





Abstract
We previously described extended-spectrum oxacillinase OXA-145 from Pseudomonas aeruginosa, which differs from narrow-spectrum OXA-35 by loss of Leu-155. The deletion results in loss of benzylpenicillin hydrolysis and acquisition of activity against ceftazidime. We report the crystal structure of OXA-145 and provide the basis of its switch in substrate specificity.
OXA-145 variants were generated by site-directed mutagenesis and purified to homogeneity. The crystal structure of OXA-145 was determined and molecular dynamics simulations were performed. Kinetic parameters were investigated in the absence and in the presence of sodium hydrogen carbonate (NaHCO3) for representative substrates.
The structure of OXA-145 was obtained at a resolution of 2.3 Å and its superposition with that of OXA-10 showed that Trp-154 was shifted by 1.8 Å away from the catalytic Lys-70, which was not N-carboxylated. Addition of NaHCO3 significantly increased the catalytic efficiency against penicillins, but not against ceftazidime. The active-site cavity of OXA-145 was larger than that of OXA-10, which may favour the accommodation of large molecules such as ceftazidime. Molecular dynamics simulations of OXA-145 in complex with ceftazidime revealed two highly coordinated water molecules on the α- or β-face of the acyl ester bond, between Ser-67 and ceftazidime, which could be involved in the catalytic process.
Deletion of Leu-155 resulted in inefficient positioning of Trp-154, leading to a non-carboxylated Lys-70 and thus to loss of hydrolysis of the penicillins. Ceftazidime hydrolysis could be attributed to enlargement of the active site and to a catalytic mechanism independent of the carboxylated Lys-70.
Introduction
Class D β-lactamases (DBL) have emerged as an important resistance mechanism against β-lactam antibiotics. They are widely disseminated among Gram-negative pathogens such as Acinetobacter spp.1,2 and Pseudomonas aeruginosa2 and display a high genetic diversity representing more than 400 enzyme variants (http://www.lahey.org/studies/webt.asp). This remarkable diversity allows this class to confer resistance to most β-lactams that are in clinical use.
Like class A (ABL) and class C (CBL), DBLs are serine-dependent enzymes. They rely on an active-site serine nucleophile to attack the β-lactam ring of the antibiotic yielding a covalent acyl-intermediate, which is then hydrolysed by an active-site water to release the free serine and the inactivated β-lactam.3,4 Both the acylation and deacylation steps require a catalytic base to promote deprotonation of the two nucleophiles. Class A enzymes use a glutamate–lysine pair as key catalytic residues.5 In CBLs, several amino acids have been shown to be important in catalysis6 although a lysine–tyrosine pair was reported as a potential acid/base catalyst.7 In contrast, DBLs use an unusual N-carboxylated lysine (Lys-70) as a general base activating the serine acylation nucleophile and the deacylating water.8,9 The crystal structures of several OXA-type enzymes (OXA-1, OXA-10, OXA-24/40, OXA-46 and OXA-48)10–15 showed that Lys-70 is carboxylated. Mutagenesis studies have confirmed the role of this residue in catalysis.16
Initially described as narrow-spectrum enzymes (i.e. OXA-1, OXA-2, OXA-10 and OXA-35) acting on the penicillins and first-generation cephalosporins, DBLs evolved into extended-spectrum enzymes (i.e. OXA-31, OXA-15 and OXA-19) enlarging their substrate profile to include late-generation cephalosporins such as ceftazidime and even carbapenems (i.e. OXA-128, OXA-24 and OXA-58).2,17 Various OXA-type enzymes have been characterized structurally but, unlike narrow-spectrum12,14,18,19 and carbapenem-hydrolysing oxacillinases13,20,21 for which several structures are available, no structures of extended-spectrum oxacillinase have been determined. Most extended-spectrum class D enzymes derive from the OXA-10 subfamily following amino acid substitutions (N73S, N143K, N143S, W154L and G157D; OXA-10 numbering) in the active site.22 However, owing to the lack of structural information, the role of these mutations is unknown.
Recently, we have described an unusual variant of the OXA-10 enzyme, OXA-145, deleted for Leu-155 (165 DBL numbering), which confers high-level resistance to ceftazidime and susceptibility to the penicillins.23 To gain insight into its catalytic mechanism, we determined the crystal structure of OXA-145 at 2.3 Å resolution. The structure provides the basis for the loss of penicillin hydrolysis and important features regarding the hydrolysis of ceftazidime.
