





Abstract
The use of genotypic resistance testing of herpes simplex virus types 1 and 2 (HSV-1 and HSV-2) is increasing because the rapid availability of results significantly improves the treatment of severe infections, especially in immunocompromised patients. However, an essential precondition is a broad knowledge of natural polymorphisms and resistance-associated mutations in the thymidine kinase (TK) and DNA polymerase (pol) genes, of which the DNA polymerase (Pol) enzyme is targeted by the highly effective antiviral drugs in clinical use. Thus, this review presents a database of all non-synonymous mutations of TK and DNA pol genes of HSV-1 and HSV-2 whose association with resistance or natural gene polymorphism has been clarified by phenotypic and/or functional assays. In addition, the laboratory methods for verifying natural polymorphisms or resistance mutations are summarized. This database can help considerably to facilitate the interpretation of genotypic resistance findings in clinical HSV-1 and HSV-2 strains.
Introduction
Within the Herpesviridae family, herpes simplex virus (HSV) belongs to the α-Herpesvirinae subfamily and the genus Simplexvirus, which is divided into the species HSV type 1 (HSV-1) and type 2 (HSV-2). The viruses have the unique property of remaining latent throughout life in sensory ganglia after newly acquired infections and may be reactivated endogenously. HSVs infect humans throughout the world. The seroprevalence of HSV-1 in Europe ranges from 50% to >90% and that of HSV-2 from 4% to 24%.1–3 Usually, the viruses cause self-limiting orolabial or genital lesions in immunocompetent individuals, but infections may also result in substantial physical and psychological morbidity. Especially in immunocompromised patients, HSV infections tend to be associated with painful, widespread and chronic exanthema and/or enanthema, leading to significant morbidity.4 After HSCT, the risk of recurrent HSV disease mainly caused by HSV-1 is ∼80%.5 Thus, this group of patients requires prolonged antiviral treatment, increasing the risk of development of drug-resistant viruses.
To date, the nucleoside analogue aciclovir and its prodrug valaciclovir have primarily been the drugs of choice for the treatment of HSV infections. Moreover, other nucleoside analogous compounds, like penciclovir and its prodrug famciclovir as well as brivudin [(E)-5-(2-bromovinyl)-2′-deoxyuridine, effective against HSV-1 and not effective against HSV-2], are available for the treatment of HSV diseases. The activation of these drugs requires three intracellular phosphorylation steps, which are catalysed by the viral thymidine kinase (TK) and cellular kinases. In the vast majority of cases, aciclovir resistance is associated with cross-resistance to the other nucleoside analogues.6 In such cases, the pyrophosphate analogue trisodium phosphonoformate (foscarnet) and the nucleotide analogue cidofovir can be recommended for alternative treatment. Both compounds act as inhibitors of the viral DNA polymerase (Pol) enzyme. In immunocompetent individuals, resistance to aciclovir has a low prevalence of <1%.7,8 Exceptions are patients with recurrent herpetic keratitis (HSV-1), in ∼6% of whom resistant isolates have been described.9,10 By contrast, the prevalence of aciclovir-resistant HSV strains has been reported in 2.5%–10.9% of immunocompromised patients, including patients with genital herpes (HSV-2),8,11–13 and several studies have described an increase in prevalence of aciclovir resistance in HSCT patients to 47% during recent years.14,15 Cross-resistance of foscarnet to aciclovir has only been found in rare cases2,16–20 and predominantly after combination treatment of immunocompromised patients.21
According to data from the literature, resistance to aciclovir is mediated in 95% of cases by frameshift mutations or by amino acid (aa) substitutions in the TK gene (UL23, 1130 bp, 376 aa), especially in active or conserved regions.6,22–24 A small amount of aciclovir resistance as well as resistance to foscarnet is related to aa substitutions within the DNA pol gene (UL30; HSV-1, 3707 bp, 1235 aa; HSV-2, 3722 bp, 1240 aa), mostly clustering in conserved loci.25 Resistance analysis is carried out conventionally by the determination of viral phenotype. Reliable but time-consuming methods like the plaque reduction assay (PRA), dye uptake assay or DNA hybridization assay allow a clear interpretation of experimental findings. However, in clinical practice phenotypic testing delays resistance-adjusted treatment. Thus, rapid genotypic testing by sequence analysis of TK and DNA pol genes has increasingly been established as the method of choice for diagnosing aciclovir and/or foscarnet resistance. However, the interpretation of genotypic findings is often difficult due to the high variability of TK and DNA pol genes26,27 and the required knowledge of resistance-related mutations.
