





Human cryptosporidiosis is caused primarily by Cryptosporidium hominis, C. parvum and C. meleagridis. To accelerate research on parasites in the genus Cryptosporidium, we generated annotated, draft genome sequences of human C. hominis isolates TU502_2012 and UKH1, C. meleagridis UKMEL1, also isolated from a human patient, and the avian parasite C. baileyi TAMU-09Q1. The annotation of the genome sequences relied in part on RNAseq data generated from the oocyst stage of both C. hominis and C. baileyi. The genome assembly of C. hominis is significantly more complete and less fragmented than that available previously, which enabled the generation of a much-improved gene set for this species, with an increase in average gene length of 500 bp relative to the protein-encoding genes in the 2004 C. hominis annotation. Our results reveal that the genomes of C. hominis and C. parvum are very similar in both gene density and average gene length. These data should prove a valuable resource for the Cryptosporidium research community.
Cryptosporidium parasites (Phylum: Apicomplexa) infect a wide range of vertebrates, from fish to humans, and are the causative agents of cryptosporidiosis in humans (Upton and Current 1985; Tzipori 1988; Widmer and Sullivan 2012). A recent, large, multicenter study of the etiology of moderate-to-severe diarrhea (MSD) in infants in the developing world found Cryptosporidium hominis to be among the four predominant pathogens associated with MSD in children under 5 years of age (Kotloff etÃÂ al.2013). Some Cryptosporidium species are capable of zoonotic transmission (Ryan, Fayer and Xiao 2014). Comparative analysis of genomes from diverse Cryptosporidium species and related protists is essential to fully understand the biology, pathology, host specificity and evolution of this genus.
The reference C. parvum IOWA II genome (Abrahamsen etÃÂ al.2004) is essentially complete, with its eight chromosomes distributed among 18 contigs, including full-length chromosomes. In contrast, the reference assembly of C. hominis, based on isolate TU502, published in 2004 (Xu etÃÂ al.2004), is a highly fragmented draft genome consisting of 1422 contigs. To accelerate research on these pathogens of public health and veterinary significance, we sequenced, assembled and annotated four Cryptosporidium genome sequences belonging to three species as part of a community White Paper undertaking. Two sequences were generated from a species infective to humans, C. hominis isolates TU502_2012 and UKH1. In addition, sequences were generated from the generalist species C. meleagridis, isolate UKMEL1, and from the TAMU-09Q1 isolate of C. baileyi, an avian-infecting parasite. All three species are enteric parasites. Cryptosporidium baileyi can complete its entire life cycle in embryonated chicken eggs, making it a useful laboratory model to address some aspects of Cryptosporidium biology. Cryptosporidium meleagridis appears to lack host specificity, as it is known to infect both avian and mammalian species (Akiyoshi etÃÂ al.2003).
Cryptosporidium hominis UKH1 and C. meleagridis UKMEL1 oocysts were isolated from fecal samples of naturally infected humans. Cryptosporidium meleagridis oocysts were propagated in immunosuppressed adult CD-1 mice, and C. hominis UKH1 in neonatal gnotobiotic pigs. Cryptosporidium hominis TU502_2012 originates from C. hominis TU502 isolate maintained by serial propagation in gnotobiotic pigs (Tzipori etÃÂ al.1994; Xu etÃÂ al.2004). Cryptosporidium baileyi oocysts were extracted from experimentally infected embryonated chicken eggs. Prior to isolating DNA, extracted oocysts were purified on density gradients (Widmer, Feng and Tanriverdi 2004) and surface-sterilized with bleach to minimize contamination with host and bacterial DNA. RNA samples were obtained from C. hominis TU502_2012 and C. baileyi TAMU-10GZ1 oocysts <4 months old, and sequenced to high coverage using strand-specific RNASeq (Parkhomchuk etÃÂ al.2009). De novo assembly of the genomic reads was performed using MaSuRCA version1.9 (Zimin etÃÂ al.2013) (Table 1).
