





Abstract
Alzheimer's disease (AD) is responsible for 60%–80% of identified cases of dementia. While the generation and accumulation of amyloid precursor protein (APP) fragments is accepted as a key step in AD pathogenesis, the precise role of these fragments remains poorly understood. To overcome this deficit, we induced the expression of the soluble C-terminal fragment of APP (C99), the rate-limiting peptide for the generation of amyloid fragments, in yeast that contain thermosensitive mutations in genes encoding proteasome subunits. Our previous work with this system demonstrated that these proteasome-deficient yeast cells, expressing C99 when proteasome activity was blunted, generated amyloid fragments similar to those observed in AD patients. We now report the phenotypic repercussions of inducing C99 expression in proteasome-deficient cells. We show increased levels of protein aggregates, cellular stress and chaperone expression, electron-dense accumulations in the nuclear envelope/ER, abnormal DNA condensation, and an induction of apoptosis. Taken together, these findings suggest that the generation of C99 and its associated fragments in yeast cells with compromised proteasomal activity results in phenotypes that may be relevant to the neuropathological processes observed in AD patients. These data also suggest that this yeast model should be useful for testing therapeutics that target AD-associated amyloid, since it allows for the assessment of the reversal of the perturbed cellular physiology observed when degradation pathways are dysfunctional.
Introduction
It is estimated that in the USA alone, 6.2 million Americans aged 65 and older live with Alzheimer's disease (AD; Alzheimer's Association 2021). Aging, genetics, and family history are AD risk factors (Tanzi et al. 1991, St. George-Hyslop 1999), yet 95% of cases arise sporadically. Given the paucity of treatments for AD, there is an urgent unmet need for therapeutics, and to intervene most effectively in a disease process, it is vital to understand the molecular mechanisms and etiology of the disease.
Regardless of the cause, all patients with AD exhibit increased amyloidogenic fragments that are generated from the cleavage of amyloid precursor protein (APP; Coughlan 2000). Although APP is ubiquitously expressed, the function of full-length APP remains poorly understood, whereas fragments of APP, such as the Aβ peptide are thought to play a central role in the development of AD (Coughlan 2000 Selkoe 2003, Selkoe and Schenk 2003). The Aβ peptide fragment is generated by two sequential cleavage events, β-secretase (BACE) cleavage of APP produces C99, the generation of which serves as the rate-limiting step for the subsequent γ-secretase cleavage that in turn releases the Aβ peptide (Hartmann et al. 1997, Sparvero et al. 2007). Other secretase enzymes, such as ζ and ε (Small 1998, Weidemann et al. 2002), can also cleave C99 to form longer fragments which similarly cause toxicity.
APP fragments can form aggregate-like deposits in the brain known as plaques (Beyreuther and Masters 1991, Small 1998, Lloret et al. 2015, Tong et al. 2015). Although these plaques may give rise to AD toxicity, we now understand that dimeric and oligomeric forms are possibly more toxic (Kuo et al. 1996, Roher et al. 1996, Garzon-Rodriguez and Sepulveda-Becerra 1997, Chromy et al. 2003, Walsh and Selkoe 2004, Tamagno et al. 2006, Harrison et al. 2007, Heo et al. 2007, Shankar et al. 2008, Viola et al. 2008). These smaller species may interfere with cellular membrane homeostasis by intercalating into the bilayer and/or producing lipid peroxides and reactive aldehydes (HNE), thereby disrupting cellular homeostasis (Yan et al. 1996, Allan Butterfield 2002, Butterfield et al. 2007, Valko et al. 2007). While the order of events is still being deciphered (Ferrari and Sorbi 2021), some events that occur in an AD brain include: (1) the excessive generation and aggregation of APP fragments to form plaques (Ashrafian et al. 2021, Koychev et al. 2021), (2) an intracellular protein—tau—becomes phosphorylated and can aggregate (Ashrafian et al. 2021, Koychev et al. 2021), (3) overzealous immune responses are initiated (Bettcher et al. 2021), (4) synapses are damaged (Sanchez-Mico et al. 2021), and (5) neurons may inappropriately re-enter the cell cycle (Galasko et al. 1998, Allan Butterfield 2002, Bossy-Wetzel et al. 2003, Giasson et al. 2003, Laferla and Oddo 2005, Guo et al. 2006, Zheng and Koo 2006, Varvel et al. 2008). Moreover, these events occur concurrent with age-related decreases in the efficiency of proteasome activity, thus reducing the ability of the cell to remove misfolded proteins. This can be accompanied by dysfunction of the mitochondrial electron transport chain further contributing to the cascade responsible for the development of AD (Mccracken and Brodsky 1996, Checler et al. 2000, Skovronsky et al. 2000, Travers et al. 2000, Fewell et al. 2001, Nunan et al. 2001, Haynes et al. 2004, Dillin and Cohen 2011, Walter and Ron 2011, Dillin et al. 2014, Walter et al. 2015). In addition, people with AD exhibit greater BACE cleavage of APP leading to greater levels of C99 and APP fragment formation, which in turn can inhibit proteasomal activity, thus propagating pathology (Lopez Salon et al. 2003, Burton et al. 2008, Hampel and Shen 2009). As proteasome function declines even further, the rate of toxic fragment generation/accumulation accelerates and/or C99 may now be cleaved by other cellular enzymes producing the longer, more toxic monomers (Koo and Squazzo 1994, Chyung et al. 1997, Cook et al. 1997, Wild-Bode et al. 1997, Greenfield et al. 1999, Dingwall 2001, Farris et al. 2003, Lopez Salon et al. 2003).
Misfolded toxic proteins in the secretory pathway, such as Aβ peptides, can be recognized and degraded by a cellular process known as Endoplasmic Reticulum Associated Degradation (ERAD; Mccracken and Brodsky 1996). This process involves chaperone-based recognition mechanisms and the retrotranslocation of proteins from the ER to the proteasome for degradation (Mccracken and Brodsky 1996). When the ERAD pathway is overwhelmed, as occurs when an excessive amount of unfolded protein is present in the ER, the Unfolded Protein Response (UPR) is activated, which in the yeast cell results in some 381 gene targets being activated by the UPR (∼5% of the genome; Travers et al. 2000). The chaperone BiP (Grp78), which is a prominent UPR target, plays an important role in regulating UPR activation (Travers et al. 2000, Fewell et al. 2001). If the UPR fails to alleviate ER stress, and/or the cellular stress causes excessive or irreversible damage to the cell, then induction of the UPR ultimately leads to apoptosis (Walter and Ron 2011, Walter et al. 2015).
As discussed, one hypothesis to explain why the ERAD/UPR quality control system fails allowing for the accumulation of misfolded proteins such as Aβ peptides in AD, is the fact that proteasomal activity and cellular stress responses decline with age (Dillin and Cohen 2011, Dillin et al. 2014). Thus, if the proteasome, which is hypothesized to play a key role in the degradation of APP fragments becomes dysfunctional, it may directly cause AD and/or accelerate progression (Skovronsky et al. 2000, Nunan et al. 2001). This hypothesis is in agreement with the established age-related decline in proteasome function in people with AD and with the fact that age is the biggest risk factor for the development of AD (Alzheimer's Association 2021, Lilienfeld and Perl 1993). The goal of this work was to investigate whether APP-derived fragments, regardless of the pathway by which they are generated, are more toxic when proteasome activity is blunted.