Materials and methods
Gene cloning and site-directed mutagenesis
The blaOXA-145 gene was cloned in pET28a(+) as described previously23 leading to pAT915. Site-directed mutagenesis was carried out by two sequential amplifications.24 Point mutations S67G, E155A and E155Q were introduced within blaOXA-145 using pAT915 as a template. The sequence of the mutated genes was determined and the mutated plasmids were transformed into Escherichia coli BL21-CodonPlus (DE3)-RIPL for protein expression.
Purification and crystallization
WT OXA-145 and mutants S67G, E155A and E155Q were produced as reported.25 The proteins were purified by size-exclusion chromatography using a Hiload 16/60 Superdex 75 (GE Healthcare) equilibrated with 50 mM Tris-HCl/0.3 M NaCl (pH 7.5) and eluted in the same buffer. The proteins were concentrated to 10 mg/mL with a Centriprep 30 concentrator (Amicon). Preliminary crystallization screening was carried out by the sitting drop vapour diffusion method with a Mosquito® (TTP Labtech) automated crystallization system. A 400 nL (1:1) mixture of protein and different crystallization solutions (672 commercially available solutions) were equilibrated against 120 μL of reservoir buffer solution in Greiner plates. The crystallization plates were stored at 18°C in an automated imaging system (Rockimager®, Formulatrix) and the protein crystallized in solutions containing polyethylene glycol from 3350 to 8000 as a precipitant. Manual optimization was performed by the hanging drop method in 24-well plates by mixing 2 μL of protein solution and 2 μL of reservoir solution equilibrated against 1 mL of the reservoir solution. The optimized conditions for crystal growth were OXA-145 (5.9 mg/mL), 20% (w/v) of polyethylene glycol 3350 and 0.2 M di-ammonium hydrogen citrate. The mixture had a final pH of 6.0. The crystal was flash-cooled in liquid nitrogen using paratone/paraffin (1:1) oil as a cryoprotectant. X-ray diffraction data were collected on beamline BM30A at Synchrotron ESRF (Grenoble, France).
Data refinement and structural analysis
Reflections were indexed, integrated and scaled using XDS and CCP4 packages.26,27 The initial model was obtained by molecular replacement with the program PHASER28 and the X-ray structure of OXA-10 (PDB: 1FOF) as a search model. The structure was automatically and manually refined with REFMAC529 and COOT30 programs, respectively. Cross-validation was used throughout and 5% of the data were used for the Rfree calculation. The stereochemical quality of the models was monitored with the PROCHECK program.29 Ramachandran plots were calculated with RAMPAGE.30 Data processing and crystallographic refinement statistics for crystal structures are listed in Table 1. The coordinates and structure factors of OXA-145 have been deposited in the RCSB Protein Data Bank, entry 4YIN.
X-ray data collection and refinement statistics of OXA-145
| X-ray data | |
| space group | P212121 |
| cell dimensions | |
| a, b, c (Å) | 46.7, 91.2, 125.3 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| resolution (Å) | 45.5–2.30 (2.42–2.30)a |
| Rmerge | 0.086 (0.403) |
| I/σI | 11.5 (2.2) |
| completeness (%) | 98.1 (91.0) |
| redundancy | 3.7 (2.6) |
| Refinement | |
| resolution (Å) | 45–2.30 |
| no. reflections | 22 869 |
| Rwork/Rfree | 0.189/0.248 |
| no. atoms | |
| protein | 3772 |
| ligand | 26 |
| water | 157 |
| B-factors | |
| protein | 35.67 |
| ligand | 38.85 |
| water | 32.15 |
| Ramachandran plot | |
| residues in favoured regions | 96% |
| residues in allowed regions | 4% |
| outliers | 0% |
| RMSDs | |
| bond lengths (Å) | 0.012 |
| bond angles (°) | 1.614 |
| X-ray data | |
| space group | P212121 |
| cell dimensions | |
| a, b, c (Å) | 46.7, 91.2, 125.3 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| resolution (Å) | 45.5–2.30 (2.42–2.30)a |
| Rmerge | 0.086 (0.403) |
| I/σI | 11.5 (2.2) |
| completeness (%) | 98.1 (91.0) |
| redundancy | 3.7 (2.6) |
| Refinement | |
| resolution (Å) | 45–2.30 |
| no. reflections | 22 869 |
| Rwork/Rfree | 0.189/0.248 |
| no. atoms | |
| protein | 3772 |
| ligand | 26 |
| water | 157 |
| B-factors | |
| protein | 35.67 |
| ligand | 38.85 |
| water | 32.15 |
| Ramachandran plot | |
| residues in favoured regions | 96% |
| residues in allowed regions | 4% |
| outliers | 0% |
| RMSDs | |
| bond lengths (Å) | 0.012 |
| bond angles (°) | 1.614 |
aValues in parentheses are for the highest-resolution shell.
