





Abstract
Carbapenemase-resistant bacteria are increasingly spreading worldwide causing public concern due to their ability to elude antimicrobial treatment. Early identification of these bacteria is therefore of high importance. Here, we describe the development of a simple and robust protocol for the detection of carbapenemase activity in clinical isolates of Enterobacteriaceae, suitable for routine and clinical applications.
The final protocol involves cellular lysis and enzyme extraction from a defined amount of bacterial cells followed by the addition of a benchmark drug (e.g. the carbapenem antibiotic imipenem or ertapenem). Carbapenem inactivation is mediated by enzymatic hydrolysis (cleavage) of the β-lactam common structural motif, which can be detected using MALDI-TOF MS.
A total of 260 strains were studied (208 carbapenemase producers and 52 non-carbapenemase producers) resulting in 100% sensitivity and 100% specificity for the KPC, NDM and OXA-48-like PCR-confirmed positive isolates using imipenem as benchmark. Differences between the benchmark (indicator) antibiotics imipenem and ertapenem, buffer constituents and sample preparation methods have been investigated. Carbapenemase activity was further characterized by performing specific inhibitor experiments. Intraday and interday reproducibility (coefficient of variation) of the observed hydrolysis results were 15% and 30%, respectively. A comparative study of our extraction method and a recently published method using whole bacterial cells is presented and differences are discussed.
Using this method, an existing carbapenemase activity can be directly read from the mass spectrum as a ratio of hydrolysed product and substrate, setting an important step towards routine application in clinical laboratories.
Introduction
The discovery of antibiotics was one of the greatest scientific achievements of the twentieth century. Thanks to the selective action of these molecules, it has become possible not only to treat diseases once considered deadly but also, at the same time, to improve the quality of life. However, the outstanding success of antibiotics has also led to their misuse. The result of this has been the natural selection of resistant pathogenic bacteria that have spread worldwide mainly as nosocomial pathogens, but nowadays also in the community.1
One important example of acquired bacterial resistance that has become a major public concern in the past years is resistance to carbapenem antibiotics caused by bacterial carbapenemases.2 Carbapenems are a class of β-lactam antibiotics with a broad spectrum of antibacterial activity that inhibit the synthesis of the bacterial cell wall by complexing penicillin-binding proteins.3,4 Among β-lactam antibiotics, carbapenems are the most potent agents against Gram-negative bacteria and they are often used as last resort medication for the treatment of severe bacterial infections.5 At present, most laboratories make use of phenotypic assays such as the modified Hodge test or disc inhibitor combination discs to confirm the presence of a carbapenemase enzyme.6,7 More sophisticated laboratories apply PCR for limited genes encoding for (known) carbapenemases. Recently, the CarbaNP test has been introduced, which is based upon a pH-dependent colour change when a carbapenemase is present.8,9 All techniques mentioned, with the exception of the CarbaNP test and PCR-based assays, are relatively time consuming, requiring 1–2 days' time-to-result.
Recently, MALDI-TOF MS has been established as an important tool in routine microbiological laboratories for the identification of bacteria and fungi, proving to be a fast and reliable procedure for standard microbiological tests.10–13 Several papers have been published describing the use of MALDI-TOF to determine β-lactamase activity and carbapenemase activity.14–24 However, less attention has been paid to the standardization of the number of cells and the description of problems derived from the use of different buffers and substrates.
Here, we describe a new protocol for the detection of carbapenemase activity in clinical isolates in which we use enzymatic extracts obtained from a defined amount of bacterial cells. The extraction protocol was applied to a total number of 260 strains consisting of 208 carbapenemase producers and 52 non-carbapenemase producers for the detection of carbapenemase activity following the increase of the hydrolysed forms of the carbapenem antibiotics ertapenem and imipenem using MALDI-TOF MS. Extracts were made to obtain a higher yield of β-lactamase enzymatic activity, thereby facilitating the MS detection. Different incubation conditions were evaluated. We also compared our method with another MS-based method that uses suspensions of intact bacterial cells.15
Materials and methods
Bacteria
In the present study, 260 strains were analysed. These strains have previously been identified in our laboratory by using MALDI-TOF MS (Biotyper; Bruker Daltonics, Bremen, Germany) and mechanisms of resistance were characterized by PCR. Among these bacteria were: 55 Klebsiella pneumoniae isolates consisting of 25 K. pneumoniae carbapenemase positive (KPC+), 8 New Delhi metallo-β-lactamases (NDM), 17 oxacillinase-48-like (OXA-48) and 5 carbapenem-susceptible ESBL; 27 Escherichia coli isolates consisting of 6 OXA-48, 5 NDM and 16 carbapenem-susceptible plasmid-encoded AmpC-type β-lactamases (AmpC); 6 Enterobacter aerogenes isolates all NDM positive; 2 Enterobacter cloacae isolates OXA-48-like positive; 1 Citrobacter freundii isolate NDM positive; 6 Acinetobacter pittii isolates consisting of 2 NDM positive and 4 carbapenem susceptible; 90 Acinetobacter baumannii isolates consisting of 6 NDM, 70 OXA-23-like, 4 OXA-24-like and 10 carbapenem susceptible; 6 Acinetobacter nosocomialis carbapenem-susceptible strains; and 66 Pseudomonas aeruginosa isolates consisting of 21 IMP-type carbapenemases, 30 VIM, 4 SPM metallo-β-lactamases and 11 ESBL carbapenem susceptible. Isolates were derived from the ErasmusMC collection or were obtained from the National Institute for Public Health and the Environment (RIVM).
Growth conditions and standardization of the bacterial inoculum
Isolates were either grown overnight in an incubator at 37°C without shaking in 10 mL of Mueller–Hinton II (MHII) broth [Becton, Dickinson and Company (BD), NJ, USA] or on blood agar plates (BD). A standard value of McFarland 3.0 was adjusted either by diluting the liquid cultures grown overnight using MHII broth or by preparing a suspension of colonies grown on solid culture medium.
Preparation of bacterial cell extract
Two different methods to obtain bacterial extracts were tested and compared. The first method consisted of mechanical disruption of the microbial cells.25 Pellets from a 15 mL end-log phase culture in Todd–Hewitt medium with an OD of 1.0 were suspended in 1 mL of 25% sucrose in 10 mM Tris-HCl buffer (pH 7.5), followed by addition of 10 µL of freshly prepared lysozyme (20 mg/mL in 10 mM Tris-HCl buffer, pH 7.5). Then 2 mL of 1.5 mM EDTA in 10 mM Tris-HCl buffer (pH 7.5) was added and incubated for 60 min on ice. After the incubation period, 15 µL (10 × concentrated) of Protea Prep Protease inhibitor (SP-820; Protea, Langdal, France) was added. The total volume was then transferred into 2 mL vials containing 18 glass beads (diameter 3 mm) and subsequently shaken in a FastPrep system (FP120 Bio 101; ThermoSavant, Holbrook, NY, USA) during 15 s at a speed of 6 m/s. The lysate was then centrifuged for 5 min at 10 000 g at room temperature, supernatant (extract) was collected and the activity was determined by MALDI-TOF MS.
The second method consisted of an enzymatic-based extraction. Two different procedures were used for liquid cultures and culture plates (i and ii, respectively).
After overnight growth at 37°C in 10 mL of MHII broth, the bacterial culture was adjusted to McFarland 3.0 in 10 mL, and cells were centrifuged for 10 min at 3400 g. The resulting pellet was then resuspended in 200 µL of B-PER II Bacterial Protein Extraction Reagent (Thermo Fisher Scientific, USA) and incubated for 1 h at room temperature after which the suspension was centrifuged for 10 min at 3400 g. The resulting supernatant was used to determine carbapenemase activity.