This review presents a database of all non-synonymous mutations of TK and DNA pol genes of HSV-1 and HSV-2 that have been reported in the literature and whose association with natural polymorphism or resistance has been determined unequivocally by phenotypic and/or functional assays. Furthermore, laboratory methods for the verification of natural polymorphisms or resistance mutations are listed. The availability of this database should promote the use of genotypic resistance testing of HSV-1 and HSV-2 and may significantly improve the interpretation of results.
Amino acid changes within the TK gene of HSV-1 and HSV-2
Natural polymorphism
The database of natural polymorphisms within the TK gene is presented in Table S1 (available as Supplementary data at JAC Online http://jac.oxfordjournals.org/) (HSV-1) and Supplementary Data (HSV-2). In addition, Figure 1(a) (HSV-1) and Figure 1(b) (HSV-2) illustrate their positions in relation to the active or highly conserved TK gene regions. In HSV-1/HSV-2, aa 51–63 correspond to a glycine-rich loop that forms the ATP-binding pocket while aa 168–176 (HSV-1) and 169–177 (HSV-2) are involved in thymine/deoxythymidine recognition.28 Furthermore, highly conserved regions have been described for aa loci 83–88 (HSV-1) or 86–89 (HSV-2), 162–164 (HSV-1) or 163–165 (HSV-2), 216–222 (HSV-1) or 216–223 (HSV-2) and 284–289 (HSV-1) or 285–290 (HSV-2).
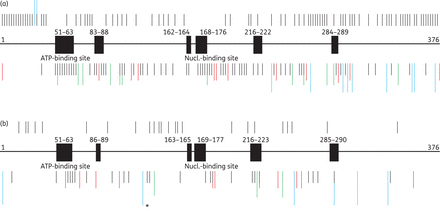
Schematic of the TK gene with conserved/active regions (black boxes) and non-conserved regions (bold line). Also shown is an overview of the positions of published aa substitutions (black bars), stop codons due to nucleotide changes (green bars), stop codons due to nucleotide insertions or deletions (red bars) and aa frameshifts without premature stop codons (blue bars) within the TK gene of HSV-1 (a) and HSV-2 (b). Bars above the TK gene show positions of natural polymorphisms and bars below indicate positions of resistance-associated substitutions. One frameshift marked with an asterisk indicates a unique exceptional deletion of 969 nucleotides that extends into the 3′-untranslated region of the TK gene. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
All natural polymorphisms have been identified within the TK gene of HSV-1 and HSV-2 strains susceptible to aciclovir, penciclovir, brivudin, foscarnet and/or cidofovir. The substitutions are clustered over the whole TK gene, predominantly outside of conserved or active regions. For HSV-1, the database contains 109 polymorphisms. Of these, 107 are aa substitutions resulting from nucleotide changes and two are deletions (Gln35deletion and Lys36deletion). Six aa substitutions arose from more than one nucleotide change. Most natural polymorphisms are located within the aa ranges 6–44 (25/109, 22.9%) and 214–376 (60/109, 55.0%); a lower density has been analysed within the aa range 61–213 (24/109, 22.0%). Three out of 109 HSV-1 polymorphisms (2.8%) have been observed within active or conserved loci. Interestingly, no natural polymorphisms have been reported in the conserved aa region 162–164 and the nucleoside-binding site of HSV-1. Black and Loeb29 reported that codon 163 plays an essential role in the function of the TK protein. Furthermore, changes of aa 173 within the nucleoside-binding site also induce a failure of enzyme function because of the crucial significance for binding nucleosides. It has been shown for aciclovir-susceptible HSV-1 strains that replacement of Pro173 cannot be tolerated.30 In conclusion, natural polymorphisms in TK of HSV-1 are mainly located outside of conserved regions (106/109, 97.2%) and very rarely inside of conserved/active gene loci (3/109, 2.8%).
