





Abstract
There is currently a global effort to reduce malaria morbidity and mortality. However, malaria still results in the deaths of thousands of people every year. Malaria is caused by Plasmodium spp., parasites transmitted through the bite of an infected female Anopheles mosquito. Treatment timing plays a decisive role in reducing mortality and sequelae associated with the severe forms of the disease such as cerebral malaria (CM). The available antimalarial therapy is considered effective but parasite resistance to these drugs has been observed in some countries. Antimalarial drugs act by increasing parasite lysis, especially through targeting oxidative stress pathways. Here we discuss the roles of reactive oxygen species and reactive nitrogen intermediates in CM as a result of host–parasite interactions. We also present evidence of the potential contribution of oxidative and nitrosative stress-based antimalarial drugs to disease treatment and control.
Introduction
Malaria is an infectious, parasitic, systemic and non-contagious disease causing high morbidity and mortality, resulting in an estimated 435 000 deaths in 2017.1 It is caused by Plasmodium spp., which are introduced into the host circulation through the bite of an infected female Anopheles mosquito. The signs and symptoms of malaria appear from Day 8 to Day 25 post-infection and are, in the uncomplicated form of the disease, non-specific (such as headache, fever and chills).2,3 However, as the disease becomes severe, the patient may present with respiratory distress, convulsions, prostration, coma, renal failure, metabolic acidosis, jaundice and hypoglycaemia, amongst other signs.4–6 Despite the global efforts to reduce malaria cases worldwide, a rise in the numbers of infected individuals has been observed in some countries in recent years.7,8 Also, severe cases of the disease continue to be registered in many countries even when antimalarial therapy is available,1,9 highlighting the need for better understanding of the mechanisms of disease and for novel and/or complementary therapeutic approaches.
Severe malaria is associated with mortality, due mostly to Plasmodium falciparum infection. Different clinical syndromes that manifest in severe malaria include cerebral malaria (CM). CM is a frequently lethal clinical syndrome of the severe form of the disease, mainly associated with P. falciparum.1,10 It is characterized by neurological complications such as coma, convulsions, abnormal muscle tone and posture, and loss of reflexes.11,12 The majority of surviving patients make a full neurological recovery from CM; however, some can suffer from irreversible neurological and/or cognitive sequelae.13–16
Evidence suggests a role for oxidative and nitrosative stresses in this clinical syndrome, as a result of host–Plasmodium interactions. In this review, the evidence will be discussed with a particular emphasis on pharmacological interventions, including adjunct therapies as potential modulators of oxidative stress and nitrosative stress in CM.
Parasite–host protein interactions
Malaria progression to CM largely depends on the interactions between host and parasite proteins, especially during blood-stage infection. Following the infection of erythrocytes, the parasite multiplies and matures, with its proteins becoming expressed on the infected erythrocyte membranes, facilitating the contact of these cells with those of the brain endothelium (Figure 1). This, in turn, contributes to the adherence of infected erythrocytes to the brain microvasculature, with subsequent disruption of the blood–brain barrier and brain inflammation.17
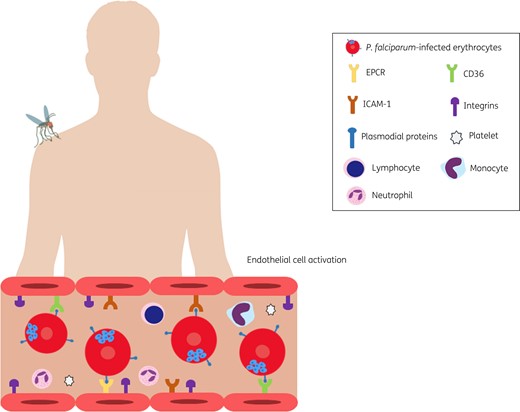
Parasite–host protein interactions. Following infection, plasmodial proteins expressed on the erythrocyte membranes facilitate their adherence to the brain microvasculature. These proteins include those of the PfEMP1 family which can interact with specific endothelial receptors such as ICAM-1, EPCR and CD36. PfEMP1 domains may also bind to integrins in the brain microvasculature. Other parasite proteins such as those of the RIFIN family and PbmaLS_05 can interact with immune cells of the host contributing to the rupture of the blood–brain barrier and endothelial cell activation. This figure appears in colour in the online version of JAC and in black and white in the printed version of JAC.