Two mL of a McFarland 3.0 bacterial suspension was obtained starting from isolated bacterial colonies. Cells were then centrifuged for 5 min at 10 000 g in a table centrifuge at room temperature. The resulting pellet was then resuspended in 40 µL of B-PER II Bacterial Protein Extraction Reagent (Thermo Fisher Scientific) and incubated for 30 min at room temperature after which the suspension was centrifuged for 5 min at 10 000 g. The resulting supernatant was used to determine carbapenemase activity.
Nitrocefin and carbapenemase activity assays
To check the presence of β-lactamase/carbapenemase activity in both extracts obtained by mechanical extraction or by enzymatic extraction, the chromogenic substrate nitrocefin was used.
For the nitrocefin reaction,26 5 µL of bacterial extract was incubated together with 10 µL of nitrocefin (1 mM in rehydration fluid) and 185 µL of 10 mM HEPES/5 mM MgCl2 buffer (pH 7.5) for 30 min in a microtitre plate. The colour change was subsequently scored visually.
Phenotypic detection of carbapenemase activity
Carbapenemase activity was determined as described previously.27 Briefly, 100 µL of extract was added to 100 µL of ertapenem (50 mg/L) and incubated for 1.5 h at 37°C, and subsequently 10 µL was spotted on to paper filter discs. These discs were incubated on to MHII agar plates before being hand-inoculated with E. coli ATCC 25922 as an indicator organism. Different dilutions (5×, 10×, 20× and 40×) were made with the same buffers in which the cell extractions were performed. The assay was regarded positive if the zone of inhibition was 5 mm smaller than the zone diameter of the control.
Assay conditions
Initially, carbapenemase activity was determined by using 16 µL of bacterial extracts, diluted 10 times (10×) in 10 mM NH4H2PO4 supplemented with 2 µL of ertapenem solution and 2 µL of ZnCl2 (50 µM). In additional experiments the bacterial extracts were 10× diluted in 10 mM NH4HCO3 supplemented with 2 µL of imipenem and 2 µL of ZnCl2 (50 µM). The addition of ZnCl2 was essential to activate the NDM enzyme, thereby showing no negative or positive effects on either KPC or OXA-48 strain carbapenem hydrolytic activity (data not shown). The mixtures were incubated for 0, 15, 30, 60 and 100 min at 37°C in a thermomixer with shaking at 400 rpm. After each time point, 180 µL of acetonitrile was added to stop the reaction. The samples were then centrifuged for 2 min at 10 000 g in a table centrifuge at room temperature. The final concentration of ertapenem and imipenem in the final incubation solution was 0.1 mg/mL. Higher concentrations (1 mg/mL and 0.5 mg/mL) were used in pilot experiments showing no significant difference in method specificity and sensitivity (data not shown). For characterization experiments the inhibitors phenyl boronic acid (PBA) at a final concentration of 2 mg/mL and 2,6-pyridinedicarboxylic acid at a final concentration of 0.05 mg/mL were used, respectively, for KPC+ and NDM strains.
MALDI-TOF analysis
Supernatants from the bacterial extracts assay were analysed by MALDI-TOF MS using a thin layer preparation. One µL of a saturated α-cyanohydroxycinnamic acid solution (10 mg/mL in acetonitrile) was pipetted on to a MALDI stainless steel plate. After these spots dried, they were covered with 1 µL of sample supernatant also in acetonitrile (see above). This procedure generates crystals of high homogeneity. MALDI-TOF spectra were recorded on an Ultraflex III MS (Bruker Daltonics) operated in the reflectron and linear mode,28 and a random laser walk with a laser fluence of 5% above threshold until 5000 shots (50 shots per raster spot) were accumulated.29 A laser repetition rate of 50 Hz was used. Additional measurements were also performed on a Microflex LT linear MALDI-TOF MS (Bruker Daltonics), i.e. the standard MALDI Biotyper (BMT) system used in our routine laboratory, to ascertain whether our method can be applied to MS devices that are currently in use in many routine microbiology laboratories. The parameters for this system were as follows: mass range 100–1000 Da, ion source 1, 19 kV, ion source 2, 17.1 kV, lens, 6 kV and pulsed ion extraction 0 ns. A total of 240 laser shots in 40 shot packages were accumulated using a laser frequency of 60 Hz.
From control experiments (0 min incubation), we could establish the amount of background hydrolysis at <0.5%, for ertapenem concentrations of 1, 0.5 and 0.1 mg/mL. At lower concentrations (e.g. 0.01 mg/mL) background hydrolysis increased to 3% (data not shown). Therefore, it was decided to use an ertapenem concentration of 0.1 mg/mL or higher. For imipenem, background hydrolysis was found to be 0.8% at a concentration of 0.2 mg/mL.
In MALDI-TOF MS, the antibiotics appear as multiple peaks due to attachment of residual salt ions (e.g. Na+ and K+). The percentage of hydrolysis was calculated from the appropriate addition of all known peaks for the antibiotic and its hydrolysed form. No extraneous salt ions were added. The percentage of hydrolysis was calculated manually from the ratio of intensities as follows: percentage hydrolysis = (hydrolysed product)/(hydrolysed product + antibiotic) × 100 using MS EXCEL (Microsoft, Redmond, WA, USA).
Results
Determination of extraction efficiency
To be able to determine which extraction method resulted in the highest yield of carbapenemase activity, two different β-lactamase extraction methods using standardized numbers of bacterial cells were tested. In one method, 15 mL of bacteria at an OD600 of 1.0 were mechanically disrupted after treatment with lysozyme. In the second one, the same number of bacterial cells was enzymatically extracted at room temperature for 1 h using a pellet, obtained from 10 mL of a McFarland 3.0 culture. The results of these procedures were then compared using two different assays. First, nitrocefin, a chromogenic substrate routinely used to detect the presence of β-lactamase enzymes, was used to test the extracts. This assay is a good indicator of a present β-lactamase activity, but it is not specific for carbapenemases. Both mechanical disruption and enzymatic extraction released considerable amounts of β-lactamase activity as determined by nitrocefin hydrolysis (data not shown). Second, a phenotypic carbapenemase specific assay (see the Materials and methods section) was used to determine the carbapenem-hydrolytic activity of these enzymes. The results of this assay showed a better efficiency of the enzymatic method compared with the time-consuming mechanical disruption procedure (Figure 1a–c). The extracts obtained with the enzymatic procedure were able to hydrolyse the carbapenem antibiotic ertapenem even up to more than 10 × dilution (Figure 1a). The extracts obtained with the mechanical procedure, on the other hand, lost their hydrolytic activity after a 10 × dilution (Figure 1b). The negative control (32199) is a carbapenem-susceptible strain and is not able to hydrolyse ertapenem (Figure 1c). Taken together, these results indicate that the enzymatic extraction method is not only more efficient but also a substantially faster and easier to handle procedure for the extraction of carbapenemases compared with the mechanical method. For this reason, all the extracts from clinical isolates were further prepared using the enzymatic protocol.

Carbapenemase activity demonstration of extracts obtained by different methods. Carbapenemase enzymes were extracted from a KPC-positive isolate (K6+) by enzymatic extraction (a) or by mechanical extraction (b). Negative control (32199) is a KPC-negative strain and is not able to hydrolyse ertapenem (c).
MALDI-TOF analysis of carbapenemase activity
Initial experiments showed that to obtain reliable and reproducible MS data in a routine clinical setup, the amount of enzymatic extract used in the protocol needs to be standardized (data not shown). To achieve standardization, a fixed volume of 10 mL of MHII containing a McFarland 3.0 suspension was used to make extracts. It was observed that the use of undiluted extracts leads to serious ion suppression in the MS used. This phenomenon is the consequence of competition for ionization occurring in the ionization source, which ultimately results in decreased sensitivity.30 Furthermore, the distribution of the undiluted sample on to the stainless steel MALDI plate did not produce homogeneous crystals. For an even distribution of the sample on the stainless steel MALDI plate and to optimize the signal-to-noise ratio in the mass spectra, a 10 × dilution of the enzyme extract was found most suitable (data not shown).
