live-cell microscopy was performed as previously described.26 Briefly, EC958 and mutant derivatives were inoculated into LB to a starting OD600 of 0.05. Cultures were incubated at 37°C with vigorous shaking for 1.5 h (OD600 ∼ 0.3). Cefotaxime was added to cultures to a final concentration of 4 mg/L and cultures were incubated for a further 1 h. Samples were collected and prepared for live-cell microscopy as previously described26 and visualized on a 2% agarose pad. Cells were observed by phase contrast using a Zeiss Axioplan 2 fluorescence microscope equipped with a Plan ApoChromat (100× NA 1.4; Zeiss) objective lens and an AxioCam MRm cooled charged-coupled-device (CCD) camera. Cell length values were quantified using AxioVision software version 4.6 (Zeiss) with default measurement settings.
Results
Identification of genes contributing to cefotaxime resistance
EC958 is resistant to cefotaxime and we exploited this by performing a genome-wide screen employing a saturated mutant library, TraDIS and resistance testing to define the set of genes that contribute to this phenotype. We used a screening procedure in which 1 million EC958 transposon mutants were exposed to a subinhibitory concentration of 4 mg/L cefotaxime for 16 h in LB before genomic DNA extraction (Figure 1a). This permitted the growth of mutants unaffected by cefotaxime, while killing mutants that were susceptible to cefotaxime. The genomic DNA from test and control samples was sequenced using our Illumina multiplexing TraDIS procedure and analysed as described previously.13 Cefotaxime-susceptible genes were identified as genes that had a significant reduction in read counts (i.e. fewer transposon insertions) in the test samples compared with the control samples (logFC ≤ −2.0; FDR ≤ 0.001). In total, 13 cefotaxime-susceptible genes were identified that satisfied this stringent criterion (Figure 1b, indicated in red). The visualization of location and orientation of insertion sites within each identified gene and its flanking regions is shown in Figure S1. Of the five β-lactamase genes present in EC958 (blaTEM-1, blaOXA-1, blaCTX-M-15, blaCMY-23 and a chromosomal ampC gene), only the blaCMY-23 gene was identified in our screen, demonstrating that blaCMY-23 is the main cefotaxime resistance determinant in EC958.
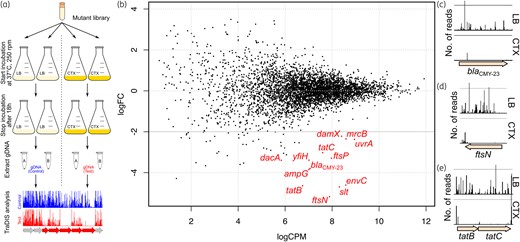
(a) Experimental design of the forward genetic screen. A highly saturated EC958 transposon mutant library was incubated in the presence of 4 mg/L cefotaxime for 16 h and then cells were harvested, genomic DNA (gDNA) was extracted and analysed by TraDIS. (b) Genes identified by TraDIS to be associated with susceptibility of EC958 to cefotaxime. Shown in red are the significant genes required for survival in the presence of cefotaxime. (c–e) Examples of the difference in Tn5 insertion for the (c) blaCMY-23, (d) ftsN and (e) tatB and tatC genes. A significant decrease in the Tn5 insertion frequency (expressed as number of reads) was observed following growth in LB containing 4 mg/L cefotaxime (bottom) compared with LB (top). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Our TraDIS screen revealed a striking association between cefotaxime resistance and genes required for cell wall synthesis and cell division (Table 1). Indeed, the set of genes identified encodes proteins that form multiple interactions with each other; they are involved in the later stages of division, or are required for either correct formation of the division protein complex, peptidoglycan synthesis or maturation of peptidoglycan.27,28 Furthermore, we also identified two genes in the Tat secretion system, a finding consistent with its role in the translocation of FtsP, a key protein required for protection of the divisome during stress29 (and also identified in our screen).