Summary statistics of whole-genome sequence and transcriptome data, assemblies and annotation.
| Cryptosporidium hominis | Cryptosporidium | Cryptosporidium | |||
|---|---|---|---|---|---|
| meleagridis | baileyi | ||||
| Isolate: DNA | TU502a | TU502_2012 | UKH1 | UKMEL1 | TAMU-09Q1 |
| gDNA Illumina library fragment size (bp) | N/A | 460 | 461 | 517 | 654 |
| No. MiSeq reads | N/A | 6,871,858 | 7,596,410 | 22,862,044 | 6,240,960 |
| No. base pairs | N/A | 1,724,836,358 | 1,906,698,910 | 6,881,475,244 | 1,566,480,960 |
| Assembly size (bp) | 8,743,570 | 9,107,739 | 9,156,091 | 8,973,200 | 8,493,640 |
| No. of contigs | 1422 | 119 | 156 | 57 | 145 |
| Contig N50 | 14,504 | 238,509 | 179,408 | 322,908 | 203,018 |
| Largest contig (bp) | 90,444 | 1,270,815 | 542,781 | 732,862 | 702,637 |
| G + C content (%) | 30.9 | 30.1 | 30.1 | 31.0 | 24.3 |
| No. protein-coding genes | 3,885 | 3,745 | 3,765 | 3,758 | 3,692 |
| Average gene length (bp) | 1,360 | 1,892 | 1,830 | 1,844 | 1,778 |
| Percent coding | 60.4% | 77.8% | 75.2% | 77.2% | 77.3% |
| Accession no. | AAEL00000000 | JIBM00000000 | JIBN00000000 | JIBK00000000 | JIBL00000000 |
| SNPs relative to TU502a synonymous : non-syn | 1303 : 2,567 | 718 : 1336 | N/A | N/A | |
| SNPs relative to TU502_2012 synonymous : non-syn | N/A | 143 : 339 | N/A | N/A | |
| Isolate: RNA | TU502_2012 | UKH1 | UKMEL1 | TAMU-10GZ1 | |
| No. HiSeq read pairs | 16,568,115 | 92,878,236 | N/A | 55,829,305 | |
| No. expressed genesb | 1,868 | 2,454 | N/A | 2,235 | |
| Accession no. | SRX481527 | SRX481475 | N/A | SRX481530 | |
| Cryptosporidium hominis | Cryptosporidium | Cryptosporidium | |||
|---|---|---|---|---|---|
| meleagridis | baileyi | ||||
| Isolate: DNA | TU502a | TU502_2012 | UKH1 | UKMEL1 | TAMU-09Q1 |
| gDNA Illumina library fragment size (bp) | N/A | 460 | 461 | 517 | 654 |
| No. MiSeq reads | N/A | 6,871,858 | 7,596,410 | 22,862,044 | 6,240,960 |
| No. base pairs | N/A | 1,724,836,358 | 1,906,698,910 | 6,881,475,244 | 1,566,480,960 |
| Assembly size (bp) | 8,743,570 | 9,107,739 | 9,156,091 | 8,973,200 | 8,493,640 |
| No. of contigs | 1422 | 119 | 156 | 57 | 145 |
| Contig N50 | 14,504 | 238,509 | 179,408 | 322,908 | 203,018 |
| Largest contig (bp) | 90,444 | 1,270,815 | 542,781 | 732,862 | 702,637 |
| G + C content (%) | 30.9 | 30.1 | 30.1 | 31.0 | 24.3 |
| No. protein-coding genes | 3,885 | 3,745 | 3,765 | 3,758 | 3,692 |
| Average gene length (bp) | 1,360 | 1,892 | 1,830 | 1,844 | 1,778 |
| Percent coding | 60.4% | 77.8% | 75.2% | 77.2% | 77.3% |
| Accession no. | AAEL00000000 | JIBM00000000 | JIBN00000000 | JIBK00000000 | JIBL00000000 |
| SNPs relative to TU502a synonymous : non-syn | 1303 : 2,567 | 718 : 1336 | N/A | N/A | |
| SNPs relative to TU502_2012 synonymous : non-syn | N/A | 143 : 339 | N/A | N/A | |
| Isolate: RNA | TU502_2012 | UKH1 | UKMEL1 | TAMU-10GZ1 | |
| No. HiSeq read pairs | 16,568,115 | 92,878,236 | N/A | 55,829,305 | |
| No. expressed genesb | 1,868 | 2,454 | N/A | 2,235 | |
| Accession no. | SRX481527 | SRX481475 | N/A | SRX481530 | |
2004 assembly (Xu etÃÂ al.2004).
Minimum 5X CDS coverage.