In a proof-of-principle experiment, i.e. to confirm whether it was possible to detect carbapenemase activity in a sensitive, specific and reproducible way, 5 K. pneumoniae KPC+, 5 NDM, 5 OXA-48 and 5 ESBL isolates were randomly chosen from our archives. The strains were incubated overnight in MHII medium and enzymatic-obtained extracts from each strain were prepared the next day according to the final procedure described in the Materials and methods section. Initially, ertapenem and later on imipenem were used as substrates at a final concentration of 1 or 0.1 mg/mL, respectively. The samples were then analysed using MALDI-TOF (Ultraflex and Biotyper).
For several antibiotics, including ertapenem and imipenem, we and others have observed that hydrolysis may immediately be followed by decarboxylation and so the hydrolysis manifests itself mainly at a peak 26 Da lower than that of the antibiotic itself,17 rather than at 18 Da higher for the unperturbed hydrolysed form. This is exemplified in Figures 2 and 3, which show hydrolysis of ertapenem by a KPC+ strain (Figure 2) and hydrolysis of imipenem by an OXA-48 strain (Figure 3) after 0 min (Figures 2a and 3a) and after 60 min (Figures 2b and 3b) incubation. From Figure 2 it is clear that the detected peaks for the carbapenemase substrate ertapenem (m/z 498 [M + Na]+, m/z 514 [M + K]+, m/z 520 [M – H + 2Na]+ and m/z 536 [M – H + Na + K]+) have been partially replaced by a peak at m/z 472, the hydrolysed and decarboxylated sodium adduct product [M + H2O–CO2 + Na]+. The protonated forms (M + H)+ for ertapenem (m/z 476) and for its hydrolysed product (m/z 450) yield weak signals only. Similarly, Figure 3 shows that the detected peaks for the substrate imipenem (m/z 300 [M + H]+ and m/z 322 [M + Na]+) have been completely replaced by peaks at m/z 274 [M + H2O–CO2 + H]+ and m/z 296 [M + H2O–CO2 + Na]+ for the hydrolysed and decarboxylated form.
![Hydrolysis of ertapenem by a KPC+ strain after 0 min (a) and after 60 min (b) incubation. Ertapenem appears at m/z 498 [M + Na]+, m/z 514 [M + K]+, m/z 520 [M – H + 2Na]+ and m/z 536 [M – H + Na + K]+. Hydrolysed species appears as a decarboxylated form at m/z 472 [M + H2O–CO2 + Na]+. Extraction method was used for carbapenemase enzyme isolation and spectra were recorded on an Ultraflex III instrument.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jac/71/10/10.1093_jac_dkw208/1/m_dkw20802.jpeg?Expires=1748099300&Signature=k3~XnBnQriT40~LfoZmN0C1o12i-C-DeWBAtI5emLcDM7dUj1S1LssEAWirHp-hy9K93UOk2MgbYvWDXPrOixNThZUyY81nmUmV3YL7gjKGfX4Tp7qoy9mPALthAVjdVKzPaLEeb7FEY9cysR-ZfJIAQygzK8wzq-P3SoAQ1YnKcWDWjuvNp4ZYBCSnGg5ZgiWMx-eRStfPDVSCNHTNEpAnnF3Cv~cJgd0e~bONT1yozQy-asZczv7M9-fu~-H~G5-z2IS0QhpztgUyFTzEz71ZBPGGkhuvam0mK2WwcWKTj~tWoRAx6nDn0zEa2ywXvPyu14cdWQ9V8LriVQ0BpJw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Hydrolysis of ertapenem by a KPC+ strain after 0 min (a) and after 60 min (b) incubation. Ertapenem appears at m/z 498 [M + Na]+, m/z 514 [M + K]+, m/z 520 [M – H + 2Na]+ and m/z 536 [M – H + Na + K]+. Hydrolysed species appears as a decarboxylated form at m/z 472 [M + H2O–CO2 + Na]+. Extraction method was used for carbapenemase enzyme isolation and spectra were recorded on an Ultraflex III instrument.
![Hydrolysis of imipenem by an OXA-48 strain after 0 min (a) and after 60 min (b) incubation. Imipenem appears at m/z 300 [M + H]+ and at m/z 322 [M + Na]+. Hydrolysed species appears as decarboxylated forms at m/z 274 [M + H2O–CO2 + H]+ and at m/z 296 [M + H2O–CO2 + Na]+. Extraction method for carbapenemase enzyme isolation was used in (a) and (b), the whole-cell method was used in (c). Spectra recorded on an Ultraflex III instrument.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jac/71/10/10.1093_jac_dkw208/1/m_dkw20803.jpeg?Expires=1748099300&Signature=IYuBrH28lXpyv~P6-Lj9K7zu9WUg3JVVv8Nl3RTbyRsrHmuKdJGuZZx5p7LOmB3NTguCvvzNjIbO6v-xBHVXQYoxfPtv2B3Uy~uVpGUgDEkYi-KtfV2bTsDRusb6qPabBsqxTSbQERXOEuCSRPHbLZ2U7vdAMmKRUOU-FFLNO-slKv9L1ZiFrJ5TXDj~5A1tB9n-hE257YRK8kV0p34p-sVVFbt5rKqUfkKCZ2zI-nOzBYcgXoBH8c9N3Uj5tHPPRhkvekRCdj8DZJphFBpNddTlj8AovvNVV0an9yk6fQFDSMMl2ZJf79lL~5S4SmOu8vnnKq1ia37imXPt~fjXlQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Hydrolysis of imipenem by an OXA-48 strain after 0 min (a) and after 60 min (b) incubation. Imipenem appears at m/z 300 [M + H]+ and at m/z 322 [M + Na]+. Hydrolysed species appears as decarboxylated forms at m/z 274 [M + H2O–CO2 + H]+ and at m/z 296 [M + H2O–CO2 + Na]+. Extraction method for carbapenemase enzyme isolation was used in (a) and (b), the whole-cell method was used in (c). Spectra recorded on an Ultraflex III instrument.
For the KPC+ strains, we were able to observe the formation of a hydrolysed form of ertapenem already 15 min after incubation. Percentages of ertapenem hydrolysis determined (n = 5) at 0, 15, 30, 60 and 100 min were <0.5% ± 0.2%, 5% ± 2%, 10% ± 4%, 35% ± 14% and 85% ± 5%, when 1 mg/mL ertapenem was used. For validation, the experiments were repeated at least three times (intraday and interday) and the method proved 100% reproducible for all the strains tested as described (Figure 4a and b). To decrease time of analysis it was decided to lower the ertapenem concentration to 0.1 mg/mL. In this case, the reaction substrate was almost completely hydrolysed already after 30 min instead of 100 min in the KPC+ isolates, with hydrolysis percentages ranging from 82% (strain 648) to 100% (strain 639, 629 and 628) (Figure 5a). Similar results were obtained for the NDM-positive strains, with hydrolysis percentages after 30 min ranging from 58% (strain C9) to 94% (strain 12649). For NDM strains, the addition of 50 µM ZnCl2 to the reaction was required (Figure 5c). Without zinc, no hydrolysis was determined (no signal above background; data not shown), as NDM needs divalent cations as cofactor.31 The method proved selective, as ESBL strains, which do not possess a carbapenemase activity, were not able to hydrolyse ertapenem (Figure 5e). Using the described method for the OXA-48-like positive strains, which are known to possess carbapenemase activity,32 it was not possible to detect any hydrolysis of ertapenem (Figure 5f).
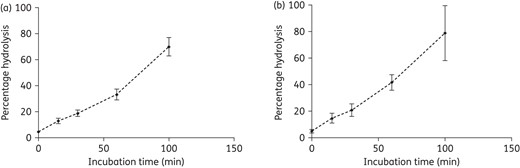
Intraday (a) and interday (b) reproducibility was obtained for a KPC+ isolate by making use of an extract obtained from a McFarland 3.0 suspension and concentrated in 200 µL of B-PER. The extract was diluted in ammonium phosphate (final concentration, 0.01 M). Used stock of ertapenem was 10 mg/mL.