List of genes identified by TraDIS that, when mutated, led to enhanced susceptibility to cefotaxime
| Gene | logFC | Function of protein |
|---|---|---|
| ftsN | −5.17 | cell-division protein, mid-cell localization, binds peptidoglycan |
| Slt | −4.70 | soluble lytic transglycosylase, cleaves the β-1,4-glycosidic bonds between GlcNAc and MurNAc in the sugar backbone of peptidoglycan |
| tatB | −4.64 | TatB subunit in TatABCE protein translocation system |
| envC | −4.61 | activates division-specific amidases AmiA and AmiB, localizes to septal ring |
| blaCMY-23 | −3.85 | β-lactamase/d-alanine carboxypeptidase, interacts with DnaK, linked to cell division and filamentation in E. coli |
| ampG | −3.80 | muropeptide transporter, involved in β-lactamase induction and peptidoglycan recycling |
| dacA | −3.39 | d-alanyl-d-alanine carboxypeptidase (PBP5), may contribute to regulation of peptidoglycan crosslinking |
| yfiH | −3.39 | polyphenol oxidase, plays a role in maintaining the peptide composition of peptidoglycan34 |
| ftsP | −3.29 | septal ring component that protects the divisome from stress, exported by the Tat system |
| tatC | −3.02 | TatC subunit in TatABCE protein translocation system |
| uvrA | −2.37 | ATPase and DNA damage-recognition protein |
| mrcB | −2.35 | glycosyl transferase with transpeptidase activity |
| damX | −2.22 | cell-division protein, localizes to septal ring, binds to peptidoglycan; mediator of reversible filamentation in UPEC55 |
| Gene | logFC | Function of protein |
|---|---|---|
| ftsN | −5.17 | cell-division protein, mid-cell localization, binds peptidoglycan |
| Slt | −4.70 | soluble lytic transglycosylase, cleaves the β-1,4-glycosidic bonds between GlcNAc and MurNAc in the sugar backbone of peptidoglycan |
| tatB | −4.64 | TatB subunit in TatABCE protein translocation system |
| envC | −4.61 | activates division-specific amidases AmiA and AmiB, localizes to septal ring |
| blaCMY-23 | −3.85 | β-lactamase/d-alanine carboxypeptidase, interacts with DnaK, linked to cell division and filamentation in E. coli |
| ampG | −3.80 | muropeptide transporter, involved in β-lactamase induction and peptidoglycan recycling |
| dacA | −3.39 | d-alanyl-d-alanine carboxypeptidase (PBP5), may contribute to regulation of peptidoglycan crosslinking |
| yfiH | −3.39 | polyphenol oxidase, plays a role in maintaining the peptide composition of peptidoglycan34 |
| ftsP | −3.29 | septal ring component that protects the divisome from stress, exported by the Tat system |
| tatC | −3.02 | TatC subunit in TatABCE protein translocation system |
| uvrA | −2.37 | ATPase and DNA damage-recognition protein |
| mrcB | −2.35 | glycosyl transferase with transpeptidase activity |
| damX | −2.22 | cell-division protein, localizes to septal ring, binds to peptidoglycan; mediator of reversible filamentation in UPEC55 |
A full list of genes is presented in Dataset S2.
logFC, logFC of number of reads mapped to each gene from screens with/without cefotaxime.
List of genes identified by TraDIS that, when mutated, led to enhanced susceptibility to cefotaxime
| Gene | logFC | Function of protein |
|---|---|---|
| ftsN | −5.17 | cell-division protein, mid-cell localization, binds peptidoglycan |
| Slt | −4.70 | soluble lytic transglycosylase, cleaves the β-1,4-glycosidic bonds between GlcNAc and MurNAc in the sugar backbone of peptidoglycan |
| tatB | −4.64 | TatB subunit in TatABCE protein translocation system |
| envC | −4.61 | activates division-specific amidases AmiA and AmiB, localizes to septal ring |
| blaCMY-23 | −3.85 | β-lactamase/d-alanine carboxypeptidase, interacts with DnaK, linked to cell division and filamentation in E. coli |
| ampG | −3.80 | muropeptide transporter, involved in β-lactamase induction and peptidoglycan recycling |
| dacA | −3.39 | d-alanyl-d-alanine carboxypeptidase (PBP5), may contribute to regulation of peptidoglycan crosslinking |
| yfiH | −3.39 | polyphenol oxidase, plays a role in maintaining the peptide composition of peptidoglycan34 |
| ftsP | −3.29 | septal ring component that protects the divisome from stress, exported by the Tat system |
| tatC | −3.02 | TatC subunit in TatABCE protein translocation system |