Summary statistics of whole-genome sequence and transcriptome data, assemblies and annotation.
| Cryptosporidium hominis | Cryptosporidium | Cryptosporidium | |||
|---|---|---|---|---|---|
| meleagridis | baileyi | ||||
| Isolate: DNA | TU502a | TU502_2012 | UKH1 | UKMEL1 | TAMU-09Q1 |
| gDNA Illumina library fragment size (bp) | N/A | 460 | 461 | 517 | 654 |
| No. MiSeq reads | N/A | 6,871,858 | 7,596,410 | 22,862,044 | 6,240,960 |
| No. base pairs | N/A | 1,724,836,358 | 1,906,698,910 | 6,881,475,244 | 1,566,480,960 |
| Assembly size (bp) | 8,743,570 | 9,107,739 | 9,156,091 | 8,973,200 | 8,493,640 |
| No. of contigs | 1422 | 119 | 156 | 57 | 145 |
| Contig N50 | 14,504 | 238,509 | 179,408 | 322,908 | 203,018 |
| Largest contig (bp) | 90,444 | 1,270,815 | 542,781 | 732,862 | 702,637 |
| G + C content (%) | 30.9 | 30.1 | 30.1 | 31.0 | 24.3 |
| No. protein-coding genes | 3,885 | 3,745 | 3,765 | 3,758 | 3,692 |
| Average gene length (bp) | 1,360 | 1,892 | 1,830 | 1,844 | 1,778 |
| Percent coding | 60.4% | 77.8% | 75.2% | 77.2% | 77.3% |
| Accession no. | AAEL00000000 | JIBM00000000 | JIBN00000000 | JIBK00000000 | JIBL00000000 |
| SNPs relative to TU502a synonymous : non-syn | 1303 : 2,567 | 718 : 1336 | N/A | N/A | |
| SNPs relative to TU502_2012 synonymous : non-syn | N/A | 143 : 339 | N/A | N/A | |
| Isolate: RNA | TU502_2012 | UKH1 | UKMEL1 | TAMU-10GZ1 | |
| No. HiSeq read pairs | 16,568,115 | 92,878,236 | N/A | 55,829,305 | |
| No. expressed genesb | 1,868 | 2,454 | N/A | 2,235 | |
| Accession no. | SRX481527 | SRX481475 | N/A | SRX481530 | |
| Cryptosporidium hominis | Cryptosporidium | Cryptosporidium | |||
|---|---|---|---|---|---|
| meleagridis | baileyi | ||||
| Isolate: DNA | TU502a | TU502_2012 | UKH1 | UKMEL1 | TAMU-09Q1 |
| gDNA Illumina library fragment size (bp) | N/A | 460 | 461 | 517 | 654 |
| No. MiSeq reads | N/A | 6,871,858 | 7,596,410 | 22,862,044 | 6,240,960 |
| No. base pairs | N/A | 1,724,836,358 | 1,906,698,910 | 6,881,475,244 | 1,566,480,960 |
| Assembly size (bp) | 8,743,570 | 9,107,739 | 9,156,091 | 8,973,200 | 8,493,640 |
| No. of contigs | 1422 | 119 | 156 | 57 | 145 |
| Contig N50 | 14,504 | 238,509 | 179,408 | 322,908 | 203,018 |
| Largest contig (bp) | 90,444 | 1,270,815 | 542,781 | 732,862 | 702,637 |
| G + C content (%) | 30.9 | 30.1 | 30.1 | 31.0 | 24.3 |
| No. protein-coding genes | 3,885 | 3,745 | 3,765 | 3,758 | 3,692 |
| Average gene length (bp) | 1,360 | 1,892 | 1,830 | 1,844 | 1,778 |
| Percent coding | 60.4% | 77.8% | 75.2% | 77.2% | 77.3% |
| Accession no. | AAEL00000000 | JIBM00000000 | JIBN00000000 | JIBK00000000 | JIBL00000000 |
| SNPs relative to TU502a synonymous : non-syn | 1303 : 2,567 | 718 : 1336 | N/A | N/A | |
| SNPs relative to TU502_2012 synonymous : non-syn | N/A | 143 : 339 | N/A | N/A | |
| Isolate: RNA | TU502_2012 | UKH1 | UKMEL1 | TAMU-10GZ1 | |
| No. HiSeq read pairs | 16,568,115 | 92,878,236 | N/A | 55,829,305 | |
| No. expressed genesb | 1,868 | 2,454 | N/A | 2,235 | |
| Accession no. | SRX481527 | SRX481475 | N/A | SRX481530 | |
2004 assembly (Xu etÃÂ al.2004).
Minimum 5X CDS coverage.