In the TK of HSV-2, 23 polymorphisms have been published. All represent aa substitutions. One polymorphism out of 23 (4.3%) polymorphisms has been found within a conserved locus (R220K). In general, the proportion of natural polymorphisms is lower in HSV-2 than in HSV-1 TK. This is predominantly due to the small number of resistance analyses of HSV-2 compared with HSV-1 strains. The HSV-2 TK exhibits only two polymorphisms within codons 252–376. Furthermore, no natural polymorphisms could be analysed in the conserved aa regions 51–63, 86–89, 163–165 and 285–290 and the nucleoside-binding site of HSV-2. One possible explanation for the lack of substitutions within aa 163–165 could be the lower enzyme activity.29 Notably, the substitution Gly39Glu was classified as a natural polymorphism of HSV-2 TK, despite its having been detected as the sole aa substitution in aciclovir-resistant HSV-2 strains.31
Resistance
The database of resistance-related non-synonymous mutations within the TK gene is summarized in Supplementary Data (HSV-1) and Supplementary Data (HSV-2) as well as in Figure 1(a) (HSV-1) and Figure 1(b) (HSV-2). Out of 134 different resistance-related mutations in HSV-1 TK, 85 (63.0%) have been characterized as nucleotide substitutions resulting in aa changes, 25 (18.7%) were due to frameshift mutations leading to stop codons, 12 (8.9%) were stop codons through nucleotide changes, 10 (7.5%) were frameshifts without premature stop codons, one (0.7%) in-frame three-nucleotide insertion led to an additional aa (Ala301insertion), and one (0.7%) in-frame three-nucleotide deletion led to an aa deletion (Ile194deletion). Five aa substitutions and two stop codons arose from more than one nucleotide change and six stop codons resulted from different nucleotide frameshifts. All corresponding viral strains but one were resistant to aciclovir and additionally to penciclovir and brivudin when tested, but susceptible to foscarnet and cidofovir when tested. One HSV-1 strain containing the aa substitution Ala168Thr within the nucleoside-binding site was considered brivudin resistant but aciclovir/penciclovir/foscarnet/cidofovir susceptible.
In accordance with data from the literature, substitutions within the first 46 (−50) aa usually are not considered responsible for drug resistance of HSV-1 strains32 with the exception of the premature stop codon Asn32stop, resulting from a nucleotide insertion. Whereas Arg41His has been characterized as a resistance-related aa substitution by van Velzen et al.,33 other authors have reported this substitution as conferring no resistance.34,35 The substitution Val120stop, arising from a deletion of guanine at nucleotide 277, has been classified as associated with aciclovir resistance without performing a phenotypic assay.27 The deletion of one nucleotide resulting in a premature stop codon can be characterized clearly as a resistance-associated substitution because of the truncated protein. A relatively high proportion of resistance mutations has been observed in two small aa regions: 51–63 (15/134, 11.2%) of the ATP-binding site and 168–176 (10/134, 7.5%) of the nucleoside-binding site. The conserved aa loci 83–88 and 216–222 (7/134, 5.2%), 162–164 (4/134, 3.0%) and 284–289 (3/134, 2.2%) were affected less often. Resistance substitutions in non-conserved regions could be analysed predominantly within the aa region 290–376 (25/134, 18.7%). In this part of the TK gene, frameshifts have been the main reasons for drug resistance. Furthermore, stop codons due to nucleotide insertion or deletion occurred most often in guanine and cytosine homopolymers in nucleotide regions 430–436, 460–464 and 548–553, which are considered as hot spots of frameshift mutations.36 Three different stops caused by insertion or deletion in nucleotide region 430–436, two different stops due to changes in nucleotides 460–464, and one stop codon as well as one frameshift without a premature stop caused by changes in the nucleotide region 548–553 have been published. A lower density of resistance mutations has been observed within codons 177–215 and 223–284 (19/134, 14.2%), 89–161 (18/134, 13.4%), 64–82 (4/134, 3.0%) and 165–167 (1/134, 0.7%). In previous studies, frameshifts in TK have been reported as the most common cause (50%) of HSV-1 resistance.36,37 However, data reviewed here show that the cause of HSV-1 resistance is mainly triggered by aa substitutions (63.0%) upon nucleotide changes in the viral TK. Interestingly, 64.9% (87/134) of the resistance-associated mutations in the TK gene of HSV-1 are located in non-conserved regions, 5.2% (7/134) in the so-called hot spots and 35.1% (47/134) within conserved/active gene loci.