X-ray data collection and refinement statistics of OXA-145
| X-ray data | |
| space group | P212121 |
| cell dimensions | |
| a, b, c (Å) | 46.7, 91.2, 125.3 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| resolution (Å) | 45.5–2.30 (2.42–2.30)a |
| Rmerge | 0.086 (0.403) |
| I/σI | 11.5 (2.2) |
| completeness (%) | 98.1 (91.0) |
| redundancy | 3.7 (2.6) |
| Refinement | |
| resolution (Å) | 45–2.30 |
| no. reflections | 22 869 |
| Rwork/Rfree | 0.189/0.248 |
| no. atoms | |
| protein | 3772 |
| ligand | 26 |
| water | 157 |
| B-factors | |
| protein | 35.67 |
| ligand | 38.85 |
| water | 32.15 |
| Ramachandran plot | |
| residues in favoured regions | 96% |
| residues in allowed regions | 4% |
| outliers | 0% |
| RMSDs | |
| bond lengths (Å) | 0.012 |
| bond angles (°) | 1.614 |
| X-ray data | |
| space group | P212121 |
| cell dimensions | |
| a, b, c (Å) | 46.7, 91.2, 125.3 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| resolution (Å) | 45.5–2.30 (2.42–2.30)a |
| Rmerge | 0.086 (0.403) |
| I/σI | 11.5 (2.2) |
| completeness (%) | 98.1 (91.0) |
| redundancy | 3.7 (2.6) |
| Refinement | |
| resolution (Å) | 45–2.30 |
| no. reflections | 22 869 |
| Rwork/Rfree | 0.189/0.248 |
| no. atoms | |
| protein | 3772 |
| ligand | 26 |
| water | 157 |
| B-factors | |
| protein | 35.67 |
| ligand | 38.85 |
| water | 32.15 |
| Ramachandran plot | |
| residues in favoured regions | 96% |
| residues in allowed regions | 4% |
| outliers | 0% |
| RMSDs | |
| bond lengths (Å) | 0.012 |
| bond angles (°) | 1.614 |
aValues in parentheses are for the highest-resolution shell.
Computational modelling and molecular dynamics simulations
The GROMACS package and the geometric and charge parameters of the OPLS-AA force field were used to carry out all energy minimizations and molecular dynamics simulations.31 Topology and RESP charges of β-lactams were obtained from ab initio calculations using GAMESS (HF/6-31G* theory level) with RED-III software, as recommended.32–34 The complexes were immersed in cubic boxes filled with TIP3P water molecules. Na+ and Cl− ions were added to neutralize the net charge of the system and reach a salt concentration of 100 mM. The first energy minimization (steepest descent with a tolerance of 100 kJ/(mol.nm), which targeted water molecules and ions alone while the configuration of protein and ligand was kept fixed, was followed by 4.2 ps MD simulation in which the protein and the ligand were kept fixed and only water molecules and ions were allowed to evolve. The system was again minimized using 500 steps of steepest descent with a conjugate gradient of 5000 steps with a tolerance of 100 kJ/(mol.nm). The protein, ions and water were then relaxed and gradually heated to 300 K with a 50 ps molecular dynamics simulation using Langevin dynamics for temperature control. The system was further equilibrated in the NVT ensemble by gradual heating of the system from 25 K to 300 K in 300 ps. During the last step of equilibration, protein, water molecules and ions were allowed to evolve and positional restraints on heavy atoms of ligands (β-lactam ring and amide group of the C7 substituent) were gradually released. An equilibrated productive dynamic simulation was then carried out for 200 ns under normal pressure (1 bar) at room temperature (300 K). The ligand position was stable as a Michaelis complex during ∼150 ns in three independent dynamic simulations. All covalent bond lengths were constrained by the SHAKE algorithm with a relative tolerance of 10−4, allowing for a time step of 2 fs. The non-bonded interactions were calculated by applying a 14 Å cut-off and periodic boundary conditions were applied in all directions. The electrostatic interactions were calculated by the particle-mesh Ewald method.35
Enzyme kinetics
Kinetic measurements were performed at 37°C in 0.1 M phosphate buffer (pH 7.0) using wavelengths and changes in extinction coefficients of β-lactams as previously described.36 Comparative activation of OXA-145 by CO2 was conducted by monitoring the rate of hydrolysis of selected β-lactams in the same buffer supplemented with 50 mM sodium hydrogen carbonate (NaHCO3) as a source of CO2. The steady-state kinetic parameters were determined from the initial rates using Hanes–Wolf linear regression. The enzyme concentrations in the reaction mixture were in the range 0.1–2 μM. All the values are the means of three independent measurements.