In this context, we highlight the importance of the erythrocyte membrane protein 1 (PfEMP1) family, encoded by approximately 60 gene variants, which contributes to the adherence of infected erythrocytes to the brain microvasculature and to further development of CM.18–20 This is due to the ability of these parasite proteins to bind to endothelial receptors such as cluster of differentiation 36 (CD36), intercellular adhesion molecule 1 (ICAM-1), integrins αVβ3 and αVβ6, and the endothelial protein C receptor (EPCR).18–21
Binding of PfEMP1 to CD36 results in the expression of integrins such as α5β1 on brain endothelial cells, making erythrocyte adhesion to the microvasculature tighter.22 Of note, this integrin is critical to the maintenance of the blood–brain barrier.23 Indeed, α5β1 expression has been associated with disruption of endothelial barriers through breakdown of VE-cadherin and claudin-5.24,25 By binding to EPCR, PfEMP1 also contributes to brain endothelial activation by reducing the levels of activated protein C, an inhibitor of thrombin generation.26,27 Excessive binding to EPCR leads to a reduction in the levels of protein C, resulting in thrombin-mediated inflammation and subsequent endothelial dysfunction.28 Also, during CM the endothelium overexpresses ICAM-1, further increasing the binding of infected erythrocytes to endothelial cells, an effect that has been associated with the availability of nitric oxide (NO).29,30
It is important to highlight that different PfEMP1 proteins bind to different endothelial receptors. PfEMP1 proteins are classified into different groups (A–E) and are characterized by large ectodomains containing multiple Duffy binding-like (DBL) and cysteine-rich interdomain region (CIDR) domains that can interact with specific endothelial receptors.31 Certain DBLβ domains found in groups A, B, and C bind to ICAM-1.32–34 CIDRα1 domains from group A bind to EPCR35,36 and CIDRα2–6 domains of groups B and C bind to CD36.37,38 Recent studies have attempted to establish a link between CM and subtypes of PfEMP1 and their domains. In this context, the ability of certain domains to bind multiple endothelial receptors has been investigated. A recent report indicated that parasites expressing DC13-containing PfEMP1 variants promote the binding of erythrocytes to both EPCR and ICAM-1 in the brain endothelium.39 More recently, in children with CM, infected erythrocytes expressing PfEMP1 with CIDRα1.4 in its CIDR were found to be more likely to bind to both EPCR and ICAM-1 in the brain microvasculature.40 Other PfEMP1 domains such as DBLδ1_D4 favour the cytoadhesion of infected erythrocytes to the integrins αVβ3 and αVβ6 expressed on endothelial and immune cells; however, the physiological relevance of these interactions to CM remains unclear.21
A subset of the RIFIN family of parasite proteins, which also become expressed on the membrane of infected erythrocytes, were found to bind to the leucocyte Ig-like receptor B1 (LILRB1)41 expressed on immune cells such as lymphocytes and monocytes.42 The RIFIN proteins of P. falciparum are encoded by 150–200 rif genes.43 Infected erythrocytes expressing these proteins were found to cause down-regulation of immune cells and to be more likely to bind to LILRB1 in patients with CM in comparison with those with the non-severe form of the disease.41
More recently, PbmaLS_05, a parasite antigen expressed during the pre- and intra-erythrocytic stages of infection, was shown to contribute to the rupture of the blood–brain barrier by activating a specific subset of CD8+ lymphocytes.44 Interestingly, deletion of PbmaLS_05 conferred protection against Plasmodium berghei-induced experimental CM (ECM) development, an effect that was associated with reduced inflammation (oedema and microhaemorrhages) and parasite load in the brain tissue of mice with the syndrome.44
Overview of oxidative and nitrosative stresses
The close interaction between host and parasite proteins triggers an inflammatory process in the vasculature, in addition to the obstruction of the brain microvasculature by infected and non-infected erythrocytes.45–47 Both responses contribute to CM progression, resulting in unbalanced inflammatory mediator release, vascular leakage, disruption of the blood–brain barrier and ultimately lead to cytotoxic effects in the brain tissue.48–53
In this context, alterations in the levels of reactive oxygen species (ROS) and reactive nitrogen intermediates (RNIs) play important roles in CM.