In the HSV-2 TK gene, 72 different resistance-related non-synonymous mutations have been reported. Of these, 34 (47.2%) have been characterized as stop codons arising from frameshift mutations, 29 (40.3%) as nucleotide substitutions resulting in aa changes, four (5.5%) as frameshift mutations without premature stops, three (4.1%) as stop codons through nucleotide changes and two (2.7%) as larger deletions within the TK gene. One large deletion at the 5′ end of the TK gene leads to an N-terminal truncation of 248 aa (1–248deletion). The other truncation represents a large deletion of 969 nucleotides starting at nucleotide 363 of the HSV-2 TK gene and extending into the downstream untranslated region (3′-deletion of 969 nucleotides resulting in the loss of aa 122–376; Figure 1b, the affected nucleotide is marked with an asterisk). This results in a protein containing the first 121 aa of the TK fused to 161 aa of the downstream non-coding region.21 Five stop codons resulted from distinct nucleotide frameshifts and four aa substitutions and one stop codon from more than one nucleotide change. With one exception, all corresponding HSV-2 strains were resistant to aciclovir and in addition to penciclovir and brivudin when tested, but susceptible to foscarnet and cidofovir. One HSV-2 strain exhibiting the aa substitution Arg51Trp has been defined as penciclovir resistant but aciclovir/foscarnet susceptible. The genotype of three isolates with stop codons due to nucleotide deletion (Met348stop by deletion of nucleotides 821–833 or nucleotides 814–826) or with a deletion of 248 aa without a premature stop codon have been classified as aciclovir-resistant because of the deficient TK protein.16 Moreover, one patient with clinical resistance after prolonged aciclovir therapy has been reported; here the substitution Gly201Asp was assumed to be resistance related without virological confirmation of the resistance phenotype.38 Variations within the first 50 aa of HSV-2 TK have been regarded very rarely as a cause of resistance. Exceptions are the 1–248deletion, the Ser32stop mutation and the Arg34Cys exchange.6,16 As for HSV-1, one reason might be the minor role of these amino acids in enzyme function. A high number of resistance-associated substitutions has been found outside of conserved loci in the aa range 224–284 (18/72, 25.0%). Stop codons arising from nucleotide insertion or deletion have been found predominantly in this gene segment. Many mutations of this type of mutation have been detected also in the non-conserved aa region 290–376 (9/72, 12.5%). Furthermore, stop codons due to nucleotide deletion or insertion within nucleotide homopolymer regions of guanine as well as cytosine at nucleotides 433–439, 463–467, 551–556 and 586–590 have been detected. Four different deletions/insertions have been published within hot-spot nucleotide regions 433–439 and 551–556, respectively, and one mutation each has been detected in the hot-spot nucleotide regions 463–467 and 586–590. The ATP-binding site (aa 51–63) is affected by drug resistance in 9.7% (7/72) followed by the conserved aa region 216–223 with 8.3% (6/72). Surprisingly, only one resistance-related substitution (Arg177Trp) could be found in aa loci 169–177 of the nucleoside-binding site (1.4%), and no resistance substitutions have been published yet in the conserved aa region 163–165. In summary, stop codons arising from nucleotide insertions or deletions, which cluster over the whole gene but predominantly over the posterior part of the enzyme, are the main cause (34/72, 47.2%) of TK-related resistance in HSV-2 strains. As was also observed in HSV-1, resistance-associated mutations of HSV-2 TK are located predominantly outside the conserved gene loci (55/72, 76.4%); 13.9% (10/72) of these are in hot spots and 23.6% (17/72) are situated inside of conserved or active gene loci.
Amino acid changes within DNA pol of HSV-1 and HSV-2
Natural polymorphism
The database of natural polymorphisms within the DNA pol gene is given in Supplementary Data (HSV-1) and Supplementary Data (HSV-2) and their locations are illustrated in Figure 2(a) (HSV-1) and Figure 2(b) (HSV-2) in relation to the conserved gene regions. In detail, DNA pol of HSV shares regions of homology that are numbered I–VII, with region I being the most conserved one.11 Additionally, the 3′–5′exonuclease domain contains three highly conserved sequence motifs, referred to as Exo I, Exo II (within region IV) and Exo III (within δ-region C). All natural polymorphisms have been identified within DNA pol of HSV-1 and HSV-2 strains susceptible to aciclovir and, with the exception of three, also to foscarnet.