Results and discussion
Overall structure of OXA-145
The apo form of OXA-145 crystallized with two independent molecules forming a homodimer in the asymmetric unit. The two active sites were located on the same face of the dimer assembly. Each monomer folds as a two-domain protein (Figure 1). The α domain (residue 56–190) comprised six α helices and the β domain (residues 20–55 and 191–264) a seven-stranded antiparallel β sheet and the N- and C-terminal helices. The catalytic site was located at the interface of the two domains. The monomers were related by a 2-fold axis, which ran parallel to helices α9 and was nearly perpendicular to the plane defined by the β4 strands from the two monomers. Dimerization of OXA-145 involved an interaction site comprising helix α9 (181–182, 185–186, 188–189), strand β4 (192–197) from each monomer and residues from two loops (85–86, 105–109). The interface corresponded to 12% of the solvent-accessible surface of each monomer. Two networks of polar interactions stabilized the dimer at each extremity of the interaction site. The first, centred on Arg-109 (Arg-109–Tyr-199, Arg-109–Ala-196), was located at the C-terminal extremity of strand β4. The second network was organized around Glu-86 and Lys-181 (Glu-86–Lys-181, Glu-86–Lys-188, Lys-181–Glu-182) and was located at the N-terminal extremity of helix α9. These interactions occurred twice owing to the 2-fold symmetry and most are conserved in the structure of OXA-10, which shared with OXA-145 a root-mean-square deviation (RMSD) of the C-α backbone atoms of 0.85 Å (Figure 1).
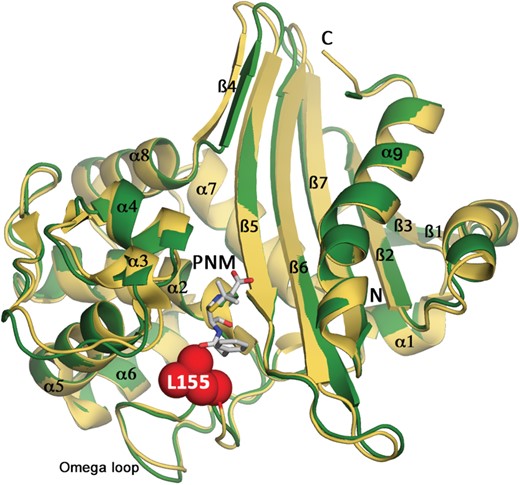
Overall structure of OXA-145. OXA-145 monomer (green) superimposed on OXA-10 (PDB: 1K54 yellow). Open form of penicillin G (PNM) is shown in white sticks and was modelled from the structure of the OXA-10 Trp154Ala mutant (PDB: 2WGI). Leu-155, absent in OXA-145, is indicated by spheres. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Active-site architecture of OXA-145 provides clues for the loss of benzylpenicillin hydrolysis
The co-crystallization of OXA-145 or trapping mutant S70G with penicillin G, oxacillin or ceftazidime was unsuccessful. The active site of OXA-145 was therefore compared with that of OXA-10 since the two structures closely resemble each other. Penicillin G was modelled from the structure of the acyl-enzyme OXA-10 W154A mutant (PDB: 2WGI) (Figure 1). The active site of OXA-145 was larger than that of OXA-10 (Figure 2a). The residues that interact with penicillin G in OXA-10 (i.e. Arg-250, Thr-206 and Phe-208) have conserved positions in OXA-145 (Figure 2b) suggesting that penicillin G would interact in the same manner as in OXA-10. In addition, the catalytic Ser-67 side chain in OXA-145 occupies a position consistent with the acyl-linkage observed in the OXA-10/W154A structure in complex with penicillin G. A major difference was conformation of the catalytic Lys-70. In OXA-10, Lys-70 is N-carboxylated whereas in OXA-145 this residue was in a free form. This could be attributed to the low pH of crystallization conditions (pH 6), which can affect the degree of carboxylation,8 but is most likely due to Leu-155 deletion. Lys-70 is housed in a relatively large hydrophobic cavity defined by Ile-112, Ala-116, Val-117 and Phe-120, which are in the same position and orientation in both enzymes (Figure 2b). Because of the Leu-155 deletion, the side chain of Trp-154 was shifted 1.8 Å from Lys-70. In