Although deficient proteasome function can lead to the generation/accumulation of APP fragments and an increased risk for AD, most studies examining this connection have treated mammalian cells with amyloid fragments while manipulating the proteasome pharmacologically. While much has been learned from these approaches, the exogenous application of amyloid assumes that it is taken up by the exposed cells. In addition, pharmacological manipulation of the proteasome assumes that the effects of these agents are on-target and direct. As such there are limitations in the interpretation of these data.
To address these deficiencies in our current knowledge the use of the yeast system has several distinct advantages (Coughlan et al. 2004, Coughlan and Brodsky 2005). The most important advantage of using yeast is the ability to disable cellular pathways using temperature sensitive or hypomorphic mutants, in a nonpharmacological manner, while expressing mammalian proteins in an inducible manner. We previously reported on the construction of an inducible C99-expressing yeast strain and the identification of the APP fragments generated when proteasome activity was compromised (Sparvero et al. 2007). Interestingly, the resulting fragments possessed striking similarities to APP fragments present in AD patients (Sparvero et al. 2007). In the current study, we determined the cellular effects of generating amyloid fragments in a proteasomal mutant strain, uncovering a myriad of cellular toxicities that are likely relevant to AD-associated neurodegeneration in humans.
Materials and methods
Yeast strains and manipulations
Cells were grown using previously published procedures (Coughlan et al. 2004). Yeast strains wild-type (WCG4, MATa PRE1 PRE2, his3-11,15 leu2-3 112 ura3) and proteasome-deficient cells (WCG4/11–21, MATa pre1-1 pre2-1 his3-11,15 leu2-3 112 ura3) in which ∼95% of the chymotrypsin activity is absent (Heinemeyer et al. 1993, Sparvero et al. 2007) were transformed using the lithium acetate procedure with the PMF vector containing a galactose-inducible promoter that can be used to drive the expression of C99, as described previously (Ito et al. 1983, Sparvero et al. 2007). To measure cellular stress and activation of the UPR the PRE1PRE2 and pre1-1pre2-1 yeast cells were co-transformed with the PMF vector to express C99 or JJB20, and with another vector that contained the Unfolded Protein Response Element (UPRE) located upstream of a lacZ (KCZY) reporter gene (Mori et al. 1996). This construct acts as a UPR reporter. A 2-day induction in 2% galactose-containing medium was shown to be optimal for inducing the expression of the C99 peptide(s) (data not shown) and this time was used for all assays presented. The cell numbers utilized for each assay are specified.
Indirect immunofluorescence microscopy
A modified version of a procedure, kindly provided by Dr J. Vogel, Department Cellular, Molecular, and Developmental Biology, Yale University, was used to perform indirect immunofluorescence microscopy. A total of 3 ml of each strain expressing C99 or containing the empty vector (i.e. JJB20) grown to an optical density measurement at 600 nm (OD600) of 0.6, was fixed in 3.7% formaldehyde and incubated for 60 min at room temperature with shaking. The cells were harvested by centrifugation, washed (500 μl per all wash steps) twice with Solution A (1.2 M sorbitol; 50 mM KPO4, and pH 7.0), resuspended in Solution A (500 μl) containing 2 μg/ml of Z100T zymolase, and incubated at 37°C for 30 min. The cells were then washed twice with Solution A, once with PBS/0.1% BSA, and twice with PBS/0.1% NP4O/0.1% BSA. Incubation with primary antibody, D4253/D4254, diluted 1:100 in PBS/0.1% BSA was performed overnight at 4°C. D4253/D4254 was generated and verified by immunoblot analysis against the same 40 C-terminal residues of APP, kindly shared by and used to generate the C-terminal antibody in the laboratory of Dr V.M.Y. Lee. Cells were washed once with PBS/0.1% BSA, once with PBS/0.1% BSA/0.1% NP4O, PBS/0.1% BSA, and were incubated for 2 h at room temperature with secondary antibody (Goat Anti Rabbit Alexa 488) diluted 1:500 in PBS/0.1%BSA. After the incubation the cells were washed once with PBS/0.1% BSA, twice with PBS/0.1% BSA/0.1%NP4O, and once with PBS/0.1% BSA. The cells were kept in the last PBS/0.1% BSA wash and added to poly-L-lysine-coated slides. Prolong Gold antifade reagent mounting medium with DAPI (Invitrogen P36931) was added to each sample, a coverslip was applied, and sealed with clear nail polish, and the slides were left at 4°C overnight to dry. Images were captured using a Zeiss Axioplan microscope with a Hammamatsu Orca CCD camera, 100× (1.3 N.A.) objective and Slidebook software (Intelligent Imaging Innovations, Denver, CO).
The unfolded protein response assay
Sucrose density gradients and measuring chaperone expression levels
Sucrose density gradients were performed as described previously (Coughlan et al. 2004). A total of 100 ml of log-phase cells (OD600 0.5–0.8) were harvested for each strain expressing C99 or containing the JJB20 vector control in triplicate. For immunoblot analyses of obtained fractions, 4%–12% Bis-Tris gradient gels and nitrocellulose membranes were used with the antiAPP antibody and an antiyeast BiP monoclonal antibody (kindly provided by Dr Mark Rose, Princeton University).
Preparation of samples for mass spectrometry
A total of 32 ml of cells grown to an OD600 of 0.5 (16 OD600 units) of each strain expressing C99 were harvested and resuspended in 200 μl of ice-cold extract buffer (10 mM Tris-HCl pH 8.0, 25 mM ammonium acetate, 500 mM iodoacetamide, 20 mM sodium azide, 20 mM EDTA, 10% TCA, and 0.45 mM cycloheximide) to which protease inhibitors had been freshly added (1 mM PMSF, 1 μg/ml pepstatin A, and 3 μg/ml leupeptin). Zirconium oxide beads were added, the mixture was agitated on a vortex mixer and the supernatant retained. Proteins were resolved using 4%–12% Bis-Tris gels (Invitrogen) and the bands stained using Flamingo Dye according to the manufacturer's instructions (BioRad). Proteins/peptides in the ∼8–10 kDa region of the gel were excised. Based on our immunoblot analyses, this was the area of the gel that showed increased amyloid staining as compared to empty vector controls. The remaining proteins in the gel were transferred to nitrocellulose for immunoblot analyses to ensure all proteins/peptides of interest had been excised. Immunoblotting was performed using antiAPP (D4254) antiserum (see above). The bound antibody was detected using goat antirabbit secondary antibody (Jackson Immunoresearch) and the signal developed using the pmole sensitive Super Signal kit (Pierce).
