





Abstract
Jacaranda mimosifolia D. Don is a deciduous tree widely cultivated in the tropics and subtropics of the world. It is famous for its beautiful blue flowers and pinnate compound leaves. In addition, this tree has great potential in environmental monitoring, soil quality improvement, and medicinal applications. However, a genome resource for J. mimosifolia has not been reported to date. In this study, we constructed a chromosome-level genome assembly of J. mimosifolia using PacBio sequencing, Illumina sequencing, and Hi-C technology. The final genome assembly was ∼707.32 Mb in size, 688.76 Mb (97.36%) of which could be grouped into 18 pseudochromosomes, with contig and scaffold N50 values of 16.77 and 39.98 Mb, respectively. A total of 30,507 protein-coding genes were predicted, 95.17% of which could be functionally annotated. Phylogenetic analysis among 12 plant species confirmed the close genetic relationship between J. mimosifolia and Handroanthus impetiginosus. Gene family clustering revealed 481 unique, 103 significantly expanded, and 16 significantly contracted gene families in the J. mimosifolia genome. This chromosome-level genome assembly of J. mimosifolia will provide a valuable genomic resource for elucidating the genetic bases of the morphological characteristics, adaption evolution, and active compounds biosynthesis of J. mimosifolia.
Significance
Jacaranda mimosifolia D. Don is an ornamental plant with great potential in environmental monitoring, soil quality improvement, and medicinal applications. Here we present a chromosome-level genome assembly of J. mimosifolia. This high-quality genome assembly will provide a valuable genomic resource for elucidating the genetic bases for elucidating the genetic bases of the morphological characteristics, adaption evolution, and active compounds biosynthesis of J. mimosifolia.
Introduction
Jacaranda mimosifolia D. Don is a deciduous tree belonging to the Bignoniaceae family that originated in South America (Mostafa et al. 2014; Ragsac et al. 2019). It is now widely cultivated in the tropics and subtropics throughout the world (Gentry 1992). It is commonly used as an ornamental plant due to its beautiful blue flowers and pinnate compound leaves (Mostafa et al. 2014; Hendra et al. 2019). In addition to its horticultural value, J. mimosifolia was found to be a useful biomonitor of soil metals and atmospheric metals (Olowoyo, van Heerden, and Fischer 2010; Olowoyo, Van Heerden, Fischer, and Baker 2010) and can also play an important role in soil quality improvement (Rigal et al. 2020). A few active compounds, including jacaric acid (Takagi and Itabashi 1981), jacaranone (Xu et al. 2003), phenylethanoids (Rana et al. 2013; Hendra et al. 2019), and flavonoids (Naz et al. 2020), are commonly isolated from different tissues of J. mimosifolia. Thus, this species has a great potential in medicinal applications due to its antioxidant, antimicrobial, and hypotensive properties (Mostafa et al. 2014). To date, studies of J. mimosifolia at the molecular level have been limited to karyotypes (Collevatti and Dornelas 2016; Cordeiro et al. 2016), plastid genomes (Zhao et al. 2019), inter-simple sequence repeat (ISSR) markers (Escandón et al. 2003), and very few nuclear genes (Kram et al. 2008). In a previous study, J. mimosifolia was confirmed to be diploid (2n = 2x = 36) (Collevatti and Dornelas 2016). However, the genome of J. mimosifolia has not yet been sequenced or reported. A recent study reported the genome sequence of its close relative Jacaranda copaia, which was found to have up to 5,869 scaffolds and an N50 size of 1.02 Mb (Burley et al. 2021). With the rapid development of sequencing technologies, more and more researchers are performing chromosome-level genome assembly using single-molecule real-time (SMRT) and chromosome conformation capture (Hi-C) technologies (Bickhart et al. 2017; Mahajan et al. 2018; Zhang et al. 2020). Here, we generated a chromosome-level genome assembly of J. mimosifolia by integrating PacBio sequencing, Hi-C sequencing, and Illumina sequencing. The high-quality genome assembly will provide a valuable genomic resource for elucidating the genetic bases of the morphological characteristics, adaption evolution, and active compounds biosynthesis of J. mimosifolia. It will also provide a reliable reference for studying the evolutionary history of Bignoniaceae and Lamiales.