OXA-type enzymes, Trp-154 interacts with the active-site carboxylated Lys-70 and stabilizes the deprotonated form of the carboxylate group.8 This promotes deprotonation of the catalytic Ser-67 to make a nucleophilic attack on the carbonyl of the β-lactam ring for the acylation step. In OXA-145, the nitrogen atom of the indole group of Trp-154 is not close enough to Lys-70 to stabilize its carboxylated form and a water molecule (W105) is present at the expected carboxylate position (Figure 2b). In addition, the hydrophobic environment surrounding Lys-70 allows reaction with CO2 by lowering its pKa thereby favouring the free base over the protonated form. The deletion of Leu-155 also resulted in a shift of the α-carbon of Glu-155 by 2 Å towards the hydrophobic cavity (Figure 2b). This shift could allow protonation of Lys-70, and thus prevent its carboxylation. Altogether, these features highlight the role of Leu-155 and explain why its deletion impairs hydrolysis of the penicillins.

Benzylpenicillin binding site. (a) Surface representation of OXA-145 (left, green) and OXA-10 (PDB: 1K54, right, yellow) active sites. (b) Stereo view of the active sites of OXA-145 and OXA-10 showing the non-carboxylated Lys-70 and the shifts of Trp-154 and Glu-155. Water molecules are indicated by spheres. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Hydrolysis of the penicillins is restored in the presence of NaHCO3
Hydrolysis of selected substrates was assayed in the presence of 50 mM NaHCO3 as a source of CO2 for the carboxylation of Lys-70 (Table 2). Kinetic parameters were compared with those obtained in the absence of NaHCO3.23 Except for ceftazidime, the addition of NaHCO3 significantly increased the catalytic efficiency against β-lactams. The greatest increases occurred for penicillin G and oxacillin (Table 2). In addition, hydrolysis curves were linear, in contrast with biphasic kinetics obtained in the absence of NaHCO3.23 However, catalytic efficiency was between 2% and 15% of that of the narrow-spectrum progenitor OXA-35.23 These findings provide indirect evidence that deletion of Leu-155 prevents carboxylation of Lys-70 in OXA-145.
Kinetic parameters of OXA-145 in the absence and in the presence of NaHCO3
| Substrate | Absence of 50 mM NaHCO3a | Presence of 50 mM NaHCO3 | ||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | |
| Penicillin G | NA | <0.05 | NA | 14.5 | 1.1 | 76 |
| Ampicillin | 5.2 | 0.3 | 52 | 7.5 | 1.1 | 148 |
| Ticarcillin | NA | <0.1 | NA | 173 | 0.5 | 2.9 |
| Piperacillin | NA | <0.02 | NA | 62.5 | 0.4 | 6.9 |
| Oxacillin | NA | <0.1 | NA | 66.2 | 2.3 | 34.7 |
| Cefalotin | 120 | 0.05 | 0.4 | 18.7 | 0.11 | 6.0 |
| Cefaloridine | 100 | 0.1 | 1.2 | 8.5 | 0.09 | 10.6 |
| Cefuroxime | 70 | 0.06 | 0.9 | 48.8 | 0.3 | 6.6 |
| Ceftazidime | 17.7 | 0.4 | 22 | 18.2 | 0.6 | 33 |
| Substrate | Absence of 50 mM NaHCO3a | Presence of 50 mM NaHCO3 | ||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | |
| Penicillin G | NA | <0.05 | NA | 14.5 | 1.1 | 76 |
| Ampicillin | 5.2 | 0.3 | 52 | 7.5 | 1.1 | 148 |
| Ticarcillin | NA | <0.1 | NA | 173 | 0.5 | 2.9 |
| Piperacillin | NA | <0.02 | NA | 62.5 | 0.4 | 6.9 |
| Oxacillin | NA | <0.1 | NA | 66.2 | 2.3 | 34.7 |
| Cefalotin | 120 | 0.05 | 0.4 | 18.7 | 0.11 | 6.0 |
| Cefaloridine | 100 | 0.1 | 1.2 | 8.5 | 0.09 | 10.6 |
| Cefuroxime | 70 | 0.06 | 0.9 | 48.8 | 0.3 | 6.6 |
| Ceftazidime | 17.7 | 0.4 | 22 | 18.2 | 0.6 | 33 |
NA, not applicable.