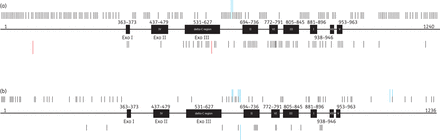
Schematic of the DNA pol gene with regions of homology I to VII as well as three Exo domains (black boxes) and non-conserved regions (bold line). Also shown is an overview of the positions of published aa substitutions (black bars) and aa frameshifts without premature stop codons (blue bars) within the DNA pol gene of HSV-1 (a) and HSV-2 (b). Bars above genes show positions of natural polymorphisms and bars below genes indicate positions of resistance-associated substitutions. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
In summary, 205 different natural aa polymorphisms have been reported in DNA pol of HSV-1. Two-hundred-and-three of them (99.0%) are aa substitutions and two (1.0%) are deletions (Glu667deletion and Gly668deletion). Seven aa substitutions (3.4%) resulted from more than one nucleotide change. Natural polymorphisms of drug-susceptible HSV-1 strains are located predominantly in the non-conserved aa regions 1–362 (65/205, 31.7%), 964–1240 (59/205, 28.8%), 628–693 (24/205, 11.7%), 846–880 (11/205, 5.4%) and 374–436 as well as 897–937 (10/205, 4.9%), 480–530 (8/205, 3.9%) and 737–771 (6/205, 2.9%). Only one substitution (Gln794His) has been reported in the non-conserved aa region 792–804 (0.5%). A low density with <2.0% of all polymorphisms has been found in the conserved regions Exo III (3/205, 1.5%) as well as in Exo II. In each of regions II and VII and in the non-conserved aa region 947–952, 1% (2/205) of natural polymorphisms have been identified. Notably, natural polymorphisms have not been detected in the loci Exo I and the regions VI, III, I and V. Overall, the overwhelming majority of natural polymorphisms within DNA pol of HSV-1 are located outside of conserved/active gene regions (196/205, 95.6%) and only a small number of polymorphisms (9/205, 4.4%) are located within such regions.
DNA pol of HSV-2 exhibits only 78 natural polymorphisms. Of these, 69 (89.6%) are aa substitutions and nine of these (10.4%) are frameshifts without premature stops. Natural polymorphisms are located predominantly in the non-conserved aa regions 1–362 (31/77, 40.3%), 628–693 (17/77, 22.1%), 964–1240 (14/77, 18.2%) and 897–937 (5/77, 6.5%). Up to three polymorphisms only were observed in each of the conserved loci Exo III (1/77, 1.3%) and region II (2/77, 2.6%), but also outside of the conserved regions within the aa stretches 792–804, 846–880 (3/77, 3.9%), 480–530 and 737–771 (1/77, 1.3%). No substitutions have been reported within the conserved gene regions Exo I, Exo II and the conserved loci referred to as VI, III, I, VII and V as well as the non-conserved aa regions 374–436 and 947–952. Frameshifts without premature stops are mostly located within aa positions 628–693 and 964–1240, respectively. Comparably to HSV-1, natural polymorphisms have been found mainly outside of conserved regions of DNA pol of HSV-2 (75/78, 96.2%) and only 3/78 (3.8%) within these regions.
According to previous studies, highly conserved region I is not supposed to be affected by genetic changes in drug-susceptible HSV strains at all.11 This also seems to be the case for regions III and V, belonging to the fingers domain and the thumb subdomain of the enzyme, respectively. The conserved region III is supposed to be involved in interaction with drugs and natural ligands39 so that mutations within this region cause predominantly drug resistance. Region V flanks the catalytic site in the palm subdomain, which contains the catalytic triad of aspartic acid residues and is essential for DNA Pol activity.40
Resistance