ROS (Figure 2) are formed as part of the host response to a range of stimuli, from oxygen inspiration to exposure to harmful stimuli such as infection. In this process, O2 can be reduced by NADPH oxidase, an enzyme largely expressed in phagocytes, to superoxide (). is then dismuted to H2O2 by superoxide dismutase (SOD).54–56 Elevated levels of H2O2 can be converted into H2O and O2 by catalase or reduced by iron to hydroxyl radicals (HO•), highly reactive products that can oxidize carbohydrates, lipids, proteins and DNA.57,58 H2O2 can also be removed by the glutathione and thioredoxin systems in a dynamic process of oxidation and reduction of glutathione and thioredoxin by the specific enzymes glutathione peroxidase (GPx) and glutathione reductase (GR), and thioredoxin reductase (TrxR), respectively.59,60
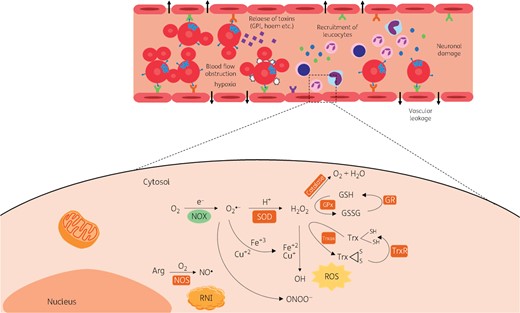
Overview of host oxidative and nitrosative stresses. ROS are formed from O2 reduction by NADPH oxidase which is largely expressed in phagocytes, to superoxide (). is then dismuted to H2O2 by SOD. Elevated levels of H2O2 can be converted into H2O and O2 by catalase or reduced by iron to HO• radicals, highly reactive products, which can oxidize carbohydrates, lipids, proteins and DNA. H2O2 can also be removed by the glutathione and thioredoxin systems. The RNI production cascade initiates with the formation of NO from arginine by NOSs. RNI products include oxidized states and adducts of the nitrogen products of NOSs (nNOS, eNOS and iNOS) such as NO•, , S-nitrosothiols and peroxynitrite (OONO−); the latter, formed by NO• and , can cause oxidation and nitration of lipids, proteins and DNA. During CM, ROS are produced in order to kill the parasites, but they also damage the endothelial cells of the cerebral vasculature, resulting in disruption of the blood-brain barrier and vascular leakage. Trx, thioredoxin; Trxox, oxidized thioredoxin. This figure appears in colour in the online version of JAC and in black and white in the printed version of JAC.
Not only the host, but also the parasite presents antioxidant machinery (Figure 3a), although differences have been observed between the host and parasite systems. P. falciparum does not express catalase or GPx and expresses two SODs (PfSOD-1 and SOD-2).61,62 Instead of GPx, the parasite expresses glutathione S-transferase (PfGST),62,63 which is suggested to be involved in resistance to antimalarial drugs.62,64
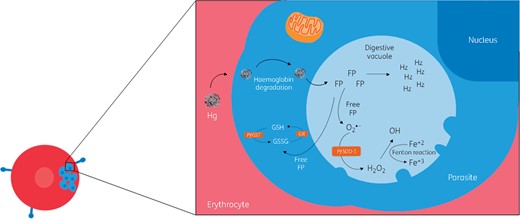
Parasite responses to oxidative stress. Following the digestion of haemoglobin in the parasite vacuole, there is accumulation of FP, a toxic product that becomes polymerized into haemozoin. Some FP (free FP) may escape polymerization needing detoxification by the antioxidant machinery of the parasite. If not removed, the free FP reacts with phospholipids of the vacuolar membrane, increasing its permeability and leading to parasite killing. The parasite expresses two SODs (PfSOD-1 and SOD-2) and PfGST as part of its antioxidant machinery. Hg, haemoglobin.This figure appears in colour in the online version of JAC and in black and white in the printed version of JAC.