Results and Discussion
Genome Sequencing, Assembly, and Quality Assessment
We generated a total of 149.21 Gb PacBio reads, 60.79 Gb Illumina reads, and 89.86 Gb Hi-C reads (supplementary table S1, Supplementary Material online). Before assembly, we obtained a genome size estimation of 739 Mb with a heterozygosity of 0.23% based on 19-mer frequency distribution analysis (supplementary fig. S1, Supplementary Material online), which was very close to the result obtained from the flow cytometry analysis (supplementary fig. S2, Supplementary Material online). The PacBio reads were preliminarily assembled into 218 contigs, with a contig N50 of 17.54 Mb and a total length of 707.32 Mb. These contigs were then anchored into 18 pseudochromosomes based on the Hi-C data (supplementary fig. S3 and table S2, Supplementary Material online). Among the 218 contigs, 101 were grouped into pseudochromosomes with a total length of 688.76 Mb, accounting for 97.36% of the entire genome assembly (fig. 1A and supplementary table S2, Supplementary Material online). The contig and scaffold N50 of the final assembly were 16.77 and 39.98 Mb (table 1 and supplementary table S3, Supplementary Material online), respectively. About 1,588 (98.39%) of the 1,614 conserved BUSCO genes were completely covered in our assembly (supplementary table S4, Supplementary Material online), suggesting the high completeness of the J. mimosifolia genome assembly.
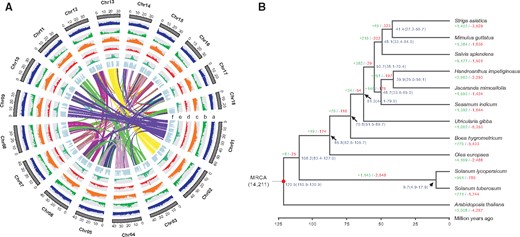
Features of the J. mimosifolia genome (A) and phylogeny and gene family evolution of J. mimosifolia (B). (A) Genome features in nonoverlapping windows of 500 Kb across the J. mimosifolia genome. Tracks from outside to inside are as follows: (a) GC content, (b) gene density, (c) repeat density, (d) LTR/Gypsy density, (e) LTR/Copia density, and (f) density of collinear blocks between J. mimosifolia and H. impetiginosus. (B) A species tree on the basis of 194 single-copy orthogroups from 12 plant species. Gene family expansion and contraction are denoted in green and red numbers, respectively. Numbers on nodes represent the inferred divergence times with 95% confidence intervals. Red dots represent the calibration time of A. thaliana–S. tuberosum divergence.
Statistics of the assembly and annotation of the J. mimosifolia genome
| Assembly | |
|---|---|
| Estimated genome size (by k-mer analysis) (Mb) | 739 |
| Total length (Mb) | 707.42 |
| GC content (%) | 33.87 |
| Contig N50 (Mb) | 16.77 |
| Longest contig (Mb) | 45.42 |
| Contig number | 290 |
| Pseudochromosome number | 18 |
| Scaffold N50 (Mb) | 39.98 |
| Longest scaffold (Mb) | 61.75 |
| Annotation | |
| Total length of repeats (Mb) | 401.81 |
| Number of protein-coding genes | 30,507 |
| Mean transcript length (bp) | 3,441.77 |
| Mean coding sequence length (bp) | 1,230.68 |
| Mean exon length (bp) | 234.87 |
| Mean intron length (bp) | 521.50 |
| Functionally annotated genes | 29,035 |
| Assembly | |
|---|---|
| Estimated genome size (by k-mer analysis) (Mb) | 739 |
| Total length (Mb) | 707.42 |
| GC content (%) | 33.87 |
| Contig N50 (Mb) | 16.77 |
| Longest contig (Mb) | 45.42 |
| Contig number | 290 |
| Pseudochromosome number | 18 |
| Scaffold N50 (Mb) | 39.98 |
| Longest scaffold (Mb) | 61.75 |
| Annotation | |
| Total length of repeats (Mb) | 401.81 |
| Number of protein-coding genes | 30,507 |
| Mean transcript length (bp) | 3,441.77 |
| Mean coding sequence length (bp) | 1,230.68 |
| Mean exon length (bp) | 234.87 |
| Mean intron length (bp) | 521.50 |
| Functionally annotated genes | 29,035 |
Statistics of the assembly and annotation of the J. mimosifolia genome
| Assembly | |
|---|---|
| Estimated genome size (by k-mer analysis) (Mb) | 739 |
| Total length (Mb) | 707.42 |
| GC content (%) | 33.87 |
| Contig N50 (Mb) | 16.77 |
| Longest contig (Mb) | 45.42 |
| Contig number | 290 |
| Pseudochromosome number | 18 |
| Scaffold N50 (Mb) | 39.98 |
| Longest scaffold (Mb) | 61.75 |