Degradation of ertapenem determined by MALDI-TOF. Extracts obtained from different clinical isolates containing different β-lactamase enzymes were investigated. For β-lactamase determinations, extracts were obtained from 10 mL McFarland 3.0 suspensions and subsequently concentrated in 200 µL of B-PER extraction buffer. Extracts were diluted in ammonium phosphate buffer (0.01 M) before determinations. The final concentration of ertapenem used was 0.1 mg/mL. NDM enzyme activity was inhibited by addition of 0.05 mg/mL 2,6-pyridinedicarboxylic acid and KPC enzyme activity was inhibited by addition of 2 mg/mL PBA (final concentration). (a) KPC; (b) KPC + inhibitor. (c) NDM. (d) NDM + inhibitor. (e) ESBL. (f) OXA.
Experiments with inhibitors of carbapenemase activity were also performed to characterize further the hydrolytic enzymatic activity present in the clinical isolates. For the KPC+ isolates 2 mg/mL PBA was used, whereas 0.1 mg/mL 2,6-pyridinedicarboxylic acid, an ion chelator, was used to inhibit K. pneumoniae NDM extracts activity by removing the mandatory divalent cofactor (Figure 5b and d). Addition of PBA reduced the activity of KPC with 50%. The NDM activity was completely inhibited by 2,6-pyridinedicarboxylic acid. Higher concentrations of 2,6-pyridinedicarboxylic acid (0.5 mg/mL) also inhibited KPC, probably through mechanisms that are independent of carbapenem resistance (Figure 6).
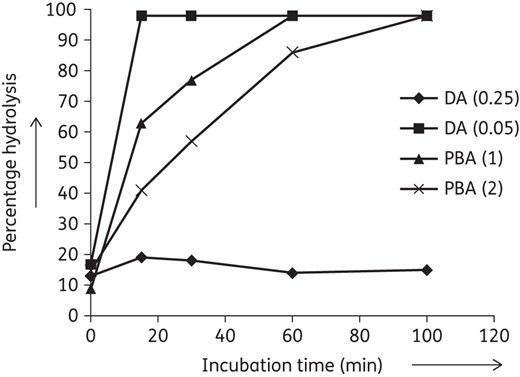
Selective effect of different concentrations of 2,6-pyridinedicarboxylic acid on KPC activity towards ertapenem. KPC carbapenemase activity determinations in the presence of 2,6-pyridinedicarboxylic acid (0.05), 2,6-pyridinedicarboxylic acid (0.25), PBA (1) and PBA (2). In parentheses, the final concentration of inhibitor is given in mg/mL. DA, 2,6-pyridinedicarboxylic acid.
Recently, it has been reported that the use of ammonium bicarbonate buffer significantly improves the sensitivity of screening methods for OXA-48.33 Further, it has been shown that these enzymes have a much higher affinity for imipenem compared with ertapenem.34,35 To overcome our initial false-negative results, it was decided to evaluate the assay for the OXA-48-like strains. For these strains, the hydrolysis assay was repeated using ammonium bicarbonate as incubation buffer, keeping the pH at 8, and providing imipenem as substrate [see Figure 3, which shows the hydrolysis of imipenem by an OXA-48 strain after 0 min (Figure 3a) and 60 min (Figure 3b) of incubation]. The percentage of hydrolysis for imipenem was calculated as follows: percentage hydrolysis = intensity (m/z 274 + m/z 296)/intensity (m/z 274 + m/z 296 + m/z 300 + m/z 322).
For imipenem, a background hydrolysis level of 0.8% in non-carbapenemase-producing strains was obtained (similar to 0.5% for ertapenem, see above). For imipenem, abundant formation of dimers and trimers was observed, which increased with the incubation time. Indeed, it is known that even in weakly acidic solutions, imipenem undergoes complex oligomerization reactions.36 For incubation periods of up to 1 h there was no effect on selectivity or specificity under the given assay conditions.
When using an imipenem concentration of 0.25 mg/mL, already after 30 min of incubation for all five OXA-48 isolates carbapenemase activity could be detected. After 90 min, the substrate was completely hydrolysed in all reaction mixtures (data not shown).
Carbapenemase detection in a clinical microbiology laboratory routine setting
With the previously described extraction method, we were already able to detect carbapenemase activity from Enterobacteriaceae in a specific and reproducible way using a combination of different buffers and substrates. This can be aided by the species identification information obtained from the MALDI-TOF. However, the aforementioned sample preparation procedure is time consuming and laborious, making its use in a routine setting cumbersome. For clinical routine application, the sample preparation procedure needs to be streamlined, faster and standardized. Accordingly, the protocol was modified by starting directly from freshly isolated bacterial colonies to make a McFarland 3.0 bacterial suspension to speed up the extract preparation. We also reduced sample-processing complexity by using only one universal buffer system and one antibiotic substrate for all isolates. The combination of ammonium bicarbonate and imipenem provided very fast and reliable results for all tested carbapenemase-producing isolates. The incubation time and the time until the samples are analysed using MALDI-TOF nevertheless had to be monitored precisely to avoid massive oligomerization. ZnCl2 was added to the reaction buffer as it is essential to activate NDM enzyme activity without disturbing non-NDM carbapenemase activity.34 With this modified protocol we were able to detect carbapenemase activity in the 5 K. pneumoniae KPC+, NDM and OXA-48 strains used in previous experiments in <2 h (from colonies to result). The ESBL control strains showed no hydrolysis of imipenem. The extracts were analysed both on an Ultraflex III MS and on a Microflex bench top MALDI-TOF MS, a routine MS now available in several clinical diagnostic laboratories.37
After these modifications and positive verification with a test panel of 20 isolates, the applicability of the current method was tested in a clinical routine laboratory. To validate our method we tested 240 additional strains acquired from the clinical routine laboratory and a reference laboratory. These included: 35 additional K. pneumoniae isolates comprising 20 KPC+, 3 NDM and 12 OXA-48-like; 27 E. coli isolates consisting of 6 OXA-48-like, 5 NDM and 16 plasmid-encoded AmpC; 6 E. aerogenes isolates all NDM positive; 2 E. cloacae isolates OXA-48-like positive; 1 C. freundii isolate NDM positive; 6 A. pittii isolates consisting of 2 NDM-positive isolates and 4 carbapenem-susceptible isolates; 90 A. baumannii isolates consisting of 6 NDM, 70 OXA-23-like, 4 OXA-24-like and 10 carbapenem susceptible; 6 A. nosocomialis carbapenem-susceptible strains; and 66 Pseudomonas aeruginosa isolates consisting of 21 IMP-type carbapenemases, 30 VIM, 4 SPM metallo-β-lactamases and 11 ESBL carbapenem susceptible.
We were able to detect carbapenemase activity in all KPC+, NDM, OXA-48-like, OXA-24-like and OXA-23-like strains tested. For KPC+ and NDM strains, the hydrolysis of imipenem was very rapid. As soon as the substrate was added to the reaction mixture at 0 min, already 50% of imipenem was promptly hydrolysed (Table 1). No hydrolysis was detected for the plasmid-encoded AmpC isolates or the susceptible A. baumannii calcoaceticus complex, A. pittii and A. nosocomialis isolates (Table 2).