Next, the excised bands were washed three times with 10 ml of 50% acetonitrile/25 mM ammonium bicarbonate. The samples were reduced and alkylated with tris (2-carboxy-ethyl) phosphine hydrochlorine (TCEP) and 55 mM iodoacetamide. After additional wash steps, the diced acrylamide pieces were dehydrated using 50% acetonitrile. A total of 300 ng trypsin (LifeTechnologies) was added to the samples overnight at room temperature. Peptides were solubilized in 1% formic acid to a total volume of 30 μl. Each sample (2 μl) was injected onto a reverse-phase column using a cooled (9°C) autosampler (Eksigent; Dublin, CA) connected to a HPLC system run at 14–18 μl/min before the split and ∼400 nl/min postsplit (Eldex; Napa, CA). A gradient of 5%–30% acetonitrile (in 0.1% formic acid) over 32 min was employed for peptide separation. The column effluent was coupled directly via fused silica capillary transfer line to a QSTAR XL™ Q-TOF tandem mass spectrometer (Sciex/Applied Biosystems; Framingham, MA) with a nanospray ion source. Data acquisition was performed using the supplied Analyst software. The LC runs were monitored by sequentially recording the precursor scan (MS, 1 s) followed by one collision-induced-dissociation (CID) acquisitions (MS/MS, 4 s each). Singly charged ions were excluded from CID selection. Normalized collision energies were employed using nitrogen as the collision gas.
The raw datafiles were converted to mascot generic format (.mgf) using the Analyst Mascot script (version 1.6b13). The de-isotoped, centroided peak lists, which were created from the Mascot script were searched against databases using an in-house Mascot™ server (Version 2.1, Matrix Science). For searches, mass tolerances were +/− 100 ppm for MS peaks, and +/− 0.3 Da for MS/MS fragment ions. Trypsin specificity was used and allowed for one missed cleavage. The modifications for the oxidation of methionine, N-terminal protein acetylation, and N-terminal peptide pyroglutamic acid formation were taken into consideration. Samples were searched against all entries in the full SwissProt (version 54.1) database using a yeast species filter. These peak lists were then again searched against Protein Prospector™, LC Batch-Tag Web™ (Version 4.25.2, University California San Francisco) with error tolerances of 100 ppm for MS peaks, and 0.15 Da for MS/MS fragment ions with an offset of −50 ppm for parent ion tolerances and the same modifications as the Mascot searches. Samples were searched against a yeast species filter using the SwissProt database (SwissProt_20070109.fasta.yeast).
The mgf files were further converted into mzXML files by use of mzStar (version 2.4.3). These files were imported into DeCyder MS (version 2.0.2586.0, GE Healthsciences, Uppsala, Sweden) using the following parameters. Retention time was cropped to look only at 22–40 min (the peptide gradient) and the proportional resampling option was selected.
Apoptosis (TUNEL) assay
A total of 10 ml of each log phase yeast strain culture (OD600 of 0.5) expressing C99 or containing the empty vector were grown for each sample utilized. These cells were fixed by adding 1 ml of 37% formaldehyde and incubated for 60 min at room temperature with shaking. For the positive control sample, 1 ml of methanol was added and incubated at room temperature with shaking before fixation with formaldehyde. The cells were harvested by centrifugation, washed three times with 500 μl PBS, resuspended in 500 μl PBS containing 35 units of lyticase (Sigma-Aldrich), and incubated at 37°C for 40 min. The cells were again washed three times with 500 μl PBS, resuspended in PBS, and 10 μl of this cell suspension was applied to a poly-L-lysine-coated microscope slide. The slides were rinsed once with 500 μl PBS, incubated in permeabilization solution (0.1% [vol/vol]) Triton X-100 and 0.1% [wt/wt] sodium citrate) for 2 min and rinsed twice with 500 μl PBS. The slides were subsequently incubated with 10 μl TUNEL reaction mixture (In Situ Cell Death Detection Kit; Fluorescein, Roche Diagnostics) for 30 min at 26°C. Finally, the slides were rinsed three times with 500 μl PBS and then mounted in Prolong Gold containing DAPI, and images were captured as described in the Immunofluoresence staining procedure, described above.
Electron microscopy
Aliquots of yeast cultures were prepared for electron microscopy as described previously (Giddings et al. 2001). Cells were harvested by vacuum filtration onto 0.45 μm Millipore filters and cryofixed by high pressure freezing in a BAL-TEC HPM-010 (BAL-TEC/RMC, Tucson, AZ). For analyses of structure without antibody labeling, samples were substituted in 2% osmium tetroxide and 0.1% uranyl acetate in acetone at −80°C, and were gradually warmed to room temperature and embedded in Epon-Araldite. Sections were stained with 2% uranyl acetate and lead citrate, then viewed in a Philips CM10 transmission electron microscope operating at 80 KV. Images were recorded with a Gatan BioScan digital camera or on Kodak 4489 electron microscope film.
Image quantification
Images for quantification were obtained with a Zeiss Axioplan2 microscope equipped with a z-step rotor, a 1.2 N.A. 100× oil objective, an excitation filter wheel, and a Hammamatsu Orca CCD camera all run using Slidebook software (Intelligent Imaging Inc). For the analysis of both the immunofluoresence staining and the apoptosis data, masks were created around the cells in the channel of opposite staining interest. For example, in the case of the immunofluorescence staining, where amyloid was detected using a secondary antibody conjugated to Alexa 488, the cells were chosen and masked using the DAPI channel only. Fluorescent intensities of the amyloid staining with 488 nm emission were determined for the cells chosen in this blind manner, removing researcher bias as to the cells chosen for analysis. For the DNA fragmentation assay, cells were stained with DAPI and those containing fragmented DNA were counted. For the immunofluorescence staining data, mean intensities of the 488 emission wavelength are reported along with the Wilcoxon–Mann–Whitney rank-sum test of the fluorescence measured for the samples. For the apoptosis data, a parametric t-test was chosen for the comparison of PRE1PRE2 + C99 versus pre1-1pre2-1 + C99 given the equal variances between mutant and wild-type cells expressing C99. Apoptosis type patterns (distinct puncta identified as TUNEL positive) were observed at intensities of approximately 1500 and above.Graphs were generated in Excel by determining bin frequencies of these data and converting them to the percentage of bin frequency data points.
Live-cell fluorescence
For the septin-GFP imaging, wild-type and proteasome-deficient cells were transformed with a URA3-marked ARS/CEN CDC3-GFP vector (Richman et al. 1999). Transformants were selected on the appropriate medium and cultures were prepared. After isolating the mid-log phase cells by centrifugation at 5000 rpm for 5 min, the cells were resuspended in water, spotted on agarose pads, and visualized using 250 ms exposures at 100% LED intensity. Micrographs were captured with an EVOSfl (Advanced Microscopy Group, Bothell, WA) all-in-one microscope equipped with an Olympus 60X oil immersion objective and a GFP LED filter cube.
Results
C99-derived peptides accumulate when C99 expression is induced in proteasome-mutant yeast cells
To determine the phenotypes of yeast cells expressing C99 when proteasome activity is compromised, we induced the expression of C99 in a proteasome-mutant strain (pre1-1, pre2-1) or in a wild-type strain (PRE1, PRE2) and compared them to the same cells carrying the empty vector control, JJB20. Indirect immunofluorescence microscopy was performed using a polyclonal antibody that recognizes the 40 C-terminal residues of C99. As shown in Fig. 1, when C99 was expressed in proteasome-deficient cells, an elongated punctate pattern of intracellular staining was apparent. This pattern was not observed in the wild-type strain expressing C99, and the background staining, as shown in the empty vector controls (JJB20), was minimal. When quantified, the production of C99 and C99-generated fragments, and oligomers thereof, were significantly increased in proteasome-mutant yeast cells expressing C99 compared to wild-type yeast cells expressing C99 (Fig. 2A and B, P < .005). These data suggest that the proteasome normally prevents these aggregates from forming inside the cell.
