





Abstract
Anaplasma phagocytophilum causes granulocytic anaplasmosis, an acute disease in humans that is also often subclinical. However, 36% are hospitalized, 7% need intensive care, and the case fatality rate is 0.6%. The biological basis for severe disease is not understood. Despite A. phagocytophilum's mechanisms to subvert neutrophil antimicrobial responses, whether these mechanisms lead to disease is unclear. In animals, inflammatory lesions track with IFN? and IL-10 expression and infection of Ifng−/− mice leads to increased pathogen load but inhibition of inflammation. Suppression of STAT signaling in horses impacts IL-10 and IFN-? expression, and also suppresses disease severity. Similar inhibition of inflammation with infection of NKT-deficient mice suggests that innate immune responses are key for disease. With severe disease, tissues can demonstrate hemophagocytosis, and measures of macrophage activation/hemophagocytic syndromes (MAS/HPS) support the concept of human granulocytic anaplasmosis as an immunopathologic disease. MAS/HPS are related to defective cytotoxic lymphocytes that ordinarily diminish inflammation. Pilot studies in mice show cytotoxic lymphocyte activation with A. phagocytophilum infection, yet suppression of cytotoxic responses from both NKT and CD8 cells, consistent with the development of MAS/HPS. Whether severity relates to microbial factors or genetically determined diversity in human immune and inflammatory response needs more investigation.
Introduction
Human granulocytic anaplasmosis (HGA) is a tick-transmitted zoonosis caused by the intraneutrophilic rickettsia, Anaplasma phagocytophilum (Dumler et al., 2007a, b; Bakken & Dumler, 2008). The infection occurs broadly across mammalian species, but disease manifestations are in part species-dependent, with humans, horses, and dogs recognized to have mild to severe clinical signs and/or symptoms with infection. Small mammals, including laboratory mice, can sustain infection without clinical signs, yet develop inflammatory histopathologic lesions nearly identical to those observed in humans, horses, and dogs, underscoring their utility as models to study disease processes (Bunnell et al., 1999; Lepidi et al., 2000; Choi et al., 2007a, b).
In humans, HGA is now the third most common vectorborne infection in the US and Europe, and is increasingly recognized as an important vectorborne infection in Asia, especially in China (Zhang et al., 2008a, b). Among cross-sectional serosurveys conducted in North America, Europe and Asia, 3.7% have A. phagocytophilum antibodies, and that proportion increases with activities predisposing to or predicting tick exposures and bites (Dumler et al., 2005, 2007a, b). However, in areas with high seroprevalences, such as in the northeast and Upper Midwest USA, active surveillance identifies far fewer clinically evident infections (Bakken et al., 1996; IJdo et al., 2000). Features of human disease include fever, myalgia, headache, malaise, thrombocytopenia, leukopenia, anemia, and mild to moderate hepatic injury sufficient to result in increased aspartate and alanine aminotransferase activities in serum (Dumler et al., 2007a, b; Bakken & Dumler, 2008). These undifferentiated clinical features result in a very broad differential diagnosis and make final definitive diagnosis of HGA difficult. This is confounded by the diversity of disease presentations in humans; while many patients have mild clinical signs and symptoms, infection results in hospitalization for 36%, ICU admission in 7%, and death in 0.6% of cases (Bakken et al., 1994, 1998; Bakken & Dumler, 2000; Chapman et al., 2006). Severe complications include septic shock-like syndromes, acute respiratory distress syndrome, immune compromise, and opportunistic infections, among others (Dumler et al., 2007a, b).
Anaplasma phagocytophilum subversion and disease pathogenesis
The underlying pathogenetic events that lead to disease manifestations are not well understood for A. phagocytophilum. Early investigations linked disease severity with pathogen load, and disease manifestations are often easily reversed with specific antimicrobial treatment (Bakken et al., 1994; Bakken & Dumler, 2006, 2008; Wormser et al., 2006; Dumler et al., 2007a, b). However, these observations were contradicted in subsequent studies where the degree of disease and pathologic injury did not correlate well with bacterial burden, and pathologic findings suggested the potential for a cytokine- or immune-mediated process (Walker & Dumler, 1995, 1996; Jahangir et al., 1998; Bunnell et al., 1999; Lepidi et al., 2000). For example, the hallmark of infection that plausibly reflects direct pathogen-mediated cytolytic injury is leukopenia, especially neutropenia, as A. phagocytophilum infects almost exclusively neutrophils in vivo. However, among infected patients, the mean infected leukocyte count is 0.3 × 109 cells L−1, whereas the mean total leukocyte count for patients with HGA is 3.7 × 109 cells L−1. As the mean leukocyte count with infection is approximately 50% of that for uninfected patients, 4.1 × 109 cells L−1, or 15-fold more leukocytes are lost than can be accounted for by direct A. phagocytophilum lysis alone (Bakken et al., 2001; Dumler et al., 2007a, b). Thus, another explanation for the hematologic derangements is needed.