Enterobacteriaceae strains used in this study and total average percentage of hydrolysis after 0 and 60 min of incubation of the extracts in ammonium bicarbonate buffer using imipenem as substrate and of the extracts in ammonium bisphosphate buffer using ertapenem as substrate
| Species | Number of strains | Percentage hydrolysis imipenem (average) 0 min | Percentage hydrolysis ertapenem (average) 0 min | Percentage hydrolysis imipenem (average) 60 min | Percentage hydrolysis ertapenem (average) 60 min | |
|---|---|---|---|---|---|---|
| KPC | Klebsiella pneumoniae | 25 | 58.4 | <1 | 99.0 | 91.7 |
| NDM | Klebsiella pneumoniae | 8 | 49.4 | <1 | 89.5 | 67.0 |
| NDM | Escherichia coli | 5 | 99.6 | <1 | 100 | 100 |
| NDM | Citrobacter freundii | 1 | 12.0 | <1 | 100 | 100 |
| NDM | Enterobacter aerogenes | 6 | 4.6 | 2.1 | 92.2 | 53.0 |
| OXA-48 | Klebsiella pneumoniae | 17 | 31.0 | 1.7 | 93.2 | 2.7 |
| OXA-48 | Escherichia coli | 6 | 5.6 | <1 | 100 | 2.3 |
| OXA-48 | Enterobacter aerogenes | 2 | 1.5 | 1.5 | 100 | 1.9 |
| ESBL | Klebsiella pneumoniae | 5 | 2.2 | <1 | 3.2 | <1 |
| AmpC | Escherichia coli | 16 | 3.6 | <1 | 3.5 | <1 |
| Species | Number of strains | Percentage hydrolysis imipenem (average) 0 min | Percentage hydrolysis ertapenem (average) 0 min | Percentage hydrolysis imipenem (average) 60 min | Percentage hydrolysis ertapenem (average) 60 min | |
|---|---|---|---|---|---|---|
| KPC | Klebsiella pneumoniae | 25 | 58.4 | <1 | 99.0 | 91.7 |
| NDM | Klebsiella pneumoniae | 8 | 49.4 | <1 | 89.5 | 67.0 |
| NDM | Escherichia coli | 5 | 99.6 | <1 | 100 | 100 |
| NDM | Citrobacter freundii | 1 | 12.0 | <1 | 100 | 100 |
| NDM | Enterobacter aerogenes | 6 | 4.6 | 2.1 | 92.2 | 53.0 |
| OXA-48 | Klebsiella pneumoniae | 17 | 31.0 | 1.7 | 93.2 | 2.7 |
| OXA-48 | Escherichia coli | 6 | 5.6 | <1 | 100 | 2.3 |
| OXA-48 | Enterobacter aerogenes | 2 | 1.5 | 1.5 | 100 | 1.9 |
| ESBL | Klebsiella pneumoniae | 5 | 2.2 | <1 | 3.2 | <1 |
| AmpC | Escherichia coli | 16 | 3.6 | <1 | 3.5 | <1 |
Enterobacteriaceae strains used in this study and total average percentage of hydrolysis after 0 and 60 min of incubation of the extracts in ammonium bicarbonate buffer using imipenem as substrate and of the extracts in ammonium bisphosphate buffer using ertapenem as substrate
| Species | Number of strains | Percentage hydrolysis imipenem (average) 0 min | Percentage hydrolysis ertapenem (average) 0 min | Percentage hydrolysis imipenem (average) 60 min | Percentage hydrolysis ertapenem (average) 60 min | |
|---|---|---|---|---|---|---|
| KPC | Klebsiella pneumoniae | 25 | 58.4 | <1 | 99.0 | 91.7 |
| NDM | Klebsiella pneumoniae | 8 | 49.4 | <1 | 89.5 | 67.0 |
| NDM | Escherichia coli | 5 | 99.6 | <1 | 100 | 100 |
| NDM | Citrobacter freundii | 1 | 12.0 | <1 | 100 | 100 |
| NDM | Enterobacter aerogenes | 6 | 4.6 | 2.1 | 92.2 | 53.0 |
| OXA-48 | Klebsiella pneumoniae | 17 | 31.0 | 1.7 | 93.2 | 2.7 |
| OXA-48 | Escherichia coli | 6 | 5.6 | <1 | 100 | 2.3 |
| OXA-48 | Enterobacter aerogenes | 2 | 1.5 | 1.5 | 100 | 1.9 |
| ESBL | Klebsiella pneumoniae | 5 | 2.2 | <1 | 3.2 | <1 |
| AmpC | Escherichia coli | 16 | 3.6 | <1 | 3.5 | <1 |
| Species | Number of strains | Percentage hydrolysis imipenem (average) 0 min | Percentage hydrolysis ertapenem (average) 0 min | Percentage hydrolysis imipenem (average) 60 min | Percentage hydrolysis ertapenem (average) 60 min | |
|---|---|---|---|---|---|---|
| KPC | Klebsiella pneumoniae | 25 | 58.4 | <1 | 99.0 | 91.7 |
| NDM | Klebsiella pneumoniae | 8 | 49.4 | <1 | 89.5 | 67.0 |
| NDM | Escherichia coli | 5 | 99.6 | <1 | 100 | 100 |
| NDM | Citrobacter freundii | 1 | 12.0 | <1 | 100 | 100 |
| NDM | Enterobacter aerogenes | 6 | 4.6 | 2.1 | 92.2 | 53.0 |
| OXA-48 | Klebsiella pneumoniae | 17 | 31.0 | 1.7 | 93.2 | 2.7 |
| OXA-48 | Escherichia coli | 6 | 5.6 | <1 | 100 | 2.3 |
| OXA-48 | Enterobacter aerogenes | 2 | 1.5 | 1.5 | 100 | 1.9 |
| ESBL | Klebsiella pneumoniae | 5 | 2.2 | <1 | 3.2 | <1 |
| AmpC | Escherichia coli | 16 | 3.6 | <1 | 3.5 | <1 |
Non-fermenting strains used in this study and total average percentage of hydrolysis after 0 and 60 min of incubation of the extracts in ammonium bicabonate buffer using imipenem as substrate and of the extracts in ammonium bisphosphate buffer using ertapenem as substrate
| Species | Number of strains | Percentage hydrolysis imipenem (average) 0 min | Percentage hydrolysis ertapenem (average) 0 min | Percentage hydrolysis imipenem (average) 60 min | Percentage hydrolysis ertapenem (average) 60 min | |
|---|---|---|---|---|---|---|
| NDM | Acinetobacter baumanii | 7 | 2.8 | <1 | 53.6 | 12.1 |
| NDM | Acinetobacter pittii | 2 | 2.5 | 1.7 | 93.5 | 25.0 |
| VIM | Pseudomonas aeruginosa | 30 | <1 | 2 | 59.4 | 90.2 |
| IMP | Pseudomonas aeruginosa | 21 | <1 | 5 | 59.9 | 86 |
| SPM | Pseudomonas aeruginosa | 4 | 2 | 75 | 65.5 | 100 |
| OXA-23 | Acinetobacter baumanii | 70 | 4.4 | <1 | 31.3 | <1 |
| OXA-24 | Acinetobacter baumanii | 4 | 39.0 | <1 | 77.0 | <1 |
| ESBL | Pseudomonas aeruginosa | 11 | 1.1 | 2 | <1 | 2 |
| Non-carbapenemase producing | Acinetobacter baumanii | 10 | 5.9 | <1 | 1 | <1 |
| Non-carbapenemase producing | Acinetobacter pittii | 4 | 1.2 | <1 | 1.5 | <1 |
| Non-carbapenemase producing | Acinetobacter nosocomialis | 6 | 1.1 | <1 | 1.5 | <1 |
| Species | Number of strains | Percentage hydrolysis imipenem (average) 0 min | Percentage hydrolysis ertapenem (average) 0 min | Percentage hydrolysis imipenem (average) 60 min | Percentage hydrolysis ertapenem (average) 60 min | |
|---|---|---|---|---|---|---|
| NDM | Acinetobacter baumanii | 7 | 2.8 | <1 | 53.6 | 12.1 |
| NDM | Acinetobacter pittii | 2 | 2.5 | 1.7 | 93.5 | 25.0 |
| VIM | Pseudomonas aeruginosa | 30 | <1 | 2 | 59.4 | 90.2 |
| IMP | Pseudomonas aeruginosa | 21 | <1 | 5 | 59.9 | 86 |
| SPM | Pseudomonas aeruginosa | 4 | 2 | 75 | 65.5 | 100 |
| OXA-23 | Acinetobacter baumanii | 70 | 4.4 | <1 | 31.3 | <1 |
| OXA-24 | Acinetobacter baumanii | 4 | 39.0 | <1 | 77.0 | <1 |
| ESBL | Pseudomonas aeruginosa | 11 | 1.1 | 2 | <1 | 2 |
| Non-carbapenemase producing | Acinetobacter baumanii | 10 | 5.9 | <1 | 1 | <1 |
| Non-carbapenemase producing | Acinetobacter pittii | 4 | 1.2 | <1 | 1.5 | <1 |