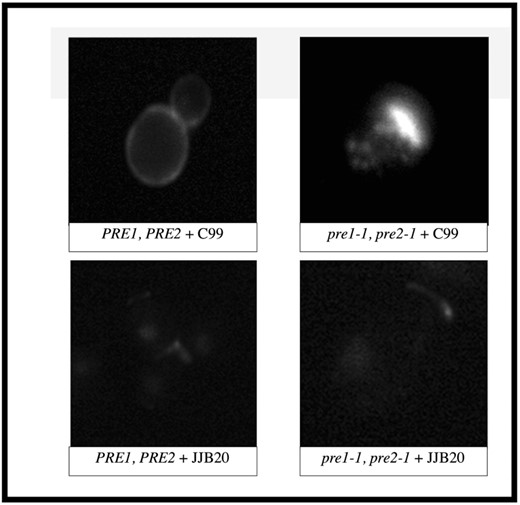
Altered subcellular localization and aggregation of C99 and its associated C-terminal fragments in proteasome-mutant yeast cells. Wild-type or proteasome-deficient (pre1-1, pre2-1) yeast cells expressing C99 or carrying an empty vector were examined by indirect immunofluorescence microscopy using an antibody that recognizes the 40 C-terminal residues of C99/APP. Localization in the mutant cells (pre1-1, pre2-1 + C99) revealed that C99 occurs in puncta that are not present in wild-type cells expressing C99 (PRE1, PRE2 + C99).
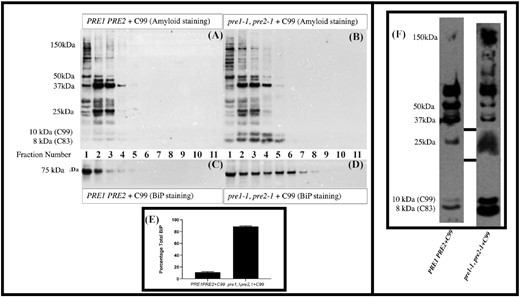
The ER chaperone BiP is mislocalized and upregulated in cells, with compromised proteasome activity, when expressing C99. Cell extracts were partially resolved by sucrose density gradients and analyzed by immunoblotting in triplicate as described in Materials and methods. Fractions 1–11 refer to the fractions taken from the top of the sucrose gradient (Fraction 1) to the last fraction at the bottom of the gradient (Fraction 11). Co-localization studies have shown that fractions 1–4 are enriched for endoplasmic reticulum while fractions 9–11 are enriched for plasma membrane. (A) Immunoblot analyses of C99 and/or its fragments in the endoplasmic reticulum and subsequent compartments of wild-type cells. (B) Expression of C99 and/or its fragments in proteasome-mutant cells. (C) BiP expression levels in wild-type cells expressing C99. (D) BiP expression levels in proteasome-mutant cells expressing C99. (E) Summary of the percentage of BiP after running triplicate samples of these gradients, in wild-type versus proteasome-mutant cells expressing C99. This implies activation of the UPR. (F) Loading controls for the sucrose gradient. While proteins/peptides in the 250–37 kDa and 25 kDa ranges sometimes showed differences between the wild-type and proteasome-mutant cells expressing C99, changes in the amount of peptide in the 8–10 kDa size range were always evident with the mutant consistently expressing more of these peptides.
Compromised proteasome activity and amyloid formation leads to cellular stress
To examine the effect of generating APP fragments in cells with compromised proteasomal activity, we performed sucrose density gradients. These experiments revealed that the levels of C99 and its fragments increased in proteasome-mutant cells and were found in an expanded range of fractions, an indication that this peptide and its fragments were present in more parts of the cell than just the endoplasmic reticulum, which is normally enriched in fractions 1–4. These data imply an overwhelmed degradative capacity when C99 was generated in mutant cells (Fig. 2A and B). By examining the ER associated fractions, BiP was found to be upregulated in proteasome-mutant cells when C99 was expressed (Fig. 2C–E). Loading controls for the sucrose gradient are shown in Fig. 2F. In Figure S1 (Supporting Information), despite using the same cell numbers for all strains, some peptides seem increased in the mutant cells expressing C99 (as shown in the Flamingo stained gel). However, in Fig. 2F while proteins/peptides in the 250–37 kDa and 25 kDa range sometimes show differences between the wild-type and mutant cells expressing C99, increases in the amount of peptide in the 8–10 kDa size range are always evident in the mutant cells expressing C99. This size range is consistent with C99 (10 kDa) and C83 fragments (8 kDa). It is possible that the accumulation of cytoplasmic C99 additionally leads to defects in proteasome activity and degradation, a phenomenon that has been shown to inhibit ERAD and to induce the UPR in a Huntington's disease model (Duennwald and Lindquist 2008)
Next, we investigated directly whether cellular stress was induced in response to the accumulation of amyloid fragments. Cell lysates were prepared from strains defective for proteasome activity and expressing C99 and an UPRE-β-galactosidase reporter assay was performed (see Materials and methods). Significantly higher levels of UPR induction were observed in proteasome-mutant strains expressing C99 (galactose growth conditions) versus proteasome-mutant strains lacking C99 (raffinose growth conditions). In addition, proteasome-mutant strains containing the vector control (pre1-1, pre2-1 + JJB20 + KCZY + galactose) showed significantly less stress compared to mutant strains expressing C99 (pre1-1, pre2-1 + C99 + KCZY grown in galactose), and wild-type strains expressing C99 (PRE1, PRE2 + C99 + KCZY grown in galactose) showed lower levels of UPR induction compared to mutant strains expressing C99 (pre1-1, pre2-1 + C99 + KCZY grown in galactose; Fig. 3) These data strongly suggest that C99, and/or the fragments generated from C99, place a burden on ER stress-associated pathways that are mitigated by the proteasome. These findings are also consistent with the notion that defects in proteasome function, in the presence of C99, leads to inhibition of the ERAD pathway and induces the UPR.
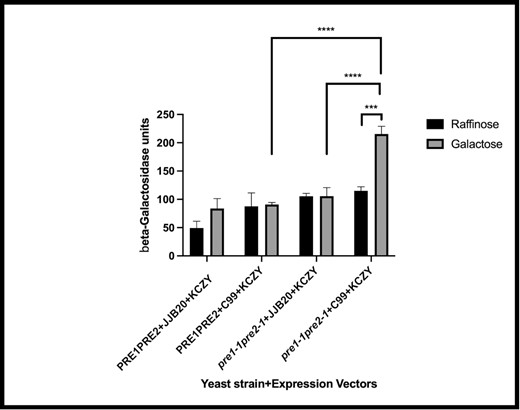
The UPR is activated in proteasome-mutant yeast that express C99. A lacZ-based reporter assay was used to measure the activation of the UPR in wild-type cells (PRE1, PRE2) and in cells mutant for proteasomal activity (pre1-1, pre2-1) expressing C99 or carrying an empty vector (JJB20). Significant differences (*** P = .0003) in the β-galactosidase units (indicative of UPR induction) were observed between pre1-1, pre2-1 + C99 + KCZY grown in raffinose (noninduced for C99 expression in the proteasome-mutant strain) versus pre1-1, pre2-1 + C99 + KCZY grown in galactose (induced expression of C99 in the proteasome-mutant strain). In addition, pre1-1, pre2-1 + JJB20 + KCZY + galactose (induced expression of empty vector in the mutant strain) was significantly different (****P < .0001) compared to pre1-1, pre2-1 + C99 + KCZY grown in galactose (induced expression of C99 in the mutant strain) and PRE1, PRE2 + C99 + KCZY grown in galactose (induced expression of C99 in the wild-type strain) was significantly different (****P < .0001) compared to pre1-1, pre2-1 + C99 + KCZY grown in galactose (induced expression of C99 in the proteasome-mutant strain).