Produced by the host, RNIs (Figure 2) include oxidized states and adducts of the nitrogen products of NO synthases [neuronal (nNOS), endothelial (eNOS) and inducible (iNOS)] including NO•, NO2−, S-nitrosothiols and peroxynitrite (OONO−). The latter, formed by NO• and , can cause oxidation and nitration of lipids, proteins and DNA.65–68 This cascade initiates with the formation of NO from arginine by NOSs, a process largely amplified by iNOS during inflammation.69
Oxidative and nitrosative stresses in CM
Role of ROS in CM
Oxidative stress has been implicated in CM, occurring as part of the host response to Plasmodium spp.,62 although its role in this form of the severe disease is not completely understood. The first evidence of the involvement of oxidative stress in CM dated from the early 1980s. By using in vitro and in vivo mouse models, the first studies suggested that during host response to infection, activated phagocytes released ROS in order to kill the parasite, but this also damaged the endothelial cells of the cerebral vasculature, resulting in disruption of the blood–brain barrier and poor disease prognosis.70–73 Subsequently, the administration of pro-oxidants in mice was found to protect against ECM by killing the intra-erythrocytic parasite.74 In parallel, human studies demonstrated that patients with CM presented with brain oedema and mononuclear cell accumulation as a result of increased vascular endothelium permeability triggered by high levels of oxidative stress, as denoted by increased lipid peroxidation in their CSF in comparison with control subjects, a response that was further exacerbated in fatal cases of the disease.75 Also, analysis of brain samples obtained post-mortem from CM patients indicated that cells such as intravascular phagocytes, perivascular macrophages and reactive glia are more likely to express the oxidative stress marker haemo-oxygenase-1 (HO-1) in haemorrhage areas.76 The increased expression of HO-1 mRNA and protein following CM was also observed in the brains of Plasmodium-infected mice.77,78 HO-1 protein expression is suggested to reach its highest levels of expression at late stages of the disease (Stage IV) and this is preceded by increased expression of GPx at Stage II.77
Similarly, increased levels of malondialdehyde and conjugated dienes were detected in the brains of mice with ECM and associated with cognitive impairment.79 Another study by Souza et al.80 showed that the parasite triggered oxidative stress via protein carbonylation in the mouse hippocampus, thus contributing to the neurological deficits and seizures observed in ECM. Interestingly, cognition was improved in mice treated with the antioxidants deferoxamine or N-acetylcysteine in combination with the antimalarial drug chloroquine; notably, neither deferoxamine nor N-acetylcysteine affected parasitaemia.79 Also, administration of the antioxidants SOD, catalase or butylated hydroxyanisole was previously shown to reduce blood–brain barrier permeability in mice with ECM.73 Another study demonstrated that superoxide and peroxynitrite mediated the apoptosis of human endothelial cells incubated with erythrocytes infected with P. falciparum.81 Indeed, both incubation with the SOD mimetic MnTBAP and transient supplementation of SOD1 conferred protection to human endothelial cells incubated with P. falciparum-infected erythrocytes.81,82
Of importance, SOD and catalase mRNA expression have been reported to become down-regulated in the host as the neurological symptoms of CM progress.77 More recently, a proteomic analysis demonstrated that antioxidant peptides related to SOD1, glutathione S‐transferase kappa 1, peroxiredoxin‐6, peroxiredoxin‐5 and mitochondrial isoform A are depleted in the brains of patients with CM.83 On the other hand, analysis of the plasma protein profile of children with uncomplicated and severe (CM and malaria anaemia) disease demonstrated that oxidative stress markers are altered in all malaria cases; however, oxidative stress was found to be highly associated with malaria anaemia whilst markers of endothelial damage were associated with CM.84
Role of RNIs in CM
Similar to the situation with ROS, alterations in the balance of NO may also contribute to CM outcome, although its definite roles in the disease are debatable. Indeed, studies have reported either a protective, a deleterious or a null role for RNIs in CM. A study by Taylor et al.85 demonstrated no link between the levels of RNIs and CM, or its clinical signs such as coma and loss of consciousness. Indeed, no consistent evidence regarding iNOS promoter haplotypes was found in Tanzanian children with CM.86 However, much evidence has indicated protective or harmful roles, especially for NO in CM. The controversial evidence of this gaseous transmitter in CM is based on the different techniques employed to measure RNIs/NO, disease time courses, experimental versus clinical studies and the choice of pharmacological tools (knockouts versus blockers/activators of involved pathways); these aspects will be now discussed.