| Non-carbapenemase producing | Acinetobacter nosocomialis | 6 | 1.1 | <1 | 1.5 | <1 |
Non-fermenting strains used in this study and total average percentage of hydrolysis after 0 and 60 min of incubation of the extracts in ammonium bicabonate buffer using imipenem as substrate and of the extracts in ammonium bisphosphate buffer using ertapenem as substrate
| Species | Number of strains | Percentage hydrolysis imipenem (average) 0 min | Percentage hydrolysis ertapenem (average) 0 min | Percentage hydrolysis imipenem (average) 60 min | Percentage hydrolysis ertapenem (average) 60 min | |
|---|---|---|---|---|---|---|
| NDM | Acinetobacter baumanii | 7 | 2.8 | <1 | 53.6 | 12.1 |
| NDM | Acinetobacter pittii | 2 | 2.5 | 1.7 | 93.5 | 25.0 |
| VIM | Pseudomonas aeruginosa | 30 | <1 | 2 | 59.4 | 90.2 |
| IMP | Pseudomonas aeruginosa | 21 | <1 | 5 | 59.9 | 86 |
| SPM | Pseudomonas aeruginosa | 4 | 2 | 75 | 65.5 | 100 |
| OXA-23 | Acinetobacter baumanii | 70 | 4.4 | <1 | 31.3 | <1 |
| OXA-24 | Acinetobacter baumanii | 4 | 39.0 | <1 | 77.0 | <1 |
| ESBL | Pseudomonas aeruginosa | 11 | 1.1 | 2 | <1 | 2 |
| Non-carbapenemase producing | Acinetobacter baumanii | 10 | 5.9 | <1 | 1 | <1 |
| Non-carbapenemase producing | Acinetobacter pittii | 4 | 1.2 | <1 | 1.5 | <1 |
| Non-carbapenemase producing | Acinetobacter nosocomialis | 6 | 1.1 | <1 | 1.5 | <1 |
| Species | Number of strains | Percentage hydrolysis imipenem (average) 0 min | Percentage hydrolysis ertapenem (average) 0 min | Percentage hydrolysis imipenem (average) 60 min | Percentage hydrolysis ertapenem (average) 60 min | |
|---|---|---|---|---|---|---|
| NDM | Acinetobacter baumanii | 7 | 2.8 | <1 | 53.6 | 12.1 |
| NDM | Acinetobacter pittii | 2 | 2.5 | 1.7 | 93.5 | 25.0 |
| VIM | Pseudomonas aeruginosa | 30 | <1 | 2 | 59.4 | 90.2 |
| IMP | Pseudomonas aeruginosa | 21 | <1 | 5 | 59.9 | 86 |
| SPM | Pseudomonas aeruginosa | 4 | 2 | 75 | 65.5 | 100 |
| OXA-23 | Acinetobacter baumanii | 70 | 4.4 | <1 | 31.3 | <1 |
| OXA-24 | Acinetobacter baumanii | 4 | 39.0 | <1 | 77.0 | <1 |
| ESBL | Pseudomonas aeruginosa | 11 | 1.1 | 2 | <1 | 2 |
| Non-carbapenemase producing | Acinetobacter baumanii | 10 | 5.9 | <1 | 1 | <1 |
| Non-carbapenemase producing | Acinetobacter pittii | 4 | 1.2 | <1 | 1.5 | <1 |
| Non-carbapenemase producing | Acinetobacter nosocomialis | 6 | 1.1 | <1 | 1.5 | <1 |
With the combination of ertapenem and ammonium phosphate buffer, we were able to detect carbapenemase activity for the NDM and KPC+ strains, but no hydrolysis of ertapenem was observed for OXA-48-like, OXA-24-like, OXA-23-like, plasmid-encoded AmpC or any of the carbapenem-susceptible strains tested (Tables 1 and 2). Further very low hydrolysis of ertapenem was observed for the Acinetobacter species after 60 min (Table 2).
The combination of ammonium bicarbonate buffer and ertapenem was also tested and hydrolysis was observed for NDM, KPC+, OXA-48-like, OXA-24-like and OXA-23-like strains with no hydrolysis in plasmid-encoded AmpC isolates, ESBL or carbapenem-susceptible strains (data not shown). However, the amount of time needed to hydrolyse the substrate was longer compared with the combination ammonium bicarbonate buffer and imipenem.
Comparison of the extraction method and the whole-cell method
We have also compared the results of our extraction method and a recently published method using whole bacterial cells.15 The results are presented in Figure 7 for Enterobacteriaceae (Figure 7a and b) and non-fermenting strains (Figure 7c and d) using imipenem as the benchmark drug. A representative mass spectrum is shown in Figure 3(c).
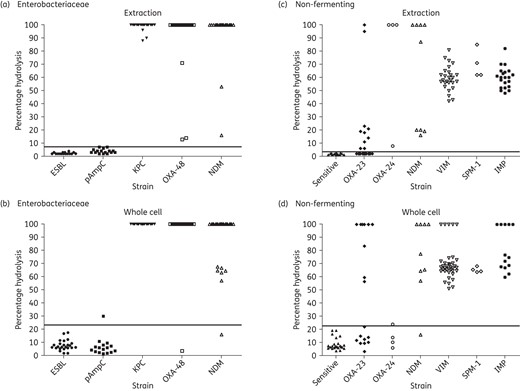
Percentage hydrolysis of various strains. (a) Enterobacteriaceae, extraction method. (b) Enterobacteriaceae, whole-cell method. (c) Non-fermenting strains, extraction method. (d) Non-fermenting strains, whole-cell method. The horizontal line represents the cut-off level, calculated as average of controls + 3 × SD.
Both sample preparation methods led to almost identical identification rates among the Enterobacteriaceae and non-fermenters tested except for OXA-23 strains, where the extraction approach led to a higher positive identification rate. The sample preparation time until data acquisition was 30 min in the whole-cell approach compared with 120 min in the extraction approach, respectively.
Discussion
The spread of antibiotic resistance poses a serious threat for public health and, as fewer new antibiotics are being introduced into the market, new strategies are needed to prevent the dissemination of lethal pathogens. One way to achieve this goal is to recognize the bacteria's specific resistance mechanism already at a very early stage. To do so, robust, fast and reproducible screening methods need to be available for clinical microbiological laboratories in their daily routine settings. The detection of the enzymatic activity against β-lactam antibiotics by using MS detection of the antibiotics' hydrolysis has been shown in several publications over the past years. Unfortunately, numerous sample preparation procedures, incubation conditions and antibiotics have been utilized for different bacterial strains. Here, we report the development and evaluation of a single, fast, robust and standardized protocol based on MALDI-TOF MS for the detection of carbapenemase activity in a broad set of clinical isolates.
The main objective of the present study was to investigate the effect of standardizing the whole hydrolysis assay methodology.
First, using an equal number of bacterial cells in each experiment should finally result in a robust method for everyday clinical routine practice. Standardization was achieved by adjusting the number of cells to a McFarland 3.0 turbidity. In our hands, similar growth conditions led to similar enzymatic activity in the bacterial isolates tested so far and use of standard amounts of bacteria led to a better reproducibility of the assay. Nevertheless, the absolute amount of active enzyme and its hydrolytic activity towards a specific antibiotic depends on each individual bacterial cell and can be influenced and altered by numerous biological and environmental factors.