| uvrA | −2.37 | ATPase and DNA damage-recognition protein |
| mrcB | −2.35 | glycosyl transferase with transpeptidase activity |
| damX | −2.22 | cell-division protein, localizes to septal ring, binds to peptidoglycan; mediator of reversible filamentation in UPEC55 |
| Gene | logFC | Function of protein |
|---|---|---|
| ftsN | −5.17 | cell-division protein, mid-cell localization, binds peptidoglycan |
| Slt | −4.70 | soluble lytic transglycosylase, cleaves the β-1,4-glycosidic bonds between GlcNAc and MurNAc in the sugar backbone of peptidoglycan |
| tatB | −4.64 | TatB subunit in TatABCE protein translocation system |
| envC | −4.61 | activates division-specific amidases AmiA and AmiB, localizes to septal ring |
| blaCMY-23 | −3.85 | β-lactamase/d-alanine carboxypeptidase, interacts with DnaK, linked to cell division and filamentation in E. coli |
| ampG | −3.80 | muropeptide transporter, involved in β-lactamase induction and peptidoglycan recycling |
| dacA | −3.39 | d-alanyl-d-alanine carboxypeptidase (PBP5), may contribute to regulation of peptidoglycan crosslinking |
| yfiH | −3.39 | polyphenol oxidase, plays a role in maintaining the peptide composition of peptidoglycan34 |
| ftsP | −3.29 | septal ring component that protects the divisome from stress, exported by the Tat system |
| tatC | −3.02 | TatC subunit in TatABCE protein translocation system |
| uvrA | −2.37 | ATPase and DNA damage-recognition protein |
| mrcB | −2.35 | glycosyl transferase with transpeptidase activity |
| damX | −2.22 | cell-division protein, localizes to septal ring, binds to peptidoglycan; mediator of reversible filamentation in UPEC55 |
A full list of genes is presented in Dataset S2.
logFC, logFC of number of reads mapped to each gene from screens with/without cefotaxime.
Validation of genes identified by TraDIS
To validate the genes identified by TraDIS, we constructed isogenic mutants containing deletions in these genes in EC958 using λ-Red-mediated homologous recombination. These mutants were tested for growth in LB in the presence of 4 mg/L cefotaxime to confirm their growth-impaired phenotype. Various levels of growth attenuation were observed; the most notable defects were found in EC958Δslt, EC958ΔblaCMY-23, EC958ΔyfiH, EC958ΔftsP, EC958ΔtatC, EC958ΔuvrA, EC958ΔmrcB and EC958ΔdamX (Figure 2).
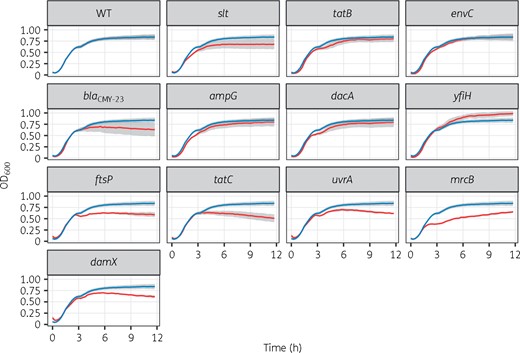
Growth curves of isogenic mutants for genes identified by TraDIS. Growth kinetics of WT (blue line) versus mutants (red line) in LB containing 4 mg/L cefotaxime. Data (n = 3) were combined from three independent experiments, presented as mean (solid line) ± SEM (grey shades). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Increased susceptibility to cephalosporins
To understand the contribution of each gene identified by TraDIS to cefotaxime resistance, we determined the MIC for each of the mutants (Table 2). These analyses revealed that mutation of blaCMY-23 resulted in the greatest increase in susceptibility, with mutation of tatB, tatC, dacA, yfiH and ftsP also leading to significant but lower levels of increased susceptibility. Mutants that did not display a difference in MIC of cefotaxime (EC958ΔenvC, EC958ΔampG, EC958ΔuvrA, EC958ΔdamX, EC958Δslt and EC958ΔmrcB) were also examined in mixed competitive 50:50 growth assays with WT EC958 in the presence of 4 mg/L cefotaxime. In these assays, only EC958Δslt was significantly outcompeted by WT EC958 (Figure S2). Next, we extended this analysis by testing our mutants for resistance to four additional cephalosporins: cefoxitin (second generation), ceftazidime (third generation), cefepime (fourth generation) and ceftaroline (fifth generation). Mutation of blaCMY-23 resulted in increased susceptibility to all four antibiotics (Table 2). All mutants exhibited increased susceptibility to at least one cephalosporin drug: mutation of tatB, tatC, dacA, yfiH and ftsP led to increased susceptibility to ceftazidime, cefepime and ceftaroline; and mutation of ampG and mrcB led to increased susceptibility to ceftazidime, cefepime and ceftaroline. There were two genes, uvrA and damX, whose mutation led to reduced susceptibility to only one drug (ceftazidime).