The database of resistance-associated non-synonymous mutations within the DNA pol gene is provided in Supplementary Data (HSV-1) and Supplementary Data (HSV-2) as well as Figure 2(a) (HSV-1) and Figure 2(b) (HSV-2). In the HSV-1 DNA pol gene, 64 different resistance-related non-synonymous mutations have been reported. Sixty-two (96.9%) have been characterized as aa substitutions and two (3.1%) are stop codons arising from nucleotide insertions or deletions. Three aa substitutions are due to more than one nucleotide change. Fifty-eight out of 64 (90.6%) viral strains were resistant to aciclovir and additionally to penciclovir and brivudin when tested. Cross-resistance to foscarnet and aciclovir could be identified in 30 viral strains (46.9%). Two viral strains with the substitutions Ala700Gly or Ala605Val were resistant to aciclovir and foscarnet but susceptible to penciclovir. Furthermore, cidofovir resistance has been identified in 11 HSV-1 strains (17.2%). Seven (10.9%) showed cross-resistance to foscarnet, brivudin or penciclovir, whereas sensitivity to aciclovir, penciclovir, brivudin or foscarnet has been analysed in 4 (4/64, 6.3%) of these 11 viral strains. Contradictory findings have been described for aciclovir in the cases of two changes (Ile1028Thr and Ser724Asn) and for three substitutions (Asp581Ala, Ser725Ala and Ile922Asn) susceptibility to aciclovir has not been tested. For the substitution Asp368Ala, aciclovir resistance has not been verified by any phenotypic assay. However, a DNA Pol functional assay has been performed by three research groups, resulting in consistent results for DNA Pol activity.41–43
Resistance mutations were detected in all conserved regions of the HSV-1 DNA pol gene, predominantly in region II (13/64, 20.3%), Exo III (12/64, 18.8%), regions III (7/64, 10.9%), VI (6/64, 9.4%), I and V (4/64, 6.3%), Exo I and region VII (1/64, 1.6%). No resistance mutations have been found within the Exo II locus. The conserved regions I, II, III and VI of the enzyme are directly or indirectly involved in the recognition and binding of nucleotides or pyrophosphate and may be associated with cross-resistance to aciclovir and foscarnet.11 Interestingly, mutations within Exo I and Exo II resulting in the inactivation of exonuclease activity have a very limited role in the development of drug resistance in HSV-1. Despite this, the activity of the enzyme is significantly reduced only by mutations within the Exo II region.42,44 Defective exonuclease activity increases mutation frequency by up to three orders of magnitude,11 which could affect the susceptibility of clinical strains to aciclovir. Less surprising is the low frequency of aa substitutions in non-conserved regions such as aa regions 1–362 and 964–1235, which have been affected very rarely, and aa 374–436 and 947–952, which have not been affected at all. To sum up, three-quarters (48/64, 75.0%) of resistance-associated substitutions in DNA pol of HSV-1 occurred in conserved gene regions and one-quarter (16/64, 25.0%) could be analysed in non-conserved loci.
For HSV-2, only 18 different resistance-related non-synonymous mutations have been published for the DNA pol gene. Seventeen (94.4%) of these have been characterized as aa substitutions. They occurred in three different conserved regions, namely region II (5/18, 27.8%), region VI (2/18, 11.1%) and region III (1/18, 5.6%). In addition, four non-conserved regions within the aa ranges 897–937 (4/18, 22.2%), 1–362 and 628–693 (2/18, 11.1%) and 846–880 (1/18, 5.6%) have been involved. In addition to the aa substitutions, there was one insertion of nucleotides GAAGAC at positions 2050–2055, resulting in no premature stop of translation. The phenotype of viral HSV-2 strains has been investigated only for aciclovir, foscarnet and cidofovir. Ten out of 18 (55.6%) isolates were resistant to aciclovir but susceptible to foscarnet. Cross-resistance to foscarnet and aciclovir but sensitivity to cidofovir, when tested, has been analysed for two viral strains (2/18, 11.1%). Moreover, five HSV-2 strains (5/18, 27.8%) showed susceptibility to aciclovir but resistance to foscarnet and cidofovir. In one strain with the aa substitution Ser725Gly, only foscarnet resistance has been verified phenotypically. In contrast to HSV-1, resistance-associated substitutions within DNA pol of HSV-2 are fairly distributed between conserved/active (10/18, 55.6%) and non-conserved loci (8/18, 44.4%).