aData are from Hocquet et al.23
Kinetic parameters of OXA-145 in the absence and in the presence of NaHCO3
| Substrate | Absence of 50 mM NaHCO3a | Presence of 50 mM NaHCO3 | ||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | |
| Penicillin G | NA | <0.05 | NA | 14.5 | 1.1 | 76 |
| Ampicillin | 5.2 | 0.3 | 52 | 7.5 | 1.1 | 148 |
| Ticarcillin | NA | <0.1 | NA | 173 | 0.5 | 2.9 |
| Piperacillin | NA | <0.02 | NA | 62.5 | 0.4 | 6.9 |
| Oxacillin | NA | <0.1 | NA | 66.2 | 2.3 | 34.7 |
| Cefalotin | 120 | 0.05 | 0.4 | 18.7 | 0.11 | 6.0 |
| Cefaloridine | 100 | 0.1 | 1.2 | 8.5 | 0.09 | 10.6 |
| Cefuroxime | 70 | 0.06 | 0.9 | 48.8 | 0.3 | 6.6 |
| Ceftazidime | 17.7 | 0.4 | 22 | 18.2 | 0.6 | 33 |
| Substrate | Absence of 50 mM NaHCO3a | Presence of 50 mM NaHCO3 | ||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | |
| Penicillin G | NA | <0.05 | NA | 14.5 | 1.1 | 76 |
| Ampicillin | 5.2 | 0.3 | 52 | 7.5 | 1.1 | 148 |
| Ticarcillin | NA | <0.1 | NA | 173 | 0.5 | 2.9 |
| Piperacillin | NA | <0.02 | NA | 62.5 | 0.4 | 6.9 |
| Oxacillin | NA | <0.1 | NA | 66.2 | 2.3 | 34.7 |
| Cefalotin | 120 | 0.05 | 0.4 | 18.7 | 0.11 | 6.0 |
| Cefaloridine | 100 | 0.1 | 1.2 | 8.5 | 0.09 | 10.6 |
| Cefuroxime | 70 | 0.06 | 0.9 | 48.8 | 0.3 | 6.6 |
| Ceftazidime | 17.7 | 0.4 | 22 | 18.2 | 0.6 | 33 |
NA, not applicable.
aData are from Hocquet et al.23
Structural basis for extended-spectrum activity of OXA-145
Kinetic analysis showed that OXA-145 possesses the same hydrolytic activity against ceftazidime in the absence or in the presence of NaHCO3 (Table 2). The mechanism by which ceftazidime was inactivated is therefore unclear, suggesting another reaction that does not involve carboxylation of Lys-70. Of note, deletion of Leu-155 frees an additional cavity, which could allow the active site to accommodate such a bulky cephalosporin (Figure 3a). In recently reported structures of class D ESBLs, P227S substitution in OXA-160 and OXA-22537 or a four-amino-acid deletion (214-RIEP-217) in OXA-16338 affect the conformation of the β5 and β6 strands, which expands the active site to accommodate ceftazidime. The mechanism of their extended activity may be therefore related to that observed in CTX-M enzymes harbouring D240G substitution in the β3 strand,39 the structural homologue of the β5 strand in class D enzymes. In OXA-145, the conformation of the β5 and β6 strands is not modified compared with OXA-10 (Figure 1). Indeed, the expanded active site of OXA-145 is due to the deletion of Leu-155, the structural homologue of residue P167 in class A enzymes. In class A TEM-, SHV- and PER-type ESBLs, the key element of the activity against ceftazidime is the conformation of the Ω loop (residues 160–181), which widens the active site and avoids steric clash between position 167 and the 2-(2-aminothiazole-4-yl)-2-oxyimino substituent of ceftazidime.40,41 The extended activity of OXA-145 is probably based on similar logic.