Considerable investigation regarding how A. phagocytophilum subverts neutrophil function to survive within this key host defense cell has been conducted (Carlyon & Fikrig, 2006; Rikihisa, 2011). However, enhanced intracellular survival or increased pathogen load does not necessarily belie clinical signs or disease. Anaplasma phagocytophilum uses several strategies (Table 1) that conceivably could also contribute to disease pathogenesis: (1) its ability to alter neutrophil mobilization of phagocyte oxidase by disorganized assembly, superoxide dismutase activity, and reduced transcription of key phagocyte oxidase genes, such as CYBB and RAC1, appear to be involved (Choi & Dumler, 2003; Carlyon et al., 2004; IJdo & Mueller, 2004); (2) its ability to induce upregulated chemokine expression and neutrophil vesicle discharge leads to the recruitment of new neutrophils hosts to support pathogen population expansion (Klein et al., 2000; Akkoyunlu et al., 2001; Scorpio et al., 2004), whereas inhibition of this process by blockade of the key chemokine IL-8 receptor (CXCR1) or pharmacologic alteration of IL-10:IFN-γ balance by dexamethasone leads to reduced pathogen numbers in tissues, but does not appear to alter or reduce host pathologic responses (Scorpio et al., 2004; Davies et al., 2011); (3) its ability to introduce microbial effectors, such as AnkA, into the neutrophil via a type IV secretion system and to alter local signaling (ROCK1 and Syk) that facilitates A. phagocytophilum endocytic entry and, over the course of infection, leads at minimum to reduced CYBB transcription and reduced microbial killing in the infected cell (Lin et al., 2007; Thomas & Fikrig, 2007; Garcia-Garcia et al., 2009b); and (4) its ability to upregulate cellular histone deacetylase transcription and activity that likely promotes transcriptional program alterations associated with pathogen survival and population expansion (Garcia-Garcia et al., 2009a). While each of these has been shown to play a role in pathogen survival in vitro, and for some, in vivo, their role in disease pathogenesis is poorly understood. For example, although blockade of CXCR1 leads to reduced pathogen load, the degree of histopathologic injury in the murine model, which correlates strongly with disease manifestations in humans and horses, is not altered, and infection in a phagocyte oxidase deficient condition does not lead to increased pathogen load but actually ameliorates inflammation (Scorpio et al., 2004, 2006).
Anaplasma phagocytophilum strategies for intracellular survival and propagation and their effect on histopathology and disease
| A. phagocytophilum-related pathogetic mechanism | Target or effector molecule | Effect on A. phagocytophilum load | Effect of gene deletion/pharmacologic/antibody inhibition on histopathologic lesions | References |
| Phagocyte oxidase | Disorganized assembly or transcriptional repression of host CYBB/RAC2 | 0 (in vivo); ↑↑↑ (in vitro) | ↓↓ (phox−/− B6 mice in vivo) | Banerjee et al. (2000a, b), Carlyon et al. (2002), von Loewenich et al. (2004), Scorpio et al. (2006) |
| Chemokine/cytokine expression | IL8, IL-10:IFNγ | ↑↑ | 0 (CXCR1 antibody-treated vs. control antibody-treated in vivo); ↓↓ (in dexamethasone-treated horses) | Akkoyunlu & Fikrig (2000), Akkoyunlu et al. (2001), Scorpio et al. (2004), Davies et al. (2011) |
| A. phagocytophilum granulocyte entry | A. phagocytophilum AnkA Host ROCK1 and Syk | ↑↑ to ↑↑↑ | ↓↓ to ↓↓↓ (vs. ROCK1 or Syk-silencing or vs. AnkA control antibody in vitro) | Lin et al. (2007), Thomas & Fikrig (2007) |
| Histone deacetylase (HDAC) | HDAC1 and 2 | ↑↑ (vs. HDAC-silenced cells or NaB or trichostatin inhibition in vitro) | Not done | Garcia-Garcia et al. (2009a, b) |
| A. phagocytophilum-related pathogetic mechanism | Target or effector molecule | Effect on A. phagocytophilum load | Effect of gene deletion/pharmacologic/antibody inhibition on histopathologic lesions | References |
| Phagocyte oxidase | Disorganized assembly or transcriptional repression of host CYBB/RAC2 | 0 (in vivo); ↑↑↑ (in vitro) | ↓↓ (phox−/− B6 mice in vivo) | Banerjee et al. (2000a, b), Carlyon et al. (2002), von Loewenich et al. (2004), Scorpio et al. (2006) |
| Chemokine/cytokine expression | IL8, IL-10:IFNγ | ↑↑ | 0 (CXCR1 antibody-treated vs. control antibody-treated in vivo); ↓↓ (in dexamethasone-treated horses) | Akkoyunlu & Fikrig (2000), Akkoyunlu et al. (2001), Scorpio et al. (2004), Davies et al. (2011) |
| A. phagocytophilum granulocyte entry | A. phagocytophilum AnkA Host ROCK1 and Syk | ↑↑ to ↑↑↑ | ↓↓ to ↓↓↓ (vs. ROCK1 or Syk-silencing or vs. AnkA control antibody in vitro) | Lin et al. (2007), Thomas & Fikrig (2007) |
| Histone deacetylase (HDAC) | HDAC1 and 2 | ↑↑ (vs. HDAC-silenced cells or NaB or trichostatin inhibition in vitro) | Not done | Garcia-Garcia et al. (2009a, b) |
↑ — increased bacterial load or survival.