Methods for HSV resistance testing
Phenotype
Phenotypic tests allow estimation of the antiviral drug concentration that inhibits virus growth by 50% (effective concentration 50%, EC50) or 90% (effective concentration 90%, EC90) in cell culture.45 Among these, the PRA has been considered the gold standard for the measurement of drug susceptibility to HSV-1 and HSV-2.24 This method is based on the enumeration of plaques that emerge from HSV-infected cells. Plaques are counted by light microscopy,46–49 plaque autoradiography46,50–52 or by the naked eye53 without staining34,54 or after staining with neutral red,53,55–58 crystal violet16,19,27,39,48,51,59–76 or methylene blue,47,77 Giemsa78,79 or unspecified staining.80–82 A second phenotypic method is the cytopathic effect reduction assay, which is based on the evaluation of the cytopathic effect on cell monolayers. The cytopathic effect is measured either directly by light microscopy83,84 or by the incorporation/metabolization of dye by living cells. In principle, the cells take up neutral red dye14,25,36,37,85,86 or metabolize red tetrazolium to yellow formazan.6,20,26,35,52,87–90 The amount of absorbed or metabolized dye is measured as OD so that the effect of the antiviral substance can be calculated. Moreover, β-galactosidase activity reflects the number of infected cells in the yield reduction assay [enzyme-linked virus inhibitory reporter assay (ELVIRA)],91–93 and luminescence represents the number of living cells in the modified Vero-ICP10-Luciferase test.70 Furthermore, the DNA reduction assay using real-time PCR10,48,94 and the DNA hybridization assay36,95,96 have rarely been implemented in HSV resistance testing. In addition, a cell growth assay in SDM79 medium with ganciclovir using recombinant Leishmania that expresses the TK gene73,97 has been carried out in a few experiments. Plating efficiency assays have been performed by some authors to determine the percentage of resistant virus in a given isolate.10,76,98 Mouse in vivo assays have been performed to investigate the reactivation properties of virus variants,38,57 to examine symptoms of an experimental infection99 and to determine virulence/pathogenicity27,100–103 as well as aciclovir resistance in vivo.104
Phenotypic tests are labour intensive and time consuming because of the need to propagate viruses in cell culture or to generate recombinant viruses. There are problems with virus isolation in cell culture, particularly if it is necessary to use blood specimens or CSF.90,105 Compared with the PRA, the dye uptake assay and DNA hybridization assay save time because the cytopathic effect can be read automatically. Although EC50/EC90 values may vary as a function of test type, cell cultures, the size of viral inoculum and the range of drug concentrations, none of the methods mentioned has been standardized to date. The PRA in particular is prone to subjective bias when very small plaques are counted,24,45,106 and some laboratories use the cytopathic effect reduction assay in place of the PRA, avoiding the problem of counting small plaques. However, phenotypic assays are well accepted and allow a clear interpretation of laboratory findings, and above all the PRA shows a high correlation with clinical parameters.90,105 The determination of the role of unknown aa changes in drug resistance based on the phenotype is problematic when several unknown substitutions or unknown substitutions combined with resistance mutations occur in the TK and DNA pol genes. In this case, recombinant viruses with only one aa change can be generated by using a set of overlapping cosmids and plasmids59,67 or by the application of bacterial artificial chromosomes (BACs).48 Subsequently, the resistance phenotype can be estimated in the customary way.
Genotype
Genotypic resistance assays are based on the identification of natural polymorphisms and resistance-associated aa substitutions by sequencing of the TK and DNA pol genes. For this purpose, the TK gene,51,64,107 the DNA pol gene100,108,109 or both genes34,68,110,111 are amplified by PCR and sequenced. Sequencing of both genes yields the most reliable results; if only one of the two genes is used aa substitutions are likely to be missed. Usually, the nucleotide sequences are compared with a reference strain, mostly HSV-1 strains 17 and KOS (accession numbers JN555585.1 and JQ673480.1) and the HSV-2 strain HG52 (accession number Z86099.2).88 Depending on the reference strain used, different natural polymorphisms can be detected. Despite this, resistance mutations can be identified regardless of the reference strain because they all are drug susceptible. This kind of test has increasing priority in the detection of resistance to anti-herpetic drugs. However, it cannot replace phenotypic tests yet due to the many uncharacterized aa substitutions. The role of these substitutions in the development of drug resistance has to be determined by phenotypic and functional assays.88 In addition, it is known that conventional Sanger sequencing technology is not sufficiently sensitive to detect all resistant HSV mutants in clinical isolates.112,113 The use of ultra-deep next-generation sequencing should be considered for the analysis of these minority sequence variants.
TK and DNA Pol function
Functional TK and/or DNA Pol assays are required especially to evaluate the significance of aa changes that cannot be clarified by phenotypic testing as being related to natural polymorphism or resistance. Previous assays measured the enzyme kinetics of DNA Pol by using radionuclide-labelled nucleoside triphosphates. To this end, [3H]TTP,114 [32P]dGTP,41 [32P]ATP,42 [3H]dCTP48 or [32P]dCTP114–117 have been incorporated in replicating viral DNA by DNA Pol. Kinetic studies concerning TK have been carried out mostly with [3H]thymidine, which is converted into [3H]thymidine monophosphate by TK.22,58,63–65,77,79,99,118–122 Subsequently, radioactivity has been measured by the diethylaminoethylcellulose method123 and by liquid scintillation counting.116 The results provided information on the enzymatic activity of DNA pol/TK mutants compared with WT. To estimate the catalytic activity of TK, the turnover number (kcat) and the Michaelis constant (Km) can be calculated.119,122,124 Additionally, the exonuclease domain of DNA pol has been studied by using radioactively labelled primers and the number of mismatched base pairs produced by DNA pol has been measured.41,43 Even though such assays yielded clear results regarding enzyme kinetics, the handling of radionuclides was restricted to certain laboratories.