To identify proteins that are altered in response to the generation of C99 and its fragments in the presence/absence of a fully functional proteasome, electrospray ionization mass spectrometry was performed as described in the Materials and methods. An abundance of peptides was found in all of the strains and expression systems used. Those represented here were ranked based on the percentage of protein sequence coverage. Peptides present in the empty-vector controls were considered to be endogenously expressed proteins and were not analyzed further. A total of six proteins were common to both the wild-type and proteasome-mutant cells expressing C99, two proteins were uniquely produced in C99-expressing wild-type cells, and 14 proteins were identified as unique to the C99-expressing mutant cells (Table 1). Interestingly, all of the species identified in the proteasome-mutant cells expressing C99 were those identified in a transcriptome analysis of yeast cells with a chemically induced UPR (Travers et al. 2000), further supporting the view that the UPR is induced under these conditions.
Proteins involved in the stress response are induced in proteasome-mutant yeast cells expressing the C99 fragment of APP. The differentially expressed proteins in the proteasome-mutant (pre1-1, pre2-1) cells expressing C99 versus the wild-type (PRE1, PRE2) cells expressing C99 were identified in the 8–10 kDa size range. These peptides were isolated using gel electrophoresis and excision, analyzed by MALDI-TOF Mass Spectrometry, and identified by Spectra taken in reflector mode with CHCA as the matrix. The findings represent post baseline correction, noise removal, and Gaussian smoothing. No MS/MS sequencing (post-source decay) could be performed on the peaks, because the absolute intensities were too low for peaks in the given mass range. Shown are the peptides, in descending order of abundance, that are uniquely generated in wild-type cells expressing C99 versus proteasome-mutant cells are shown.
| pre1-1pre2-1 + C99 | Where in the cell? | Main cellular function |
|---|---|---|
| SSA1 | Nucleus, cytoplasm, and cell wall | Chaperone, disassembles aggregates, protein import into nucleus, and folds newly translated proteins |
| SSA2 | Cytoplasm, vacuolar membrane, and cell wall | Same as SSA1, also involved in vacuolar import and responds to stress |
| SSB1 | Cytoplasm | Ribosome associated chaperone, folds newly translated proteins |
| SSB2 | Cytoplasm and polysome | Same as SSB1, ribosome associated |
| SSE1 | Cytoplasm | Same as SSA1, also acts as a holdase preventing protein misfolding |
| SSE2 | Cytoplasm | Same as SSE1 |
| SAT | Cytoplasm | Trafficking in the secretory pathway, ARF activity, ER to Golgi and intra-Golgi, and sterol esterification |
| Proteinase inhibitor I(B)2 | Cytoplasm and vacuole | Reduces endopeptidase activity and promotes vacuolar fusion |
| RPPO | Large ribosomal subunit | Involved in large ribosomal assembly |
| MET17 | Cytoplasm | Methionine and cysteine synthesis and responds to toxins and/or drugs |
| YBR025C/OLA1 | Cytoplasm | Interacts with the proteasome |
| Alcohol dehydrogenase 1 | Cytosol | Converts acetaldehyde to ethanol |
| Alcohol dehydrogenase 2 | Cytoplasm | Converts ethanol to acetaldehyde |
| PGAM1/MPGM1 | Mitochondria and cytoplasm | Yeast monophosphate glycerate kinase, generates 3-phosphoglycerate in the glycolysis pathway |
| PRE1PRE2 + C99 | ||
| YNR034W-A | Cytoplasm | Unknown, expression regulated by Msn2p/Msn4p; transcriptional activators that bind stress response elements |
| HMF1 | Cytoplasm, nucleus, and cytosol | Heat shock inducible, similar to the mitochondrial matrix factor Mmf1p |
| Both | ||
| ATP synthase α | Mitochondria | Required for ATP synthesis |
| ATP synthase β | Mitochondria | Required for ATP synthesis |
| RP51A | Cytoplasm | Ribosome assembly |
| RP51B | Cytoplasm | Ribosome assembly |
| Fructose-bisphosphate aldolase | Mitochondria, cytoplasm, and cytosol | Required for glycolysis and gluconeogenesis |
| L38 | Ribosome | Translation |
| pre1-1pre2-1 + C99 | Where in the cell? | Main cellular function |
|---|---|---|
| SSA1 | Nucleus, cytoplasm, and cell wall | Chaperone, disassembles aggregates, protein import into nucleus, and folds newly translated proteins |
| SSA2 | Cytoplasm, vacuolar membrane, and cell wall | Same as SSA1, also involved in vacuolar import and responds to stress |
| SSB1 | Cytoplasm | Ribosome associated chaperone, folds newly translated proteins |
| SSB2 | Cytoplasm and polysome | Same as SSB1, ribosome associated |
| SSE1 | Cytoplasm | Same as SSA1, also acts as a holdase preventing protein misfolding |
| SSE2 | Cytoplasm | Same as SSE1 |
| SAT | Cytoplasm | Trafficking in the secretory pathway, ARF activity, ER to Golgi and intra-Golgi, and sterol esterification |
| Proteinase inhibitor I(B)2 | Cytoplasm and vacuole | Reduces endopeptidase activity and promotes vacuolar fusion |
| RPPO | Large ribosomal subunit | Involved in large ribosomal assembly |
| MET17 | Cytoplasm | Methionine and cysteine synthesis and responds to toxins and/or drugs |
| YBR025C/OLA1 | Cytoplasm | Interacts with the proteasome |
| Alcohol dehydrogenase 1 | Cytosol | Converts acetaldehyde to ethanol |
| Alcohol dehydrogenase 2 | Cytoplasm | Converts ethanol to acetaldehyde |
| PGAM1/MPGM1 | Mitochondria and cytoplasm | Yeast monophosphate glycerate kinase, generates 3-phosphoglycerate in the glycolysis pathway |
| PRE1PRE2 + C99 | ||
| YNR034W-A | Cytoplasm | Unknown, expression regulated by Msn2p/Msn4p; transcriptional activators that bind stress response elements |
| HMF1 | Cytoplasm, nucleus, and cytosol | Heat shock inducible, similar to the mitochondrial matrix factor Mmf1p |
| Both | ||
| ATP synthase α | Mitochondria | Required for ATP synthesis |
| ATP synthase β | Mitochondria | Required for ATP synthesis |
| RP51A | Cytoplasm | Ribosome assembly |
| RP51B | Cytoplasm | Ribosome assembly |
| Fructose-bisphosphate aldolase | Mitochondria, cytoplasm, and cytosol | Required for glycolysis and gluconeogenesis |
| L38 | Ribosome | Translation |
Proteins involved in the stress response are induced in proteasome-mutant yeast cells expressing the C99 fragment of APP. The differentially expressed proteins in the proteasome-mutant (pre1-1, pre2-1) cells expressing C99 versus the wild-type (PRE1, PRE2) cells expressing C99 were identified in the 8–10 kDa size range. These peptides were isolated using gel electrophoresis and excision, analyzed by MALDI-TOF Mass Spectrometry, and identified by Spectra taken in reflector mode with CHCA as the matrix. The findings represent post baseline correction, noise removal, and Gaussian smoothing. No MS/MS sequencing (post-source decay) could be performed on the peaks, because the absolute intensities were too low for peaks in the given mass range. Shown are the peptides, in descending order of abundance, that are uniquely generated in wild-type cells expressing C99 versus proteasome-mutant cells are shown.