MICs (mg/L) of EC958 mutants against cephalosporins
| Strain | CTX | FOX | CAZ | FEP | CPT |
|---|---|---|---|---|---|
| EC958 | 12 | 32 | 16 | 0.75 | 6 |
| Δslt | 12 | 48 | 3–4 | 0.38–0.5 | 3–6 |
| ΔtatB | 4–6 | 32–48 | 2–3 | 0.19–0.25 | 1.5–2 |
| ΔenvC | 12 | 48 | 2–4 | 0.5 | 6 |
| ΔblaCMY-23 | 0.75–1 | 8 | 0.25–0.38 | 0.38–0.5 | 0.75–2 |
| ΔampG | 8–12 | 32 | 4 | 0.38–0.5 | 3–4 |
| ΔdacA | 6–8 | 24–32 | 3 | 0.19–0.38 | 3 |
| ΔyfiH | 8 | 32–48 | 3 | 0.25–0.38 | 2–3 |
| ΔftsP | 6–8 | 48 | 2 | 0.25 | 2 |
| ΔtatC | 6 | 32–48 | 2–3 | 0.25 | 2 |
| ΔuvrA | 12–16 | 32 | 3–4 | 0.75 | 3–6 |
| ΔmrcB | 12 | 32 | 3–4 | 0.5 | 3–4 |
| ΔdamX | 16 | 48 | 4 | 0.75 | 6 |
| Strain | CTX | FOX | CAZ | FEP | CPT |
|---|---|---|---|---|---|
| EC958 | 12 | 32 | 16 | 0.75 | 6 |
| Δslt | 12 | 48 | 3–4 | 0.38–0.5 | 3–6 |
| ΔtatB | 4–6 | 32–48 | 2–3 | 0.19–0.25 | 1.5–2 |
| ΔenvC | 12 | 48 | 2–4 | 0.5 | 6 |
| ΔblaCMY-23 | 0.75–1 | 8 | 0.25–0.38 | 0.38–0.5 | 0.75–2 |
| ΔampG | 8–12 | 32 | 4 | 0.38–0.5 | 3–4 |
| ΔdacA | 6–8 | 24–32 | 3 | 0.19–0.38 | 3 |
| ΔyfiH | 8 | 32–48 | 3 | 0.25–0.38 | 2–3 |
| ΔftsP | 6–8 | 48 | 2 | 0.25 | 2 |
| ΔtatC | 6 | 32–48 | 2–3 | 0.25 | 2 |
| ΔuvrA | 12–16 | 32 | 3–4 | 0.75 | 3–6 |
| ΔmrcB | 12 | 32 | 3–4 | 0.5 | 3–4 |
| ΔdamX | 16 | 48 | 4 | 0.75 | 6 |
Bold text indicates a significant change compared with WT EC958.
CTX, cefotaxime; FOX, cefoxitin; CAZ, ceftazidime; FEP, cefepime; CPT, ceftaroline.