Second, to reduce the assay complexity, the use of a single buffer system and a single benchmark antibiotic for all carbapenemase strains tested is favourable. We therefore evaluated ammonium phosphate and ammonium hydrogen carbonate incubation buffer solutions. Two common carbapenem antibiotics, ertapenem and imipenem, were compared. Taken together, the use of ammonium hydrogen carbonate buffer together with imipenem greatly improved the sensitivity towards most carbapenemases, including OXA-23 and OXA-48, and strongly decreased the duration of the assay with typical incubation times <60 min. For KPC+ and NDM strains the reaction was also very fast as observed for E. coli's strains (incubation time <60 min; Table 1). Table 3 shows the performances of the two tested benchmark antibiotics, imipenem and ertapenem. The major advantage of imipenem lies in its sensitivity towards OXA-48 as opposed to the negative result for ertapenem. In addition, the rate of hydrolysis for imipenem in the given incubation time is longer than that for ertapenem by a factor of 5.0 ± 1.7 (ratio for hydrolysis of imipenem and of ertapenem, n = 8), allowing for a more rapid assay. As the β-lactamase assay specifically detects the carbapenemase activity, antibiotic hydrolysis can be directly correlated with enzymatic presence. Therefore, a complete hydrolysis is not required to determine the presence of an active carbapenemase in a strain. This further decreases the theoretical assay time, as total hydrolysis is not required to select a suitable threshold. A drawback of using imipenem is the lower signal intensities obtained. MS signals for imipenem are about a factor 5 lower than that for ertapenem, demanding thorough assay preparation and workflows and therefore well-trained laboratory staff. Compared with ertapenem, imipenem is less stable in aquatic solution; indeed imipenem undergoes rapid dimerization,36 easily detected by a peak at m/z 599 in the mass spectrum. Carbapenemase activities towards the dimer are not known. Imipenem is further very prone to auto-hydrolysis in the presence of water and at low pH values. We found that the best mass spectra for the calculation of hydrolysation ratios were obtained when a fresh solution of imipenem is prepared and not used longer than for 2days. However, the sensitivity of imipenem towards OXA-48, we argue, outweighs its disadvantages and so we propose to use imipenem as a benchmark antibiotic for routine laboratories in combination with a precise operation procedure in a clearly defined workflow sequence.
Comparison of the benchmark antibiotics imipenem and ertapenem
| Observables | Imipenem | Ertapenem |
|---|---|---|
| Specificity | + | + |
| Sensitivity (excluding OXA) | + | + |
| Sensitivity for OXA-48 | + | − |
| In-use stability | − | + |
| MS response | − | + |
| Hydrolysis rate | + | − |
| Observables | Imipenem | Ertapenem |
|---|---|---|
| Specificity | + | + |
| Sensitivity (excluding OXA) | + | + |
| Sensitivity for OXA-48 | + | − |
| In-use stability | − | + |
| MS response | − | + |
| Hydrolysis rate | + | − |
Comparison of the benchmark antibiotics imipenem and ertapenem
| Observables | Imipenem | Ertapenem |
|---|---|---|
| Specificity | + | + |
| Sensitivity (excluding OXA) | + | + |
| Sensitivity for OXA-48 | + | − |
| In-use stability | − | + |
| MS response | − | + |
| Hydrolysis rate | + | − |
| Observables | Imipenem | Ertapenem |
|---|---|---|
| Specificity | + | + |
| Sensitivity (excluding OXA) | + | + |
| Sensitivity for OXA-48 | + | − |
| In-use stability | − | + |
| MS response | − | + |
| Hydrolysis rate | + | − |
The rationale behind the use of cell extracts resides in the fact that β-lactamases are captured in the periplasmic space.38 Therefore, extracts contain a higher yield of enzymes in an incubation assay and this should result in a higher activity towards the antibiotic. In addition, some strains have modified cell walls or other resistance mechanisms masking the hydrolytic activity. Extraction of enzymes should lead to a detectable enzymatic activity also in those strains. We evaluated a mechanical and an enzymatic lysis approach.
Using enzymatic lysis resulted in most reliable results compared with a mechanical cell disruption, combining both ease of use and reduced hands-on time. The enzymatic lysis extraction furthermore resulted in a higher enzymatic activity. This enabled us to dilute the extracts, resulting in a better MS signal-to-noise ratio and improving the reproducibility and detection of substrates and hydrolysed products. Dilution of the extracts by a factor of 10 led to considerable hydrolysis of the carbapenem antibiotics in a time frame of 60 min.
In a recent publication the disappearance of the substrate imipenem after 20 min incubation at 37°C has been correlated with the emergence of an unidentified metabolite at m/z 254 corresponding to the loss of 46 Da from imipenem.22 In the present study the same peak was also observed, but only when using whole cells; on the other hand this particular unidentified metabolite was never present when using β-lactamase extracts. The mechanism of formation of this potential metabolite remains unclear. Further, an intense 254 Da peak together with the 300 Da peak corresponding to imipenem was also observed in the spectra of carbapenem-susceptible strains when using bacterial suspensions instead of extracts. We could find only a weak correlation between the intensity of the m/z 254 peak and observed amount of hydrolysis, see Figure S1 (available as Supplementary data at JAC Online). In effect, the cut-off values are exceedingly large using this potential metabolite. We conclude not to consider this mass for calculating imipenem hydrolysis.
To investigate the specificity of the reaction, we used inhibitors for KPC and NDM strains.39,40 In our study we used 2,6-pyridinedicarboxylic acid (also known as ‘dipicolinic’ acid), a known inhibitor of metallo-β-lactamases.38 This molecule is a strong inhibitor of metallo-β-lactamase activity and efficiently blocked the hydrolysis of ertapenem by NDM strains at a concentration of 0.05 mg/mL. A higher concentration of 0.25 mg/mL inhibited additionally the β-lactamase activity of KPC extracts. KPC belongs to class A carbapenemases, which do not need divalent cations as cofactors. It is possible that high concentrations of dipicolinic acid have a secondary effect on other components of the extracts, resulting in inhibition of KPC carbapenemases. A different effect has been observed for PBA, which is used as an inhibitor for KPC strains.39,41,42 The mechanism of action consists of the formation of a reversible bond between the boron and the β-lactamase, which is not hydrolysed by the enzyme.42,43 The addition of PBA at a concentration of 2 mg/mL can slow the activity of KPC strains down to 50% through inhibition, but not lower. As inhibition in this case is based on a competition mechanism, probably the amount of PBA used in our assay was not sufficient to cope with the carbapenemases present in KPC extracts, as a lower concentration of PBA (1 mg/mL) has a lower inhibitory effect. As the assay just uses intensity ratios, total inhibition is not necessary. A simple calculation of a suitable cut-off value is already sufficient to distinguish clearly the inhibited carbapenemase assays from the assays not inhibited. This value can also be set on an empirical basis or using controls in case of the whole-cell approach.
Third, data processing is usually a very time consuming and error-prone step in clinical laboratory routine, particularly in MS. Data processing was based on the ratio of hydrolysis observed from the intensity ratios of intact and hydrolysed antibiotic forms (including respective adducts). To calculate the ratios it was assumed that the intact antibiotic and its enzymatically hydrolysed and further decarboxylated forms have similar ionization efficiencies in MALDI. That is to say, it is assumed that ionization efficiency of the respective adduct (e.g. Na+ and K+) are comparable for both the substrate and product forms. If the ratio exceeds a certain threshold, the test can be interpreted as positive. The threshold can be therefore set precisely and allows reproducible analysis results, which can also be easily electronically stored, making this method more favourable than, e.g. colorimetric assays having a strong human bias and which are difficult to store in a routine workflow. Data calculation and interpretation could further be automated by using dedicated software. Using an optimized algorithm could reduce the hydrolysis rate required for a correct identification, and thereby the incubation time.