Therefore, the development and use of non-radioactive functional assays was an important step to improve the analysis of TK and DNA pol mutations. Initially, a lactate dehydrogenase–pyruvate kinase-coupled assay was implemented in which the oxidation of NADH to NAD+ was measured spectrophotometrically.120,122 To this end, mutated TK genes have been expressed in Escherichia coli and purified. This method allows the indirect estimation of TK phosphorylation activity on the basis of quantifying the amount of released ADP that emerges from exhausted ATP as a result of the action of TK. Information on the enzymatic function of TK can also be obtained by the modified use of the DiviTum™ test (Biovica, Uppsala, Sweden)35,89 and the ADP-Glo™ kinase assay (Promega, Charbonnières-les-Bains, France).125 Both assays are commercially available but neither of them applies to the actual TK substrate (thymidine) nor the antiviral drug aciclovir. The former test is an ELISA that uses bromodeoxyuridine (BrdU) as substrate for recombinant TK proteins. This substrate is phosphorylated by TK to BrdU monophosphate. The latter assay provides ADP as a substrate for the recombinant enzyme and estimates the amount of ATP that is generated in a luciferase/luciferin reaction. These functional tests do not use aciclovir as a substrate for TK, so that false-positive results are possible because altered substrate specificity cannot be detected. Therefore, TK functional assays with thymidine and/or aciclovir as substrate have been developed on the basis of MS33,126 and HPLC.122,127 MS detects metabolized aciclovir monophosphate by analysing samples based on their mass-to-charge ratio, while HPLC separates the analytes regarding their hydrophobicity. Both methods allow differentiation between fully functional, deficient and partially deficient TK activity as well as altered substrate specificity. Nevertheless, variations in the results of different functional assays have been observed, and results should therefore be interpreted with caution. However, non-radioactive functional TK assays represent a useful tool to evaluate the significance of aa substitutions in the resistance of clinical HSV strains. A major limitation is often the difficulty of defining TK activity values resulting in viral sensitivity or resistance to nucleoside analogues. Furthermore, aa changes occurring in the DNA pol gene cannot be addressed by these novel non-radioactive assays. Thus, direct testing of replication-competent viruses using BAC technology or a set of overlapping cosmids and plasmids, as described above, is necessary to overcome these problems.
For further understanding of substitutions in the TK and DNA pol genes, structural analyses can be performed in addition to phenotypic and functional assays. In 1998 Kussmann-Gerber et al.120 applied molecular modelling based on crystal structures of HSV-1 TK that were clarified in 1995.79,128 In a recent publication, Hussin et al.61 demonstrated crystal structure models of HSV-1 TK and DNA pol.129 It should be noted that structural analyses alone are inappropriate for the characterization of aa substitutions regarding their associations with resistance. However, they are suited to the illustration of how aa changes may affect protein stability and thus enzyme function.
Conclusions and perspectives
The information presented in this article about the polymorphisms and resistance mutations in the HSV TK and DNA pol genes can facilitate considerably the interpretation of genotypic resistance findings in clinical HSV-1 and HSV-2 strains and thus the diagnosis of drug resistance. This relates in particular to non-synonymous nucleotide substitutions outside of active or conserved gene regions. In addition, the overview of the technologies used to evaluate phenotypic and genotypic HSV resistance presented in this article can help to standardize a methodical approach for resistance testing. The establishment of genotypic resistance testing as method of choice requires a further considerable amount of research. Until then, the evaluation of phenotypic resistance will remain an indispensable procedure.
Funding
The study was supported in parts by the Robert Koch-Institute, Berlin, Germany, within the context of the national reference laboratory network.
Transparency declarations
None to declare.
Acknowledgements
We thank Christa Rothmann and Heike Urban for technical assistance. After publication, an updated version of the database will be available online at www.virologie.uniklinikum-jena.de/.
References
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}