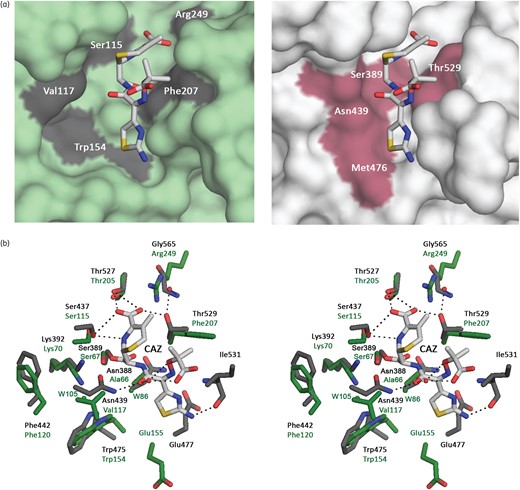
Modelling of ceftazidime binding site. (a) Surface representation of OXA-145 (left, green) and BlaR1 (PDB: 1XKZ, right, grey) active sites interacting with ceftazidime. (b) Stereo view of the active sites of OXA-145 (green) and OXA-10 (grey) showing interactions of ceftazidime (white sticks) with canonical active-site residues. Water molecules are indicated by green and red spheres, respectively, for OXA-145 and BlaR1. CAZ, ceftazidime. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
In the absence of a complex structure of the OXA-145 S67G mutant with ceftazidime, we studied the binding of the antibiotic by modelling its interaction with OXA-145 based on the structure of the sensor domain of BlaR1 from Staphylococcus aureus.42 The fold of this domain is similar to that of OXA-10-type β-lactamases and a superposition with OXA-145 results in an RMSD of the C-α backbone atoms of 1.29 Å. The superimposition revealed conservation of important active-site residues including Ser-67, Lys-70, Ser-115, Thr-205 and Trp-154 (Ser-389, Lys-392, Ser-437, Thr-527 and Trp-475, respectively, in BlaR1) (Figure 3b). Moreover, most of the residues interacting with ceftazidime in BlaR1 were conserved in OXA-145, except Asn-439, Asn-388 and Thr-529, which were replaced by, respectively, Val-117, Ala-66 and Phe-207 in OXA-145 (Figure 3b). Although these residues share several hydrogen bonds with ceftazidime that could be lost in OXA-145, ceftazidime might bind OXA-145 in the same position as in BlaR1. BlaR1 was shown to promote acylation of the active-site Ser-389 by a carboxylated Lys-392, as occurs in DBLs.42 However, it is unclear how OXA-145 is capable of hydrolysing ceftazidime with a non-carboxylated Lys-70.
To understand this activity, we performed molecular dynamics simulations of OXA-145 in complex with ceftazidime. The data showed that the side chain of Glu-155, which is solvent oriented in the structure of the apoenzyme, could be repositioned close to the active-site Ser-67 for ∼50% of the duration of the simulation (Figure 4). In this conformation, Glu-155 and Ser-67 can maintain a water molecule (W1) by hydrogen bonds near the β-lactam ring. This configuration is similar to that observed in the class A enzymes for the catalytic Glu-166 residue, which is known to be responsible for activation of the catalytic Ser-70 (ABL numbering) during the β-lactam acylation step of the catalytic process and the activation of the catalytic water molecule during the deacylation step.43 Thus, we hypothesize that hydrolysis of ceftazidime by OXA-145 could involve an alternative catalytic process based on the Glu-155 residue and the water molecule W1 located on the α-face of the acyl ester bond. However, mutation of Glu-155 to Ala or Gln did not impair ceftazidime hydrolysis. The catalytic efficiencies of E155A and E155Q mutants were 2–7-fold lower than that of the WT owing mainly to 2–5-fold higher KM values (Tables 2 and 3). This indicated that Glu-155 plays a role in ceftazidime binding. The E155A mutant led to a substantial increase in penicillin G hydrolysis, suggesting that Glu-155 contributes to lowering the hydrophobicity of the active-site cavity, which leads to Lys-70 decarboxylation and loss of penicillin G hydrolysis.