| Annotation | |
| Total length of repeats (Mb) | 401.81 |
| Number of protein-coding genes | 30,507 |
| Mean transcript length (bp) | 3,441.77 |
| Mean coding sequence length (bp) | 1,230.68 |
| Mean exon length (bp) | 234.87 |
| Mean intron length (bp) | 521.50 |
| Functionally annotated genes | 29,035 |
| Assembly | |
|---|---|
| Estimated genome size (by k-mer analysis) (Mb) | 739 |
| Total length (Mb) | 707.42 |
| GC content (%) | 33.87 |
| Contig N50 (Mb) | 16.77 |
| Longest contig (Mb) | 45.42 |
| Contig number | 290 |
| Pseudochromosome number | 18 |
| Scaffold N50 (Mb) | 39.98 |
| Longest scaffold (Mb) | 61.75 |
| Annotation | |
| Total length of repeats (Mb) | 401.81 |
| Number of protein-coding genes | 30,507 |
| Mean transcript length (bp) | 3,441.77 |
| Mean coding sequence length (bp) | 1,230.68 |
| Mean exon length (bp) | 234.87 |
| Mean intron length (bp) | 521.50 |
| Functionally annotated genes | 29,035 |
Genome Annotation
We estimated that 401.81 Mb (56.80%) of the J. mimosifolia genome was composed of repetitive elements (supplementary table S5, Supplementary Material online). Long terminal repeats (LTRs) were the most abundant repeat class, accounting for 49.50% of repetitive sequences representing 28.12% of the genome. Within the LTR family, Copia and Gypsy were the two most abundant subfamilies, representing 14.87% and 12.34% of the genome, respectively.
A total of 30,507 protein-coding genes were predicted, with mean transcript size of 3,442 bp, an average coding sequence size of 1,231 bp, and an average of 5.24 exons per gene. The annotated protein-coding genes covered 91.82% of the 1,614 conserved BUSCO genes (supplementary table S4, Supplementary Material online), indicating the high completeness of the gene annotation. Overall, 29,035 (95.17%) of the protein-coding genes could be annotated by at least one database, and 24,261 (79.53%) genes could be classified by gene ontology (GO) terms (supplementary table S6, Supplementary Material online).
Evolution of Gene Families
A total of 194 single-copy orthogroups were identified across the 12 plant genomes (supplementary table S7, Supplementary Material online). As expected, the species tree constructed through coalescent-based analysis on the basis of these single-copy genes confirmed that J. mimosifolia was mostly related to Handroanthusimpetiginosus (fig. 1B). The divergence time between J. mimosifolia and H. impetiginosus was estimated to be 39.9 million years ago (Ma), slightly after the separation between Bignoniaceae and Pedaliaceae at 48.7 Ma.
Gene family clustering analysis identified a total of 29,545 gene families comprising 354,790 genes among J. mimosifolia and 11 other species, of which 5,118 families were present in all 12 species. Among the 12 species, 481 gene families consisting of 2,034 genes were unique to J. mimosifolia (supplementary table S7, Supplementary Material online). GO enrichment analysis revealed that these unique genes were mainly related to “cellular process,” “pteridine-containing compound metabolic process,” “isoflavonoid biosynthetic process,” “positive regulation of response to water deprivation,” and “zinc ion binding” (supplementary fig. S4, Supplementary Material online). We also identified 103 gene families comprising 1,268 genes that were significantly expanded in J. mimosifolia (P < 0.01). GO enrichment analysis revealed that these genes were highly enriched in “brassinosteroid metabolic process,” “defense response,” “pigment biosynthetic process,” and “detoxification” (supplementary fig. S5, Supplementary Material online). By contrast, a total of 16 gene families comprising 71 genes were significantly contracted in the J. mimosifolia genome. These genes were highly enriched in “response to symbiotic bacterium,” “mucilage extrusion from seed coat,” “proteolysis,” “plant-type cell wall modification,” and “GTP binding” (supplementary fig. S6, Supplementary Material online).