Evidence of a detrimental role for NO in CM
It was initially hypothesized that NO is generated during infection following the production of TNF as a consequence of the contact of infected erythrocytes with the cerebral blood vessels, contributing to neuronal damage and the subsequent neurological signs of CM, in addition to increased intracranial pressure.87 Later, inhibitors of NO were administered to mice with CM; the study showed these drugs did not stop disease progression.88,89
Further studies in humans demonstrated that patients with longer coma duration and those in deeper coma presented with raised levels of RNIs in their serum in comparison with those with lighter coma; also, fatal cases presented with higher levels of these mediators than survivors.90 In another study, the CSF concentrations of NO were found to be higher in children who died from CM in comparison with those who survived the disease;91 the same study suggested that NO derived from sources other than leucocytes was detrimental to CM. Supporting a deleterious role for NO in CM, it was suggested that in the absence of TNF, NO levels are reduced and mice are protected from fatal ECM.92
On the other hand, neither oral administration of the iNOS inhibitor aminoguanidine nor ablation of iNOS influenced ECM in mice.93 Also, reports from Thai adults demonstrated that patients with CM presented with increased brain iNOS expression during the acute phase of the syndrome.94 Inhibition of NO generation was found to protect the endothelial cells against Plasmodium-induced apoptosis.81
Evidence of a protective role for NO in CM
Despite evidence indicating that NO production may be harmful to patients with CM, other studies have shown an opposite role for this transmitter in the syndrome. Accordingly, Tanzanian CM children were found to present with lower levels of leucocyte-dependent NO in their plasma.95 Also, a polymorphism in the iNOS gene of Tanzanian and Kenyan children was associated with increased circulating levels of NO and a protective phenotype against CM.96 This finding was supported by a later study by Trovoada et al.97 who identified polymorphisms in the iNOS promoter and associated them with increased NO levels and protection against CM in Angolan children.
Analysis of the plasma levels of the NO synthesis substrate l-arginine demonstrated it to be reduced in Tanzanian children with CM in comparison with healthy controls; the low levels of l-arginine were associated with limited NO production and fatality.98 Also, the systemic l-arginine levels in children who recovered from CM were found to be comparable to those of healthy subjects of similar age; CM children presented low levels of NO at the time of hospital admission.99 The low availability of systemic NO in CM has been attributed to increased conversion of l-arginine to ornithine and urea, instead of NO, by M2 monocytes100 and also to low systemic levels of tetrahydrobiopterin (BH4), an enzyme cofactor required for NO synthesis from l-arginine.101
In 2006, Gramaglia et al.48 showed that low NO availability contributes to ECM in mice and that injection of an exogenous NO donor reduces plasma cytokine levels and brain oedema formation. Similarly, endothelial dysfunction in Indonesian individuals with severe malaria was found to be reversed by l-arginine infusion.102 Of importance, polymorphisms of eNOS may enhance its expression and subsequent NO production, protecting adult patients against CM.103 The same group also showed that polymorphisms decreasing NO are associated with low NO production and development of CM.104 More recently, it was suggested that both eNOS and nNOS functions are impaired in mouse ECM; these alterations contribute to cerebrovascular dysfunction and reduced NO availability, which are partially restored by BH4 treatment.50 In another study, repeated treatment with the NO donor dipropylenetriamine NONOate delayed CM-induced death, improved cerebrovascular blood flow and vascular tone, especially in the microvasculature, and decreased leucocyte accumulation in these vessels.105 A similar result in survival was found in animals receiving prophylactic inhaled NO40 or l-arginine infusion.106
Oxidative and nitrosative stress-based antimalarial drug development