Kinetic parameters of OXA-145 E155A and E155Q mutants
| Substrate | E155A | E155Q | ||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | |
| Penicillin G | 112 | 2.4 | 21.4 | 67.2 | 0.68 | 10.1 |
| Ceftazidime | 93.3 | 0.3 | 3.2 | 40.1 | 0.4 | 10.0 |
| Substrate | E155A | E155Q | ||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | |
| Penicillin G | 112 | 2.4 | 21.4 | 67.2 | 0.68 | 10.1 |
| Ceftazidime | 93.3 | 0.3 | 3.2 | 40.1 | 0.4 | 10.0 |
Kinetic parameters of OXA-145 E155A and E155Q mutants
| Substrate | E155A | E155Q | ||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | |
| Penicillin G | 112 | 2.4 | 21.4 | 67.2 | 0.68 | 10.1 |
| Ceftazidime | 93.3 | 0.3 | 3.2 | 40.1 | 0.4 | 10.0 |
| Substrate | E155A | E155Q | ||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | KM (μM) | kcat (s−1) | kcat/KM (mM−1.s−1) | |
| Penicillin G | 112 | 2.4 | 21.4 | 67.2 | 0.68 | 10.1 |
| Ceftazidime | 93.3 | 0.3 | 3.2 | 40.1 | 0.4 | 10.0 |
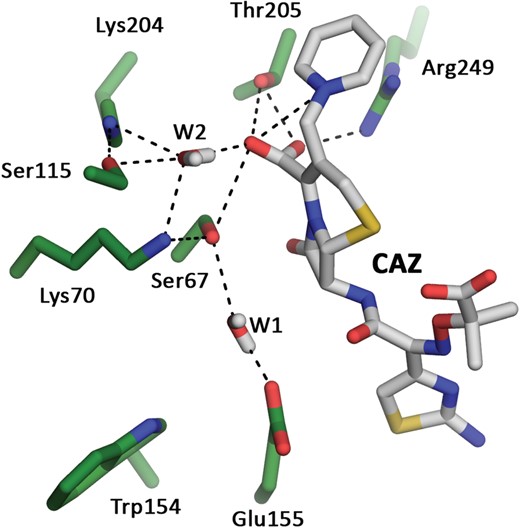
Molecular dynamics simulations of OXA-145 in complex with ceftazidime. Water molecules W1 and W2 are located, respectively, on the α- and β-face of the acyl ester of ceftazidime. CAZ, ceftazidime. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
We also observed from molecular dynamics simulations a second water molecule (W2) closely maintained on the β-face by a network of electrostatic interactions involving Ser-67, Lys-70, Ser-115, Lys-204, Thr-205 and the C2 carboxylate and the pyridinium function of ceftazidime (Figure 4). This conformation suggests that the catalytic process is assisted by ceftazidime and a water molecule (W2) located on the β-face of the acyl ester bond, as usually observed in CBLs. These class C enzymes do not appear to rely on a single residue as the catalytic general base, but rather on a hydrogen-bonding network acting as a charge relay system with assistance from the β-lactam substrate.6 In light of the mutagenesis of Glu-155 and in the absence of a complex structure of OXA-145 with ceftazidime, it is tempting to speculate that OXA-145 depends on the hydrogen-bonding network to activate Ser-67 for the acylation step.
Conclusions
In conclusion, the structure of OXA-145, which is the first structure of an extended-spectrum DBL from the OXA-10 family, provides an unusual example of the evolution of substrate specificity in class D enzymes. The study further highlights the role of the carboxylated Lys-70 in their mechanism of catalysis but also their ability to bypass the N-carboxylated Lys-70 and hydrolyse ceftazidime by another mechanism probably similar to that of CBLs.
Funding
This project was funded by an unrestricted grant from Reckitt Benckiser and by Institut Pasteur.
Transparency declarations
None to declare.
Acknowledgements
Preliminary work of this study was presented at the Fifty-first Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 2011 (Abstract C1-599).
We are grateful to ESRF (Grenoble, France) for provision of synchrotron facilities and thank the staff of beamline BM30A for assistance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}