| pre1-1pre2-1 + C99 | Where in the cell? | Main cellular function |
|---|---|---|
| SSA1 | Nucleus, cytoplasm, and cell wall | Chaperone, disassembles aggregates, protein import into nucleus, and folds newly translated proteins |
| SSA2 | Cytoplasm, vacuolar membrane, and cell wall | Same as SSA1, also involved in vacuolar import and responds to stress |
| SSB1 | Cytoplasm | Ribosome associated chaperone, folds newly translated proteins |
| SSB2 | Cytoplasm and polysome | Same as SSB1, ribosome associated |
| SSE1 | Cytoplasm | Same as SSA1, also acts as a holdase preventing protein misfolding |
| SSE2 | Cytoplasm | Same as SSE1 |
| SAT | Cytoplasm | Trafficking in the secretory pathway, ARF activity, ER to Golgi and intra-Golgi, and sterol esterification |
| Proteinase inhibitor I(B)2 | Cytoplasm and vacuole | Reduces endopeptidase activity and promotes vacuolar fusion |
| RPPO | Large ribosomal subunit | Involved in large ribosomal assembly |
| MET17 | Cytoplasm | Methionine and cysteine synthesis and responds to toxins and/or drugs |
| YBR025C/OLA1 | Cytoplasm | Interacts with the proteasome |
| Alcohol dehydrogenase 1 | Cytosol | Converts acetaldehyde to ethanol |
| Alcohol dehydrogenase 2 | Cytoplasm | Converts ethanol to acetaldehyde |
| PGAM1/MPGM1 | Mitochondria and cytoplasm | Yeast monophosphate glycerate kinase, generates 3-phosphoglycerate in the glycolysis pathway |
| PRE1PRE2 + C99 | ||
| YNR034W-A | Cytoplasm | Unknown, expression regulated by Msn2p/Msn4p; transcriptional activators that bind stress response elements |
| HMF1 | Cytoplasm, nucleus, and cytosol | Heat shock inducible, similar to the mitochondrial matrix factor Mmf1p |
| Both | ||
| ATP synthase α | Mitochondria | Required for ATP synthesis |
| ATP synthase β | Mitochondria | Required for ATP synthesis |
| RP51A | Cytoplasm | Ribosome assembly |
| RP51B | Cytoplasm | Ribosome assembly |
| Fructose-bisphosphate aldolase | Mitochondria, cytoplasm, and cytosol | Required for glycolysis and gluconeogenesis |
| L38 | Ribosome | Translation |
| pre1-1pre2-1 + C99 | Where in the cell? | Main cellular function |
|---|---|---|
| SSA1 | Nucleus, cytoplasm, and cell wall | Chaperone, disassembles aggregates, protein import into nucleus, and folds newly translated proteins |
| SSA2 | Cytoplasm, vacuolar membrane, and cell wall | Same as SSA1, also involved in vacuolar import and responds to stress |
| SSB1 | Cytoplasm | Ribosome associated chaperone, folds newly translated proteins |
| SSB2 | Cytoplasm and polysome | Same as SSB1, ribosome associated |
| SSE1 | Cytoplasm | Same as SSA1, also acts as a holdase preventing protein misfolding |
| SSE2 | Cytoplasm | Same as SSE1 |
| SAT | Cytoplasm | Trafficking in the secretory pathway, ARF activity, ER to Golgi and intra-Golgi, and sterol esterification |
| Proteinase inhibitor I(B)2 | Cytoplasm and vacuole | Reduces endopeptidase activity and promotes vacuolar fusion |
| RPPO | Large ribosomal subunit | Involved in large ribosomal assembly |
| MET17 | Cytoplasm | Methionine and cysteine synthesis and responds to toxins and/or drugs |
| YBR025C/OLA1 | Cytoplasm | Interacts with the proteasome |
| Alcohol dehydrogenase 1 | Cytosol | Converts acetaldehyde to ethanol |
| Alcohol dehydrogenase 2 | Cytoplasm | Converts ethanol to acetaldehyde |
| PGAM1/MPGM1 | Mitochondria and cytoplasm | Yeast monophosphate glycerate kinase, generates 3-phosphoglycerate in the glycolysis pathway |
| PRE1PRE2 + C99 | ||
| YNR034W-A | Cytoplasm | Unknown, expression regulated by Msn2p/Msn4p; transcriptional activators that bind stress response elements |
| HMF1 | Cytoplasm, nucleus, and cytosol | Heat shock inducible, similar to the mitochondrial matrix factor Mmf1p |
| Both | ||
| ATP synthase α | Mitochondria | Required for ATP synthesis |
| ATP synthase β | Mitochondria | Required for ATP synthesis |
| RP51A | Cytoplasm | Ribosome assembly |
| RP51B | Cytoplasm | Ribosome assembly |
| Fructose-bisphosphate aldolase | Mitochondria, cytoplasm, and cytosol | Required for glycolysis and gluconeogenesis |
| L38 | Ribosome | Translation |
C99 expression in cells with dysfunctional proteasome activity accumulate electron-dense material in the nuclear envelope, display an abnormal DNA staining pattern, and undergo apoptosis
To determine the biological consequences of C99 expression in pre1-1pre2-1 cells, aliquots of yeast cultures were prepared for transmission electron microscopy (TEM) as described in the Materials and methods section and in (Giddings et al. 2001). Nuclear blebbing was evident in the presence or absence of C99 expression (denoted by arrows, Fig. 4). Only in the mutant cells expressing C99 was there additional electron dense material in the nuclear envelope or extra nuclear inclusions (indicated by black arrows). Because the nuclear envelope is continuous with the ER membrane, one could hypothesize that deposition of material in this region of the cell might trigger the induction of pathology.
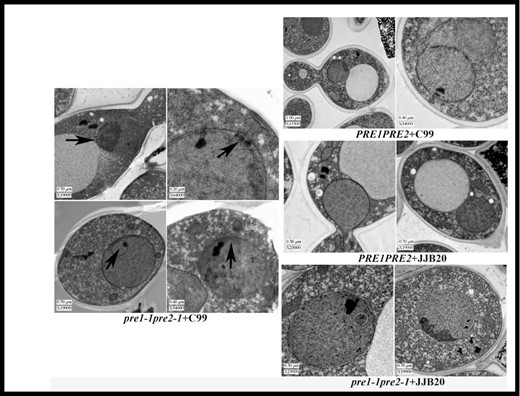
Accumulation of electron dense material in the nuclear envelope/ER of C99-expressing proteasome-mutant yeast cells. TEM images of wild-type or proteasome-mutant yeast cells expressing C99 or carrying an empty vector. Black arrosws indicate electron-dense material and intranuclear inclusions in the nuclear envelope.