MICs (mg/L) of EC958 mutants against cephalosporins
| Strain | CTX | FOX | CAZ | FEP | CPT |
|---|---|---|---|---|---|
| EC958 | 12 | 32 | 16 | 0.75 | 6 |
| Δslt | 12 | 48 | 3–4 | 0.38–0.5 | 3–6 |
| ΔtatB | 4–6 | 32–48 | 2–3 | 0.19–0.25 | 1.5–2 |
| ΔenvC | 12 | 48 | 2–4 | 0.5 | 6 |
| ΔblaCMY-23 | 0.75–1 | 8 | 0.25–0.38 | 0.38–0.5 | 0.75–2 |
| ΔampG | 8–12 | 32 | 4 | 0.38–0.5 | 3–4 |
| ΔdacA | 6–8 | 24–32 | 3 | 0.19–0.38 | 3 |
| ΔyfiH | 8 | 32–48 | 3 | 0.25–0.38 | 2–3 |
| ΔftsP | 6–8 | 48 | 2 | 0.25 | 2 |
| ΔtatC | 6 | 32–48 | 2–3 | 0.25 | 2 |
| ΔuvrA | 12–16 | 32 | 3–4 | 0.75 | 3–6 |
| ΔmrcB | 12 | 32 | 3–4 | 0.5 | 3–4 |
| ΔdamX | 16 | 48 | 4 | 0.75 | 6 |
| Strain | CTX | FOX | CAZ | FEP | CPT |
|---|---|---|---|---|---|
| EC958 | 12 | 32 | 16 | 0.75 | 6 |
| Δslt | 12 | 48 | 3–4 | 0.38–0.5 | 3–6 |
| ΔtatB | 4–6 | 32–48 | 2–3 | 0.19–0.25 | 1.5–2 |
| ΔenvC | 12 | 48 | 2–4 | 0.5 | 6 |
| ΔblaCMY-23 | 0.75–1 | 8 | 0.25–0.38 | 0.38–0.5 | 0.75–2 |
| ΔampG | 8–12 | 32 | 4 | 0.38–0.5 | 3–4 |
| ΔdacA | 6–8 | 24–32 | 3 | 0.19–0.38 | 3 |
| ΔyfiH | 8 | 32–48 | 3 | 0.25–0.38 | 2–3 |
| ΔftsP | 6–8 | 48 | 2 | 0.25 | 2 |
| ΔtatC | 6 | 32–48 | 2–3 | 0.25 | 2 |
| ΔuvrA | 12–16 | 32 | 3–4 | 0.75 | 3–6 |
| ΔmrcB | 12 | 32 | 3–4 | 0.5 | 3–4 |
| ΔdamX | 16 | 48 | 4 | 0.75 | 6 |
Bold text indicates a significant change compared with WT EC958.
CTX, cefotaxime; FOX, cefoxitin; CAZ, ceftazidime; FEP, cefepime; CPT, ceftaroline.
Changes in cell morphology
To further characterize the effect of cefotaxime on EC958 and each of the respective mutants, we investigated their cell morphology using live-cell imaging. Cefotaxime is known to cause filamentation by inhibiting PBP3 (FtsI)30 and our analysis revealed visible morphology changes upon treatment with cefotaxime for the ΔtatB, ΔtatC, ΔenvC, ΔblaCMY-23, ΔftsP, Δslt and ΔdacA mutants (Figure 3). In addition, we also observed mid-cell bulging in the Δslt and ΔdacA mutants (Figure 3).
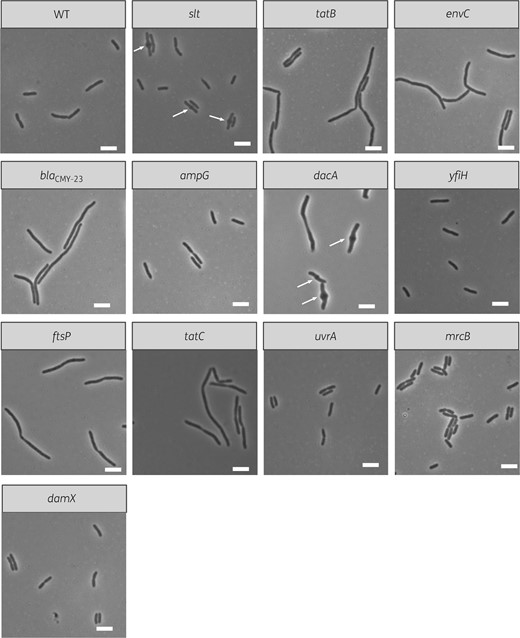
Live-cell images of isogenic mutants for genes identified by TraDIS. Each image depicts a field of view for cells from each isogenic mutant after treatment with 4 mg/L cefotaxime for 1 h and is representative of the cell population. The white scale bar represents 5 μm. Arrows indicate the location of mid-cell bulges.