Conclusions
In this study, we present a high-quality chromosome-level genome assembly of J. mimosifolia using PacBio single-molecule long-read sequencing, supplemented with Illumina short-read sequencing and Hi-C sequencing. The final genome assembly was ∼707.32, 688.76 Mb (97.36%) of which could be grouped into 18 pseudochromosomes. This assembly is highly contiguous with contig and scaffold N50 values of 16.77 and 39.98 Mb, respectively. A total of 56.80% of the genome was composed of repeat sequences. A total of 30,507 protein-coding genes were predicted, 95.17% of which could be functionally annotated. Phylogenetic analysis on the basis of 194 single-copy orthogroups across 12 plant species confirmed the close genetic relationship between J. mimosifolia and H. impetiginosus. The divergence time between these two species was estimated to be 39.9 Ma. Gene family clustering revealed 481 unique, 103 significantly expanded, and 16 significantly contracted gene families in the J. mimosifolia genome. The high-quality chromosomal genome assembly of J. mimosifolia will provide a valuable genomic resource for elucidating the genetic bases for blue flower color development, the biosynthesis of active compounds, and environmental adaption as well as for studying the evolutionary history of Bignoniaceae and Lamiales.
Materials and Methods
Sample Preparation and Sequencing
We sequenced a single adult tree of J. mimosifolia from Sichuan University in Sichuan Province, southwestern China. Fresh leaves were collected and frozen immediately in liquid nitrogen. High-quality genomic DNA was extracted using the cetyl trimethylammonium bromide (CTAB) method (Doyle and Doyle 1987). For PacBio single-molecule real-time (SMRT) sequencing, at least 10 µg of sheared DNA was used to generate 40-kb SMRTbell libraries according to the manufacturer’s protocol. Subsequently, the SMRTbell libraries were sequenced on the PacBio Sequel II Platform (Pacific Biosciences). A paired-end PCR-free library with an insert size of ∼350 bp was generated and sequenced on an Illumina HiSeq 2500 platform. For Hi-C sequencing, more than 2 g of young leaves were frozen in liquid nitrogen prior to the Hi-C experiment. The Hi-C libraries were then constructed as described previously (Louwers et al. 2009), including chromatin extraction and digestion, DNA ligation, and purification. The DNA was sheared to a mean fragment size of ∼350 bp and sequenced on an Illumina HiSeq 2500 platform.
To aid in gene annotation, we performed RNA sequencing for fresh tissues of the leaf, stem, and peel from the same plant used for genome sequencing using an Illumina HiSeq 2500 platform.
Genome Size Estimation
The genome size of J. mimosifolia was estimated using two different approaches. First, a k-mer depth distribution (k = 19) was obtained with Jellyfish v2.2.9 (Marçais and Kingsford 2011) based on the Illumina short reads, and the final result was plotted in GenomeScope. Second, a flow cytometry analysis was performed using Glycine max as a reference standard and propidium iodide as the stain.
Genome Assembly and Assessment
The PacBio reads were self-corrected and de novo assembled using CANU (Koren et al. 2017) with a genomeSize parameter of 730 m. Primary contigs were polished using Pilon v1.22 (Walker et al. 2014) with the filtered PacBio reads. Then, Illumina short reads were mapped to the assembly using BWA v0.7.17 (Li and Durbin 2009) and Pilon was used to improve the contigs. Potential allelic haplotigs were identified and removed with purge_dups (Guan et al. 2020). The contigs were finally anchored into chromosomes using 3D-DNA software (Dudchenko et al. 2017) based on the Hi-C data. The genome quality was evaluated by examining the coverage of highly conserved genes using BUSCO v3.0.2 (Simão et al. 2015) with the “embryophyta_odb10” ortholog set.