Quinolines, such as chloroquine and amodiaquine, are antimalarial drugs conventionally used to treat uncomplicated and severe forms of malaria including CM. These drugs bind to ferriprotoporphyrin IX (FP), a product of the digestion of haemoglobin by the parasite, which becomes polymerized into haemozoin.107–110 Thus, such therapies impair haemozoin formation, leading to the accumulation of an important quantity of FP in the parasite digestive vacuole, which then reacts with phospholipids of the vacuolar membrane, increasing its permeability and ultimately leading to killing of the parasite.110 Derivatives of quinolines such as piperaquine also increase free FP,111,112 whilst primaquine induces ROS formation in erythrocytes leading to lipid peroxidation and cell haemolysis.113
As a toxic product, free FP needs to be detoxified and this is achieved by the glutathione systems of both the host and the parasite.114,115 Other derivatives of quinolines include lipophilic quinoline/methanol drugs such as lumefantrine and halofantrine, which act by reducing FP detoxification in the parasite, thus inducing ROS production.114,116 By augmenting free FP or ROS, quinolines and derivatives demand increased antioxidant activity (especially by the glutathione system) in order for the parasite to survive; resistance to this group of drugs has been linked to increased parasite GST activity.117,118
In this scenario, artemisinin and its synthetic derivatives (dihydroartemisinin, artesunate, artemether and arteether) emerged as a novel class of antimalarial drugs; notably, dihydroartemisinin is the active metabolite of this class of drug, meaning the others need to be metabolized into this compound in order to have antimalarial activity. Artemisinin-based drugs contain an endoperoxide bridge that is responsible for their antimalarial activity and has to be activated to generate free radical species. They act through different mechanisms in order to cause parasite death. These mechanisms include the induction of free FP and ROS production119–122 and the rapid depolarization of the membrane potential of the parasite, an effect inhibited by ROS scavengers and iron chelators.121 Also, the excessive induction of ROS by artemisinin drugs can cause destruction and/or inhibition of the parasite mitochondria by acting on molecules involved in the electron transport chain of the parasite, such as the cytochrome-c oxidase and the NADH:quinone oxidoreductase (PfNDH2) system.121,123 Importantly, lack of PfNDH2 is suggested to confer resistance to artemisinins.123 Artemisinin-based drugs can also induce parasite dormancy, which has been linked to resistance to these therapies.124
In order to deal with parasite resistance, combination therapy based on artemisinins was introduced in the 1990s. Five of these combinations were recommended for malaria treatment: (i) artemether/lumefantrine; (ii) artesunate/mefloquine; (iii) artesunate/amodiaquine; (iv) dihydroartemisinin/piperaquine; and (v) artesunate/sulfadoxine/pyrimethamine. However, resistance has been observed even to these combined therapies.125
In recent years, the WHO126 has recommended combinations of quinoline derivatives and artemisinin derivatives (artemether/lumefantrine, artesunate/amodiaquine, artesunate/mefloquine or dihydroartemisinin/piperaquine). Although these combinations have increased treatment efficacy, resistance or loss of susceptibility of P. falciparum to such therapies has also been observed in some countries.127–131 The overall effects of the currently available antimalarial therapies are depicted in Figure 4.
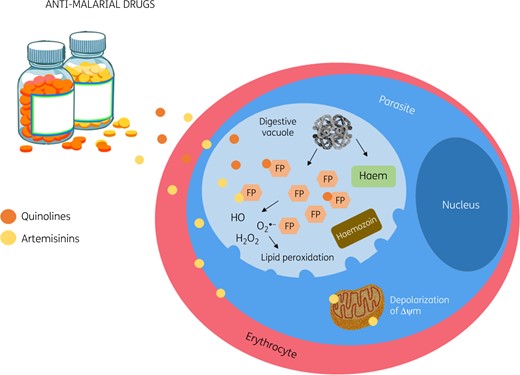
Oxidative stress-based antimalarial drugs. Quinolines bind to FP, impairing haemozoin formation. This leads to the accumulation of FP in the parasite digestive vacuole and results in the lysis of the parasite. Quinolines derivatives increase FP or induce ROS formation in the erythrocytes, leading to lipid peroxidation and cell haemolysis. Artemisinin-based drugs induce free FP and ROS production, the rapid depolarization of the membrane potential of the parasite and destruction and/or inhibition of the parasite mitochondria. Δψm, mitochondrial membrane potential. This figure appears in colour in the online version of JAC and in black and white in the printed version of JAC.