Recently, results comparable to those presented in this work were obtained by making use of whole bacterial cell suspensions in a MALDI-TOF-based approach.22 We finally compared both methods with respect to specificity, sensitivity, background levels, assay duration and possibility for quantification for kinetic experiments. A total of 260 strains were compared for both methods. As can be seen from Figure 7, both methods lead to similar carbapenemase identification rates in a binary (yes-or-no) way, with the extraction method (method I) resulting in a significantly lower cut-off level, probably because the spectra obtained by this method are ‘cleaner’ leading to better signal-to-noise ratios.
Standardization of the number of cells followed by extraction resulted in negligible hydrolysis in the respective negative samples and in extensive hydrolysis in positive samples using the β-lactamase assay. Using whole cells for carbapenemase detection resulted in negative controls showing some hydrolysis, possibly due to enhanced auto-hydrolysis catalysed by unknown cellular components, as can be inferred from Figures 2 and 3. In contrast to the extraction approach, normalization to dedicated positive and negative control strains is therefore required for the whole-cell approach to compensate for this effect of an assay-to-assay variation (decrease in substrate and increase in a hydrolysed product).
Using A. baumannii isolates in a 60 min incubation assay resulted in a heterogeneous sensitivity: OXA-23 strains showed a reduced hydrolysis rate in both the extraction and the whole-cell assay, with the extraction assay showing better hydrolysis results. OXA-24 strains were positively identified using the extraction method, whereas the whole-cell assay produced significantly less hydrolysis and therefore only few positive identifications. Probable availability of the substrate for carbapenemases was the reason in the case of the extracts. OXA-23 and OXA-24 strains were therefore not included in our assessment of specificity and sensitivity. The comparative results of both methods are given in Table 4. Nevertheless the assay itself showed a superior sensitivity compared with most phenotypic assays currently used in routine settings.6,7 Prolonged incubation times and further modified assay conditions such as pH value could further improve analytic sensitivity.
Comparative results for method I (extraction; enzymatic lysis) and method II (whole cell)
| Properties/observables | Enterobacteriaceae strains (n = 91) | Non-fermenting strains (n = 169) | ||
|---|---|---|---|---|
| method Ia | method IIb | method Ia | method IIb | |
| Specificityc | 100% (80.8–100) | 95.2% (74.1–99.8) | 100% (86.3–100) | 100% (86.3–100) |
| Sensitivityc | 100% (93.5–100) | 97.1% (89.1–99.5) | 100% (96.6–100) | 99.3% (95.4–100) |
| Variation in background leveld | 1%–7% | 1%–23% | 1%–3% | 3%–23% |
| Assay time | − | + | − | + |
| Quantification | + | − | + | − |
| Properties/observables | Enterobacteriaceae strains (n = 91) | Non-fermenting strains (n = 169) | ||
|---|---|---|---|---|
| method Ia | method IIb | method Ia | method IIb | |
| Specificityc | 100% (80.8–100) | 95.2% (74.1–99.8) | 100% (86.3–100) | 100% (86.3–100) |
| Sensitivityc | 100% (93.5–100) | 97.1% (89.1–99.5) | 100% (96.6–100) | 99.3% (95.4–100) |
| Variation in background leveld | 1%–7% | 1%–23% | 1%–3% | 3%–23% |
| Assay time | − | + | − | + |
| Quantification | + | − | + | − |
aExtraction method.
bMethod using whole cells.
cValues in parentheses refer to specificity and sensitivity with 95% CI.
dPercentage hydrolysis of negative controls.
Comparative results for method I (extraction; enzymatic lysis) and method II (whole cell)
| Properties/observables | Enterobacteriaceae strains (n = 91) | Non-fermenting strains (n = 169) | ||
|---|---|---|---|---|
| method Ia | method IIb | method Ia | method IIb | |
| Specificityc | 100% (80.8–100) | 95.2% (74.1–99.8) | 100% (86.3–100) | 100% (86.3–100) |
| Sensitivityc | 100% (93.5–100) | 97.1% (89.1–99.5) | 100% (96.6–100) | 99.3% (95.4–100) |
| Variation in background leveld | 1%–7% | 1%–23% | 1%–3% | 3%–23% |
| Assay time | − | + | − | + |
| Quantification | + | − | + | − |
| Properties/observables | Enterobacteriaceae strains (n = 91) | Non-fermenting strains (n = 169) | ||
|---|---|---|---|---|
| method Ia | method IIb | method Ia | method IIb | |
| Specificityc | 100% (80.8–100) | 95.2% (74.1–99.8) | 100% (86.3–100) | 100% (86.3–100) |
| Sensitivityc | 100% (93.5–100) | 97.1% (89.1–99.5) | 100% (96.6–100) | 99.3% (95.4–100) |
| Variation in background leveld | 1%–7% | 1%–23% | 1%–3% | 3%–23% |
| Assay time | − | + | − | + |
| Quantification | + | − | + | − |
aExtraction method.
bMethod using whole cells.
cValues in parentheses refer to specificity and sensitivity with 95% CI.
dPercentage hydrolysis of negative controls.
In contrast, hydrolysis rates in NDM strains were better assessed using the whole-cell approach compared with the extraction approach. This is probably due again to the fact that NDM needs divalent cations as cofactors, which are obviously less effective in the extraction approach although supplemented with the buffer. Again, by using a dedicated threshold for the detection of a positive hydrolysation reaction, a total hydrolysis is not required to identify a carbapenemase strain, therefore enabling both methods to produce highly sensitive and reproducible results. Notably, both methods give very high specificities and sensitivities (extraction method: 100%; whole-cell method >95%).
Making use of a standardized number of cells and subsequent extraction makes this method less technician dependent, but, on the other hand, using whole cells is less laborious and faster, with possible hands-on times about 30 min compared with 120 min for method I. However, due to the standardization of method I, it allows quantitative measurements for kinetic experiments on more sophisticated (and expensive) MS systems for specific questions, as shown in Figure 5 and inhibition experiments as seen in Figure 6.
Recently, it has been suggested that a MALDI-TOF-based method for carbapenemase detection would not be adequate for a clinical routine laboratory.44 By using a standardized protocol combined with a linear MALDI-TOF like the MBT, an instrument present in many clinical laboratories, sufficient data for application of this assay were produced (data not shown), making this procedure applicable and affordable for many microbiology users. Furthermore, the presence or absence of carbapenemase activity, using whole cells or extracts, can be obtained in 1 or 2 h, thereby providing important information for the optimization of the antimicrobial therapy.
In conclusion, the present study shows that carbapenemase activity can be detected in a reproducible way by a standardized extraction method coupled with an MS analysis of the sample using the substrate and the hydrolysed mass peak of imipenem. All KPC, NDM and OXA-48 positive isolates tested were confirmed to possess high yields of carbapenemase activity in contrast to the negative ESBL and plasmid-encoded AmpC controls. Further characterization of carbapenemase activity can be achieved by adding specific enzyme inhibitors in the reaction mixture. This is an important step towards the improvement of characterization of carbapenemase-producing strains in a routine setting. In comparison with results obtained by PCR confirmation, the method in which extracts are applied results in 100% sensitivity and 100% specificity (Table 4). However, more importantly our technique represents a comprehensible and robust assay that can lead to improved patient care. Owing to its relative ease of use and time savings the presented method can be readily introduced in to the daily clinical microbiology laboratory workflow.
Funding
This research received funding from the Netherlands Organisation for Scientific Research (NWO) in the framework of the Technology Area COAST.
Transparency declarations
None to declare.
Acknowledgements
The Rijksinstituut voor Volksgezondheid en Milieu (RIVM, Bilthoven, the Netherlands) is acknowledged for providing reference isolates.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}