Next, we determined the cell length distribution of WT EC958 and each of the mutants in the absence and presence of cefotaxime from the live-cell images using AxioVision software. In LB in the absence of cefotaxime, the ΔtatB, ΔenvC, ΔampG, ΔyfiH, ΔftsP, ΔtatC, ΔmrcB and ΔdamX mutants possessed a significant (but small) difference in cell length compared with WT EC958 (Figure 4). In cefotaxime-treated samples, increased cell length was more pronounced, as expected when (directly or indirectly) targeting PBP3 (FtsI) function (Figure 4). The most significant filamentation was observed for the ΔblaCMY-23 mutant, which increased in length to 10–20 μm upon treatment with cefotaxime compared with the untreated control (cell length distribution of 2–5 μm). Overall, all but two of the mutants (ΔmrcB and ΔdamX) showed significant changes in their cell length compared with resistant WT EC958 in the presence of cefotaxime (Figure 4). Interestingly, the Δslt, ΔenvC, ΔampG and ΔuvrA mutants displayed a significant increase in cell length, suggesting restoration of cefotaxime function via PBP3 (FtsI) targeting, despite the fact that their MIC of cefotaxime was unchanged compared with WT EC958 (Figure 4).
![Cell length distribution of WT EC958 and isogenic mutants for genes identified by TraDIS. The distribution of bacterial cell length of the WT (control) and each isogenic mutant was determined in LB [CTX(−), right] or LB supplemented with 4 mg/L cefotaxime [CTX(+), left]. On average, 95 cells (range 32–135, SD = 29.12) were measured per sample. The Kruskal–Wallis rank sum test followed by Dunn’s test was applied to identify mutants with significant changes in cell length compared with the WT at each respective treatment. P values are coded as: ***P < 0.0001, **P < 0.001 and *P < 0.01.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jac/75/6/10.1093_jac_dkaa023/1/m_dkaa023f4.jpeg?Expires=1750212893&Signature=A0qkKV1C5eUHQPuKzgiMk0qD4AMZc98P7nJpKwejVeYJh1oHJbrohI1HDd-ncD2wHRQe0ZqxaYb4KVfWrQcHecwDzrYxdnHw947uefIKaBZPl6HKGbxbkWeDsWySpJSVJ6t~YtHUVl1RxnfKmrFjZPXoDlIa1gL7EIFFpnOVi0ZryYZWvPOtW3e4XnINpPqtEBOSgYOuZJr8UA~1S~EIm9ArL7C9TbF47VROgPUQs6LEYdkzthIJXY~tfPPc2rfcRw~j8t14zORpDmX6gJOl2jYEp-MlC11pe4tnoOZweQGGOU-YeOHCmVINKva9Q6Mg1WM5axPALSH7OFfPE7zUlg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cell length distribution of WT EC958 and isogenic mutants for genes identified by TraDIS. The distribution of bacterial cell length of the WT (control) and each isogenic mutant was determined in LB [CTX(−), right] or LB supplemented with 4 mg/L cefotaxime [CTX(+), left]. On average, 95 cells (range 32–135, SD = 29.12) were measured per sample. The Kruskal–Wallis rank sum test followed by Dunn’s test was applied to identify mutants with significant changes in cell length compared with the WT at each respective treatment. P values are coded as: ***P < 0.0001, **P < 0.001 and *P < 0.01.
Discussion
The globally disseminated UPEC ST131 clone was first recognized by its association with the cefotaxime resistance gene blaCTX-M-1531,32 and genomic analyses have revealed that this gene is found in the subclade of fluoroquinolone-resistant ST131 that also contains the greatest number of resistance-associated genes.3,6 In this study, we used a high-throughput genetic screening approach employing a saturated mini-Tn5 mutant library and TraDIS to discover genes required for cefotaxime resistance in the E. coli strain EC958, a well-characterized fluoroquinolone- and cefotaxime-resistant ST131 strain.33 Our screen identified 13 genes associated with cefotaxime resistance; this included eight genes involved in cell wall synthesis and cell division (ftsN, slt, envC, ampG, dacA, ftsP, mrcB and damX), the tatB and tatC genes that encode the Tat secretion system, uvrA (involved in DNA damage repair), yfiH (which encodes a polyphenol oxidase involved in maintaining the composition and integrity of peptidoglycan)34 and the blaCMY-23 cephalosporin-resistance gene. The identification of blaCMY-23 confirmed a previous finding that although EC958 contains the blaCTX-M-15 gene on its pEC958 plasmid, its primary mechanism of cefotaxime resistance occurs through the chromosomally located blaCMY-23 gene.17 EC958 also contains additional β-lactamase genes, including a chromosomal ampC gene and plasmid-encoded blaTEM-1 and blaOXA-1 genes; however, these were not required for cefotaxime resistance under the conditions employed in this study.