All the genomes except C. hominis UKH1 were annotated using a semi-automated approach. We trained Augustus (Stanke etàal.2004) using a set of previously manually curated genes. Consensus predictor EVidence Modeler, EVM (Haas etàal.2008), was used to generate annotations based on predictions from Augustus and GeneMark-ES (Borodovsky and Lomsadze 2011), transcripts assembled from RNAseq reads and matches to a set of highly conserved eukaryotic genesâÂÂthe Core Eukaryotic Genes Mapping Approach genes (Parra, Bradnam and Korf 2007). In addition, 394 genes (â¼10% of all genes) in the C. hominis TU502_2012 genome were manually annotated using Web Apollo (Lee etàal.2013). The manually curated genes are thought to encode antigens (Ifeonu et al., in preparartion). The C. hominis genes TU502_2012 were mapped to the C. hominis UKH1 assembly using GMAP (v2015-12-31), and filtered to include only matches that extend at least over 95% of the sequences and have âÂÂ¥95% alignment identity at the amino acid level. The final assembly attributes are listed in Table 1. This Whole Genome Shotgun project has been deposited in DDBJ/EMBL/GenBank under the accession numbers listed in Table 1 and the sequences are accessible at CryptoDB (http://CryptoDB.org). These are the first versions of genome sequence assemblies and annotations for each isolate.
The genome of C. hominis isolate TU502 has been sequenced previously (Xu etÃÂ al.2004). We resequenced the genome of this isolate, after multiple passages, in an attempt to improve the reference genome assembly and gene set for this species. The resulting C. hominis TU502_2012 genome assembly consists of only 119 contigs, a 10-fold reduction relative to the 2004 assembly. The genome assembly is now more complete, and roughly the same size as that of C. parvum, which is also 9.1 Mbp in length (Abrahamsen etÃÂ al.2004). The genes in the new annotation are on average 500 bp longer than their counterparts in the original 2004 annotation, resulting in an increase of 17% in the fraction of the genome that encodes for proteins. In order to determine if this gene structural annotation is more accurate than the one published in 2004, we compared the length of all C. parvum IOWA II proteins with their orthologs in either C. hominis TU502 or C. hominis TU502_2012. The distribution of length differences based on the comparison to the 2012 reannotation indeed has lower variance, with an additional 500 genes similar in length between the two species (Fig. 1). Also, there are 538 C. parvum genes without orthologs in the C. hominis TU502 2004 annotation compared to only 288 such cases in the 2012 annotation. Interestingly, while the original C. hominis annotation had a preponderance of genes shorter than their C. parvum orthologs, the current gene set is skewed in the opposite direction (Fig. 1). Whether this difference is real, or a result of remaining gene structure errors in one or both species, remains to be determined. The C. hominis TU502_2012 annotation contains 206 predicted protein-coding genes with no orthologs in C. parvum IOWA II. Of the 3745 predicted protein-coding genes in C. hominis TU502_2012, only 63% are also found in all other annotated Cryptosporidium genomes available to date: C. parvum IOWA II, C. meleagridis UKMEL1, C. baileyi TAMU-09Q1 and C. muris RN66 (Fig. 1). Finally, 110 predicted protein-coding genes are present in the three newly sequenced genomes, but homologs are absent in the current C. parvum predicted proteome. These significant differences in gene content among species are, in all likelihood, due mostly to the limitations of the semi-automated annotation approach used, rather than to true instances of gene gain/loss. An intense, manual curation effort of the genome annotation of each species is ongoing, and will be essential to validate these results.
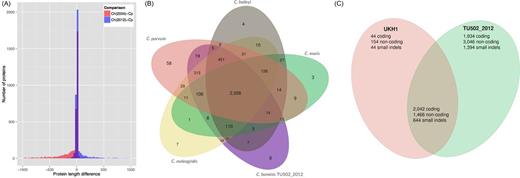
Inter- and intraspecies genome-wide comparisons of genome composition. (A) Comparison of protein length between C parvum and the 2004 and 2012 versions of the C. hominis TU502. (B) Distribution of orthologous gene clusters in five Cryptosporidium species. (C) Distribution of SNPs and short indels among three C. hominis isolates, TU502, TU502_2012 and UKH1. DNA sequence reads from the C. hominis TU502_2012 and UKH1 were mapped against the reference genome assembly of C. hominis TU502, as well as against each other, using BWA (Li and Durbin 2009). SNPs and small indels were identified using GATK (McKenna etàal.2010). Identified variants were further filtered for reliability, according to the following parameter values: (DP < 12) âÂÂ¥ (QUAL < 50) âÂÂ¥ (SB > âÂÂ0.10) âÂÂ¥ (MQ0 > = 2 && (MQ0/(1.0 * DP)) > 0.1). SNPs were categorized as coding and non-coding, given the assembly and the annotation, using VCFtools.