As in other cell types, several stresses induce an apoptosis-like program in yeast (Carmona-Gutierrez et al. 2010). To assay for apoptosis-like responses upon C99 expression, we examined DNA integrity in each of the cell types used (Fig. 5A–E). The fraction of cells positive for DNA fragmentation was scored for at least 280 cells. We found that a total of 31.2% of C99-expressing pre1 pre2 cells had fragmented DNA (n = 525), compared to 11.3% (n = 300) of the pre1 pre2 cells carrying the empty vector, 4.6% (n = 500) of wild-type cells carrying the empty vector, and 7.5% (n = 280) of wild-type cells expressing C99 (Fig. 5A–E). We also examined the cells using a TUNEL assay (Neale and Keeney 2009, Kavakçıoğlu and Tarhan 2018, Lee and Lee 2018; Fig. 5F–H). The mean intensities of the PRE1PRE2 + C99 versus pre1-1pre2-1 + C99 were 903.1 and 990 respectively (P < .0001), as determined by a parametric t-test. Apoptosis type patterns (distinct punctate identified as TUNEL positive) were observed at intensities of approximately 1500 and above and were found exclusively in the pre1-1pre2-1 + C99 (7.37% of cells). The data are consistent with the induction of apoptosis upon expression of C99 in the pre1-1pre2-1 mutant cells.
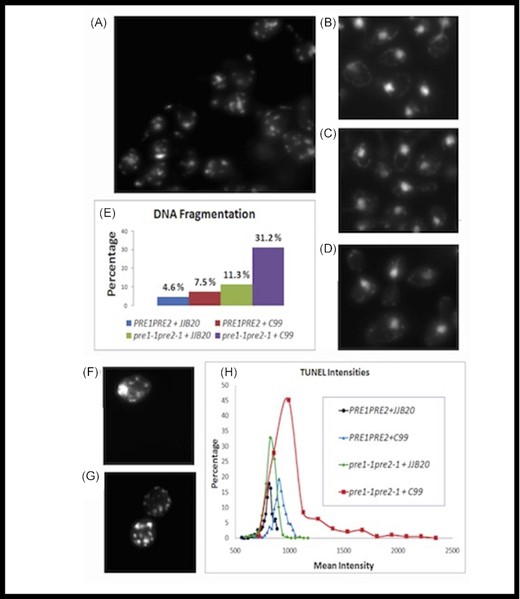
C99 expression induces apoptotic phenotypes in proteasome-mutant yeast cells. In order to examine potential apoptotic phenotypes, we used DAPI staining to examine DNA fragmentation (A)–(E) and performed TUNEL assays (F)-(H) in wild-type and proteasome-mutant yeast cells expressing C99 or carrying an empty vector. The percentage of cells positive for DNA fragmentation, as determined by fragmented DAPI staining, shows that (A) 7.5% (n = 280) of wild-type cells expressing C99 (B) 31.2% of proteasome-mutant cells expressing C99 (n = 525), (C) 4.6% (n = 500) of wild-type cells carrying the empty vector, and (D) 11.3% (n = 300) of proteasome-mutant cells carrying the empty vector, exhibited DNA fragmentation (E) Excel graphs were generated by determining the bin frequencies of these data and converting them to the percentage of bin frequency data points. (F) Only the proteasome-mutant yeast cells (pre1-1, pre2-1) expressing C99 showed increased TUNEL-positive staining. (G) Positive control for the TUNEL assay (mutant cells incubated in methanol for 30 min). (H) Percent TUNEL positivity for wild-type (PRE1, PRE2) and proteasome-mutant (pre1-1pre2-1) yeast cells expressing C99 or carrying an empty vector (JJB20). Significant levels of apoptosis were observed in the proteasome-mutant cells expressing C99 compared to wild-type cells expressing C99 (7.4%, P < .046) as determined by the Wilcoxon–Mann rank-sum test.
A protein required for cell division exhibits wild-type localization in proteasome-deficient yeast expressing the APP fragment
The proteasome has known roles in clearing aggregation-prone proteins (Brodsky and Mccracken 1999, Hanna et al. 2019). We considered the possibility that C99 aggregation in proteasome-deficient cells could represent a fate common to many proteins in the mutant cells. To test this possibility, we examined the localization of a septin protein. Septin proteins form hetero-oligomeric complexes in the cytosol that polymerize at the cell cortex into cytoskeletal filaments (Mostowy and Cossart 2012). In yeast cells, native septins form a distinct ring at the mother-bud neck (Oh and Bi 2011). Human septins are found in pathology-associated protein aggregates (Mostowy and Cossart 2012), and a human septin co-purifies with human beta-amyloid (Oláh et al. 2011). Moreover, misfolded mutant yeast septins are prone to forming punctate aggregates in thermally stressed wild-type cells (C.M.C. and M.A.M, personal communication). Thus, yeast septin localization represented a sensitive readout of general protein folding in proteasome-deficient cells expressing C99. As in control cells (pre1 pre2 carrying the empty vector, or wild-type cells expressing C99), a GFP-tagged copy of the septin Cdc3 localized normally to a ring at the mother-bud neck in proteasome-mutant cells expressing C99 (Fig. 6). Thus, the localization patterns we observed for C99 in proteasome-mutant cells do not reflect general defects in proteostasis.
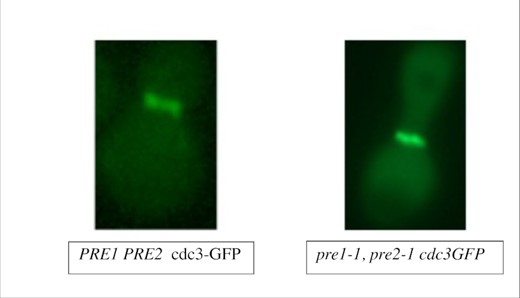
A GFP-tagged septin/Cdc3 localizes normally to a ring at the mother-bud neck in proteasome-mutant yeast cells expressing C99. Wild-type or proteasome-deficient yeast cells were transformed with a plasmid for expression of GFP-tagged septin (CDC3-GFP; Richman et al. 1999). Micrographs of log-phase cells were captured to examine CDC3-GFP localization to the ring in the mother-bud neck. Scale bar corresponds to 5 µm.
Discussion
Our data demonstrate that the generation of C99 and its fragments is toxic in cells with compromised proteasome activity as indicated by the aggregation of protein inside the cells, induction of the UPR, increased chaperone expression, accumulation of electron dense material in the nuclear envelope/ER, DNA fragmentation, and apoptosis. This represents the first report of a direct link between the C99 amyloidogenic species, cellular toxicity, and proteasome dysfunction. Given that treatment with proteasomal inhibitors alone is sometimes sufficient to induce apoptosis (Zanotto-Filho et al. 2010, Nam et al. 2015), the use of a yeast strain, i.e. solely deficient in proteasome activity offers a clear advantage for identifying and characterizing these pathologies.