Based on the existing therapies and the evidence gathered on the dual role of ROS and NO in CM, it has become clear that preventing the progression of malaria infection to severe disease depends on treatment timing and host response to infection, i.e. whilst ROS and free FP are important to kill the parasite, they may also harm the host, causing haemolysis and excessive ROS formation.113,132
Targeting of RNIs has also shown promise. Artemisinin/NO donor hybrid compounds have been successfully tested in ECM.133 Prophylactic treatment with the NO donor S-nitrosoglutathione decreased the incidence of ECM by reducing brain oedema formation, leucocyte accumulation and haemorrhage, in addition to inhibiting parasite growth.134 Also, continuous infusion of l-arginine, the substrate for NO synthesis, alone or in combination with artesunate, improved survival of mice with ECM.106 Importantly, a randomized double-blind placebo-controlled trial of inhaled NO demonstrated that neuroprotection can be achieved by increasing the bioavailability of this gaseous transmitter in children with severe malaria.135
In this context, with the discovery of new oxidative stress- and NO-controlling pathways, novel avenues have been explored in malaria treatment. For instance, targeting of host proteins such as transient receptor potential vanilloid 1 (TRPV1), a non-selective cation channel expressed on neuronal and non-neuronal cells of the host (such as immune and endothelial cells),136 and a recently discovered oxidative stress sensor and regulator of NO and ROS137–139 protected P. berghei ANKA-infected mice from ECM.140 This response was associated with increased oxidative stress (high levels of H2O2 and increased protein nitrotyrosine and carbonyl protein residues). Although blockage of TRPV1 was promising in experimental malaria, the clinical use of such drugs was suggested to be limited as it increases mortality in bacterial infections,137,139,141 which are frequent in areas with high malaria incidence.142–145
Conclusions
Overall, clinical and experimental data demonstrate that anti-ROS and FP therapies are potentially important for malaria treatment, both historically and in future treatment regimens, and are likely to prove useful in reducing disease-associated mortality and morbidity. This has been combined with increased surveillance and improved timeliness of treatment in susceptible areas. However, mortality and sequelae are still serious threats for patients infected with Plasmodium spp. strains that are resistant to the available drugs and also those not able to effectively fight infection through a balance between oxidant and antioxidant-dependent responses. Studies on modulators of NO availability in rodent models indicate that the targeting of RNIs is a promising research field. Indeed, the clinical use of inhaled NO has emerged as a novel therapy to aid CM treatment.135 An interesting approach would be the combination of RNI-targeting drugs with quinoline derivatives and/or artemisinin derivatives. However, the clinical safety and effectiveness of such therapies remain to be addressed.
All of this evidence clearly indicates that controlling the prognosis of CM depends on fine tuning between treatment timing and targeting of host/parasite oxidative and nitrosative responses. Thus, drugs targeting these pathways could be employed as adjuncts to antimalarial combination therapy in order to achieve greater reductions in mortality and morbidity in CM patients.
Funding
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES; grant number 3325/2013; finance code 001), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; 309046/2016 -5), Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; 2018/20468-0), Fundação de Amparo à Pesquisa e ao Desenvolvimento Científico e Tecnológico do Maranhão (FAPEMA; grant numbers UNIVERSAL-01119/16 and BEPP-04089/15), and Programa INCT-INOVAMED (grant number 465430/2014-7). D.M.S. Pereira received a PhD studentship from CAPES.
Transparency declarations
None to declare.
References
WHO. World Malaria Report 2018.
WHO. World Malaria Report 2010.

{kind=link}
{kind=link}
{kind=link}
{kind=link}