Repeat Annotation
Both homolog-based and de novo approaches were used to determine repetitive elements in the J. mimosifolia genome. Initially, a repeat library of green plants (Viridiplantae) was derived from the Repbase database v22.11 using RepeatMasker v4.0.7 (Tarailo-Graovac and Chen 2009). Then a de novo repeat library was generated using RepeatModeler v1.0.11 (Price et al. 2005), with default parameters. Finally, RepeatMasker was used to map the J. mimosifolia genome against these two repeat libraries.
Gene Annotation
We performed gene annotation in the repeat-masked genome by using a combination of de novo, homology-based, and transcriptome-based prediction methods. The de novo prediction was conducted using Augustus v3.2.3 (Stanke et al. 2006) with pretrained parameters from Arabidopsis thaliana. For the homology-based prediction, protein sequences from five sequenced plant species, namely, H. impetiginosus (Silva-Junior et al. 2018), Olea europaea (Unver et al. 2017), Sesamum indicum (Wang et al. 2014), Solanum tuberosum (Potato Genome Sequencing Consortium 2011), and A. thaliana (Kaul et al. 2000), were aligned to the J. mimosifolia genome using TBLASTN v2.2.26 (Camacho et al. 2009). GeneWise v2.4.1 (Birney et al. 2004) was then used for the accurate prediction of gene models based on the aligned genomic regions. For the transcriptome-based prediction, both de novo and genome-guided approaches were used to assemble the RNA-seq data with Trinity v2.5.1 (Grabherr et al. 2011). All of the assembled transcripts were aligned to the genome assembly, and the gene structures were improved using PASA v2.2.0 (Haas et al. 2008). Finally, all predicted gene models were integrated into a consensus gene set using EVM v1.1.1 (Haas et al. 2008).
Functional annotation of protein-coding genes was achieved with BLASTP against the SwissProt and TrEMBL databases (Bairoch and Apweiler 2000). Protein motifs and domains were annotated using InterProScan v5.31 (Hunter et al. 2009) by searching against multiple publicly available databases. GO IDs for each gene were retrieved with Blast2GO v2.5 (Conesa et al. 2005). The pathways in which the genes might be involved were annotated using the KAAS (KEGG Automatic Annotation Server).
Gene Family Analysis and Phylogenetic Analysis
Gene family clustering analysis was performed using BLASTP and OrthoMCL v2.0.9 (Li et al. 2003) based on the annotated genes of J. mimosifolia and 11 other sequenced plant species, namely, 8 Lamiales (H. impetiginosus, S. indicum, Salvia splendens, Mimulus guttatus, Striga asiatica, Utricularia gibba, Boea hygrometrica, and O. europaea), 2 Solanales (S. tuberosum and Solanumlycopersicum), and 1 rosid plants (A. thaliana). We identified 194 single-copy orthogroups across the 12 plant genomes. For each orthogroup, protein sequences were aligned using MAFFT v7.313 (Katoh et al. 2002) and conserved sites were extracted from the alignments using Gblocks v0.91b (Castresana 2000). The conserved sequences were concatenated into a single multiple sequence alignment. A maximum-likelihood phylogeny was constructed using RAxML v8.2.11 (Stamatakis 2014) under the PROTGAMMAILGX model with 100 bootstrap replicates. Then MCMCTREE in PAML v4.9e (Yang 2007) was applied to estimate the divergence time among 12 species based on the phylogenetic tree with the main parameters of burnin = 1,000,000, nsample = 20,000, and sampfreq = 150. The divergence time between A. thaliana and S. tuberosum (111–131 Ma) was obtained from the TimeTree database (http://www.timetree.org, last accessed May, 12, 2021) and used as fossil calibration point. Gene family evolution was examined across the time-calibrated phylogenetic tree using CAFÉ v3.1 (De Bie et al. 2006) with the main parameters -p = 0.05 and lambda -s.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Acknowledgments
This work was supported by startup funds provided by Chengdu University.
Data Availability
Raw sequencing data and genome assembly have been deposited at the NCBI under the BioProject PRJNA678551 with biosample accession number SAMN16807716. The annotations and predicted peptides are available on FigShare at the link: https://doi.org/10.6084/m9.figshare.14379086.v1.

{kind=link}