The data presented in this work strongly suggest that C99, and/or the fragments generated from C99, place a burden on ER stress-associated pathways that are mitigated by the proteasome. Given that BiP is a known regulator of the UPR (Kopp et al. 2019), that Aβ peptide has been shown to induce the UPR, that BiP can bind to APP, and that BiP expression correlates with AD progression (Kohno et al. 1993, Bertolotti et al. 2000, Sato et al. 2000, Gupta et al. 2006, Pincus et al. 2010), the generation of C99 and its fragments when proteasomal activity is compromised may be responsible for the UPR and upregulated BiP expression. This can result if BiP binds to the APP fragments, freeing Ire1 to induce the UPR. These findings suggest that defects in proteasome function, in the presence of C99, result in the formation of aggregates that either overwhelm the ERAD pathway or block the proteasome (Bence et al. 2001), in turn inducing the UPR (Travers et al. 2000, Okada et al. 2002). In addition, all of the proteins we identified as induced in our system (Table 1) were also reported in studies examining the spectrum of UPR-induced genes in yeast (Travers et al. 2000). The induction of these pathways, and the role of the UPR, are most likely activated to help the cell manage the excessive production of APP-derived fragments in response to an ERAD defect, which arises from the absence of a fully functional proteasome.
The induction of ER stress and the UPR led us to hypothesize that apoptosis, which contributes to AD and neurodegeneration (Madeo et al. 1997, 1999, Kruman 2004, Ueberham and Arendt 2005, Wilson 2005, Chong et al. 2006, Hoozemans et al. 2006, Kudo et al. 2006, Mosch et al. 2007, Nagy 2007, Ahn et al. 2008, Konstantinova et al. 2008, Perrone et al. 2008, Huang and Chen 2009, Lee et al. 2009, Motegi et al. 2009, Granic et al. 2010, Devalle et al. 2012, Bushman and Chun 2013, Núñez et al. 2015), may also occur in our model. Using DAPI staining and a TUNEL assay, we observed that 31.2% of the cells appeared to be aneuploid and that 7.37% of these cells were TUNEL-positive. Hence, cellular stress appeared to significantly affect cellular homeostasis. This may be due either to an upset in the cell cycle or to cell death by some other mechanism, such as an induction of apoptosis. Based on the important role of the nuclear envelope for maintaining cellular viability (Wilson 2005), the electron dense and extra nuclear material we observed deposited in the nuclear envelope/ER and nucleus in the C99-expressing proteasome-defective cells (Fig. 4) may have lethal consequences. In addition, reports that AD neurons re-enter but do not necessarily complete the cell cycle, which leads to apoptosis (Kruman 2004, Ueberham and Arendt 2005, Chong et al. 2006), led us to hypothesize that amyloid-associated protein accumulation may similarly hinder the cell cycle. Cell cycle defects are known to occur in ∼20% of neurons in vulnerable brain areas early in disease progression (Hoozemans et al. 2006, Mosch et al. 2007, Nagy 2007, Lee et al. 2009). Cell cycle re-entry can also be caused by expressing mutant forms of APP (e.g. APPswe), APP oligomers, and/or APP fragments (Esteras et al. 2012, Xu et al. 2013), which may in turn explain the aneuploidy-related staining we observed (Fig. 5). Interestingly, although the role of the proteasome with regard to the regulation of cell cycle check-point proteins is well documented (Konstantinova et al. 2008, Huang and Chen 2009, Motegi et al. 2009), we did not observe abnormal DNA staining patterns in the proteasome-mutant cells transformed with the empty vector. This implies that the abnormalities we noted are instead caused by the generation of APP fragments in mutant cells. However, it is notable that amyloid itself may cause aneuploidy (Granic et al. 2010), in turn generating more vulnerable cell populations that are more responsive to the toxic effects of amyloid. In the central nervous system, aneuploidy also results in stem cells that are less viable and regenerative (Devalle et al. 2012, Bushman and Chun 2013), possibly removing replacements for those cells that die.
In summary, this work provides new insights into the physiological effects that result when proteasome activity is compromised and C99 is expressed and processed. Our findings help elucidate the link between the production of APP fragments and neurodegeneration and shed light on the impact of a failing proteasome in aged AD patients. Because cleavage of C99 into amyloidogenic fragments is more prevalent in cells with compromised proteasome activity (Sparvero et al. 2007), we postulate that losing the ability to degrade/process C99 by the proteasome either allows a greater concentration of substrate to accumulate (in this case C99) that then becomes available for other proteolytic enzymes (Burton et al. 2008), which in turn generate fragments and aggregates that are toxic, and/or inhibits the capacity to remove fragments that are toxic. If the types of fragments observed are endogenously produced, we conclude that a functional proteasome protects cells against the toxicity of APP fragments. It also points to the cellular mechanisms that may be affected under conditions that cause the observed toxicity. Importantly, this work also provides evidence that yeast can serve as a valuable model system for research and testing therapeutics targeted for diseases that result from amyloid fragment accumulation when proteostasis is dysfunctional, and the utility of this type of model system for the investigation of other neurodegenerative diseases.
Funding
C.M.C acknowledges support of an American Heart Association postdoctoral fellowship during the early stages of this study. A subsequent Faculty Research Fund (FRF), two Professional Research Opportunity Grants for Faculty PROF-88292 and PROF-88750, and Partnership in Scholarship monies awarded to C.M.C. as a Faculty partner for Sarah Patz, Trinh Nguyen, Abram D'Amato, John Galvin, Alicia Goossens, Elizabeth Curran, and Francisco Arteaga, which allowed this work to come to fruition. J.L.B. acknowledges support from the Pittsburgh Center for Kidney Research (DK79307) and grant GM131732 from the National Institutes of Health. This work was also supported in part by a grant (R00 GM086603) from the National Institutes of Health to M.A.M.
ACKNOWLEDGEMENTS
The authors wish to thank Thomas Giddings and Mark Winey (MCDB, UC Boulder) for performing, analyzing, and covering some of the cost of the TEM work described in this paper. We would like to thank the Brodsky lab for their technical advice and support, especially Jennifer Goeckeler. We are also grateful to Anna Sher for helpful discussions regarding the statistical analysis, Robert Fuller, (University of Michigan) for the JJB20 construct, Alicia Todd (University of Melbourne, Australia) for the KCZY vector, Jack Kinnamon for the use of his microscope, Robert Doms and Don Pijak (University of Pennsylvania) who provided the APP constructs, and V.M.Y Lee and Susan Leight (University of Pennsylvania) for providing the peptide sequence for the generation of the D4253 and D4254 antibodies used in this work. We would like to mention Joseph Angleson (University of Denver) for his support throughout this project, the use of his microscope to obtain the apoptosis data, and for helping with the analysis of the fluorescent data obtained for both the IF and apoptosis experiments. We thank all who provided feedback on this manuscript and all the students in the lab who helped with other yeast experiments including Connie Sun, Sarah Patz, Trinh Nguyen, Abram D'Amato and Erika Ross, which in turn aided in determining optimal growth conditions for the strains used in this work.
Conflicts of interest
None declared.
References
Author notes
John Galvin, Elizabeth Curran, Francisco Arteaga and Alicia Goossens are Contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}