We have previously shown that the mini-Tn5 used in this study is capable of driving expression of a downstream gene via read-through from the promoter of the chloramphenicol-resistance gene cassette.35,36 However, we did not find any insertion or intergenic region exhibiting significant orientation bias (Datasets S2 and S3), which is an indicator of possible overexpression or disruption of a downstream gene. Although our analysis by edgeR only used the differences in total mapped reads to identify significant hits, a closer look at the positions of individual insertions within the 13 genes identified by TraDIS (Figure S1) confirmed that the number of insertion sites was also significantly reduced in the cefotaxime-treated pool. All genes identified possessed multiple insertions in both orientations in the LB control pool, confirming that the reduction in insertion sites is specific to cefotaxime treatment.
Of the 13 genes identified, we successfully generated defined mutations in 12 genes (all except ftsN). These 12 mutants were subjected to phenotypic testing to confirm their role in cefotaxime resistance. Growth curves of these mutants using the same concentration of cefotaxime employed in our TraDIS screen revealed that eight mutants (67%; Δslt, ΔblaCMY-23, ΔyfiH, ΔftsP, ΔtatC, ΔuvrA, ΔmrcB and ΔdamX) possessed visible growth defects compared with WT EC958 (Figure 2). Furthermore, six of the mutants (ΔtatB, ΔblaCMY-23, ΔdacA, ΔyfiH, ΔftsP and ΔtatC) were more susceptible to cefotaxime (Table 2). Overall, the combination of growth analysis in the presence of cefotaxime and MIC determination confirmed the involvement of 10 genes (83%) in resistance to cefotaxime. We also expanded the susceptibility testing of these mutants to second-, third-, fourth- and fifth-generation cephalosporins (Table 2). Significantly, the mutants containing deletions in the genes identified in our study showed reduced MIC of at least one of these cephalosporins and all mutants exhibited a reduced MIC of ceftazidime.
The β-lactam class of antibiotics targets PBPs and exerts a variety of effects on E. coli, including filamentation, induction of a spherical cell morphotype and rapid cell lysis due to dysregulation of cell wall integrity.37,38 Because the main target of cefotaxime in E. coli is the cell division-associated PBP3 (FtsI), and its inhibition leads to cell filamentation,15,39 we employed live-cell imaging to assess changes in the cell morphology of WT EC958 and mutants generated in this study in the absence and presence of cefotaxime. In the absence of cefotaxime, the cell morphology of several mutants displayed subtle differences compared with WT EC958, in agreement with other studies that have shown that mutation of these genes in different strains affects cell morphology.34,40–43 However, in the presence of cefotaxime these differences became much more pronounced for all mutants except ΔmrcB and ΔdamX (Figure 4). The filamentation morphotype of the ΔenvC mutant in the presence of cefotaxime was similar to that previously reported for E. coli strain PM61, which contains a mutation in envC and possesses an oversized PBP3 (FtsI) due to its unprocessed C-terminal peptide.44 Likewise, bulging of the Δslt mutant is consistent with a similar phenotype reported in an E. coli K12 strain following mutation of slt and subsequent exposure to β-lactams specific for PBP3 (FtsI).43 Finally, the EC958 ΔdacA mutant examined in this study formed mid-cell bulges in the presence of cefotaxime, possibly due to the uncoupling of peptidoglycan synthesis and turnover. In this respect, it is notable that mutation in dacA (encoding PBP5) in E. coli K12 (strain CS109) also exhibited cell branching and other aberrant cell morphologies following treatment with aztreonam, a β-lactam antibiotic that targets PBP3 (FtsI).45,46
The TatB and TatC proteins oligomerize and assemble to form integral components of the inner membrane Tat secretion system.47 In E. coli, there are 36 known and predicted substrates of the Tat translocation system,48 including FtsP.49 Mutation of the Tat system results in a phenotype that may arise from the mislocalization of an entire set of Tat substrates.50 Therefore, the filamentation observed in the ΔtatB or ΔtatC mutants may occur indirectly due to their inability to translocate FtsP and other substrates to their correct locations. FtsP is a divisomal protein required during cellular stress and an ΔftsP mutant exhibits filamentation at 42°C.41 FtsP is recruited to the septal ring by FtsN.29 Although ftsN was identified in our TraDIS screen, we were unable to generate an independent mutant, likely due to its previous description as an essential gene.23 Multicopy of FtsP can suppress the cell division defect due to mutation in PBP3 (FtsI),51 the target protein of cefotaxime. Taken together, our data suggest that mutations in ftsP, tatB and tatC could potentiate cefotaxime treatment via reduced compensatory effects on PBP3 (FtsI) inhibition.