Genetic differences among C. hominis isolates were identified by read mapping, followed by calling and filtering of single nucleotide polymorphisms (SNPs) and small insertions/deletions (indels). A total of 10ÃÂ 526 sequence variants were identified in C. hominis TU502_2012 relative to the reference C. hominis TU502 assembly; in contrast, only 4394 sequence variants were found between C. hominis UKH1 and the reference C. hominis. Interestingly, the vast majority of the differences relative to the reference TU502 genome are shared between the two new isolates (Fig. 1). A plausible explanation, which remains to be verified, is that these SNPs common to both new isolates are in fact sequencing errors in the original C. hominis TU502 assembly, which was based on low-coverage Sanger sequencing. This, however, does not explain the fact C. hominis TU502_2012 has more differences relative to TU502 than does UKH1. It is possible that during the approximate 20 passages in gnotobiotic pigs which C. hominis TU502_2012 isolate has experienced between 2004 and 2012, the make-up of the parasite population has shifted. In the absence of methods for cloning and expanding single Cryptosporidium sporozoites, the isolates sequenced to date are likely to be heterogeneous populations (Grinberg and Widmer 2016). In fact, high-throughput sequencing of a polymorphic locus demonstrated the presence of multiple alleles in laboratory and natural Cryptosporidium isolates (Widmer etÃÂ al.2015).
We generated RNAseq data for two of the species, C. hominis and C. baileyi. These data are strand specific, a tremendous advantage when attempting to generate accurate gene-specific expression values in highly gene-dense genomes, where neighboring transcriptional units often overlap (Tretina, Pelle and Silva 2016). The quantity of RNAseq data generated for C. hominis UKH1 was six times than that for the TU502_2012 isolate (Table 1). Despite this difference, the relative expression values for each gene are remarkably similar for the two isolates (r2 â¼ 0.96; Fig. 2), which supports the strength of the relative expression results. The RNAseq data generated from oocysts indicate that â¼50% and â¼60% of protein-coding genes are expressed in C. hominis TU502_2012 and C. baileyi, respectively, during this stage of the life cycle (Table 1). Gene expression is also positively correlated between species (r2 â¼ 0.51; Fig. 2), with lactate/malate dehydrogenase (LDH), a GDP-fucose transporter, agrin and the ubiquitous heat shock protein 90 (HSP90) being among the most highly expressed genes in both species. LDH and HSP90 have been shown to be among the top nine most highly expressed genes in C. parvum oocysts (Zhang etàal.2012). Genes preferentially expressed in one or the other species may provide a good starting point to investigate biological differences between taxa. Among the genes that differ most in expression level between the two species are pyridine nucleotide-disulphide oxidoreductase, which has a higher level of expression in C. hominis, and AhpC/TSA family protein, WD repeat-containing protein 82 and DNA mismatch repair protein msh-2, all of which have higher expression levels in C. baileyi.
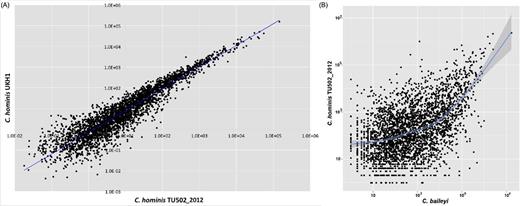
Gene expression in Cryptosporidium oocysts is correlated within and between species. (A) Correlation in oocyst gene expression is highly correlated between two isolates of C. hominis (r2 â¼ 96%). (B) Correlation in oocyst gene expression is correlated between C. hominis and C. baylei (r2 â¼ 51%), particularly among the most highly expressed genes.
The work on Cryptosporidium genomes and their respective annotations with particular emphasis on the manual curation of the structure and function of all protein-coding genes is continuing. Together with the identification of genes unique to each species and genes with species-specific expression profiles, this work will facilitate the identification of genes responsible for host specificity and other phenotypes relevant to the understanding of cryptosporidiosis.
The authors would like to thank Saul Tzipori for facilitating the use of gnotobiotic piglets and to the staff of the animal facility for managing the animals.
FUNDING
Thisworkwas funded by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIAID, NIH), under contract number HHSN272200900009C. Genome sequencing was carried out by the Genome Resource Center at the Institute for Genome Sciences, University of Maryland School of Medicine.
Conflict of interest.None declared.
REFERENCES

{kind=link}
{kind=link}