A previous study by Liu et al.52 defined the susceptibility profile for ∼4000 single-gene knockouts in the KEIO collection against 22 antibiotics, including three β-lactams: ampicillin, cefradine and cefoxitin. Six genes found in our study (dacA, yfiH, ftsP, tatB, tatC and mrcB) overlapped with the genes identified in this previous study, with the differences observed likely due to: (i) different cellular effects of cefotaxime, a third-generation cephalosporin, compared with ampicillin, cefradine and cefoxitin; and (ii) the different strains used in the studies (BW25113 versus EC958). Indeed, cefotaxime binds to PBP3 (FtsI) while cefoxitin binds to PBPs 4, 7 and 8,15 which could dictate the set of genes required for survival against cefotaxime compared with cefoxitin. As a demonstration of the distinct resistome differences between the two E. coli strains, the four mutants in BW25113 that displayed increased susceptibility to cefoxitin in the Liu et al. study52 (ΔdacA, ΔyfiH, ΔtatC and ΔmrcB) did not show any changes in MIC of cefoxitin in the EC958 strain used in this work. In fact, none of the mutants in our study showed reduced MIC of cefoxitin except for ΔblaCMY-23. This highlights a fundamental difference in the two studies, namely that the test strain EC958 in our study is resistant to cephalosporins. As a consequence, mutations in the genes identified in our study were capable of sensitizing the cells to third-generation cephalosporin treatment despite the presence of the cephalosporinase gene blaCMY-23.
As a result of the activity of the cephalosporinase CMY-23 in EC958 and mutants derived from this strain (except for the ΔblaCMY-23 mutant), we would predict that the cells were exposed to a lower intracellular concentration of cefotaxime than strains lacking cephalosporinase activity. This concentration, despite being lower and non-lethal, resulted in a significant increase in cell length in WT EC958 compared with exposure to LB alone (Figure 4), indicative of low-level PBP3 (FtsI) inhibition. Therefore, the phenotype of the mutants examined in this study following exposure to cefotaxime (except the ΔblaCMY-23 mutant) would have most likely been caused by the combined effect of low-level PBP3 (FtsI) inhibition and mutation of each respective gene, possibly resulting in altered peptidoglycan architecture. For example, the reduced cefotaxime MIC and filamentation of the ΔdacA mutant upon cefotaxime exposure can be attributed to mutation of dacA together with PBP3 (FtsI) inhibition. As a consequence, we hypothesize this sensitization to cefotaxime could be achieved using a combination of cefotaxime and a DacA (PBP5) inhibitor. Recently, non-β-lactam inhibitors of PBPs were shown to be highly promising agents that could counter the emergence of β-lactam resistance,53 especially cyclic boronates that inhibit serine-β-lactamases, MBLs and PBP5.54 Our data suggest that the use of such inhibitors in combination with cefotaxime might rescue the use of cefotaxime against resistant pathogens.
In conclusion, this study identified the intrinsic resistome against cefotaxime for EC958, a representative strain of the globally dominant UPEC ST131 clone. The mutation of these genes potentiated the effect of cefotaxime despite the presence of a cephalosporinase, providing a basis for the development of novel inhibitors. Therefore, we suggest the genes identified in this study represent potential targets for combinatory therapy against β-lactamase-producing Enterobacteriaceae.
Funding
This work was supported by a Project grant from the Australian National Health and Medical Research Council (NHMRC) (GNT1106590) and a Discovery Project grant from the Australian Research Council (DP150102062). M.A.S. is supported by an NHMRC Senior Research Fellowship (GNT1106930). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Transparency declarations
None to declare.
Supplementary data
Figures S1 and S2 and Datasets S1 to S3 are available as Supplementary data at JAC Online.

{kind=link}
{kind=link}
{kind=link}
{kind=link}