↓ — decreased or ‘0’ no change in histopathologic inflammation compared with unmodified control.
Anaplasma phagocytophilum strategies for intracellular survival and propagation and their effect on histopathology and disease
| A. phagocytophilum-related pathogetic mechanism | Target or effector molecule | Effect on A. phagocytophilum load | Effect of gene deletion/pharmacologic/antibody inhibition on histopathologic lesions | References |
| Phagocyte oxidase | Disorganized assembly or transcriptional repression of host CYBB/RAC2 | 0 (in vivo); ↑↑↑ (in vitro) | ↓↓ (phox−/− B6 mice in vivo) | Banerjee et al. (2000a, b), Carlyon et al. (2002), von Loewenich et al. (2004), Scorpio et al. (2006) |
| Chemokine/cytokine expression | IL8, IL-10:IFNγ | ↑↑ | 0 (CXCR1 antibody-treated vs. control antibody-treated in vivo); ↓↓ (in dexamethasone-treated horses) | Akkoyunlu & Fikrig (2000), Akkoyunlu et al. (2001), Scorpio et al. (2004), Davies et al. (2011) |
| A. phagocytophilum granulocyte entry | A. phagocytophilum AnkA Host ROCK1 and Syk | ↑↑ to ↑↑↑ | ↓↓ to ↓↓↓ (vs. ROCK1 or Syk-silencing or vs. AnkA control antibody in vitro) | Lin et al. (2007), Thomas & Fikrig (2007) |
| Histone deacetylase (HDAC) | HDAC1 and 2 | ↑↑ (vs. HDAC-silenced cells or NaB or trichostatin inhibition in vitro) | Not done | Garcia-Garcia et al. (2009a, b) |
| A. phagocytophilum-related pathogetic mechanism | Target or effector molecule | Effect on A. phagocytophilum load | Effect of gene deletion/pharmacologic/antibody inhibition on histopathologic lesions | References |
| Phagocyte oxidase | Disorganized assembly or transcriptional repression of host CYBB/RAC2 | 0 (in vivo); ↑↑↑ (in vitro) | ↓↓ (phox−/− B6 mice in vivo) | Banerjee et al. (2000a, b), Carlyon et al. (2002), von Loewenich et al. (2004), Scorpio et al. (2006) |
| Chemokine/cytokine expression | IL8, IL-10:IFNγ | ↑↑ | 0 (CXCR1 antibody-treated vs. control antibody-treated in vivo); ↓↓ (in dexamethasone-treated horses) | Akkoyunlu & Fikrig (2000), Akkoyunlu et al. (2001), Scorpio et al. (2004), Davies et al. (2011) |
| A. phagocytophilum granulocyte entry | A. phagocytophilum AnkA Host ROCK1 and Syk | ↑↑ to ↑↑↑ | ↓↓ to ↓↓↓ (vs. ROCK1 or Syk-silencing or vs. AnkA control antibody in vitro) | Lin et al. (2007), Thomas & Fikrig (2007) |
| Histone deacetylase (HDAC) | HDAC1 and 2 | ↑↑ (vs. HDAC-silenced cells or NaB or trichostatin inhibition in vitro) | Not done | Garcia-Garcia et al. (2009a, b) |
↑ — increased bacterial load or survival.
↓ — decreased or ‘0’ no change in histopathologic inflammation compared with unmodified control.
Evidence for immunopathogenesis of disease with A. phagocytophilum
The immunologic responses to A. phagocytophilum include both humoral and cellular; however, immune control of A. phagocytophilum is still under investigation. Anaplasma phagocytophilum is cleared with kinetics that mimic rising antibody titers (Bakken et al., 1996, 2002); yet there are clear examples of patients with high antibody titers, substantial bacteremia, and disease (Bakken et al., 1994, 1996). While SCID mice cannot control infection, neither do they develop clinical signs, suggesting a dichotomous relationship between immune processes that protect and those that may cause injury (Hodzic et al., 1998; Bunnell et al., 1999). In contrast, A. phagocytophilum tissue and blood loads do not change with genetic deletion of iNOS (Nos2−/−), TLR2 or MyD88, and only variably so for phagocyte oxidase (Cybb−/−) (Banerjee et al., 2000a, b; von Loewenich et al., 2004). Yet, activation of these same innate immune pathways by infection leads to hepatic inflammatory injury, in part dependent upon mouse strain genetic background (Scorpio et al., 2006).
The cellular immune hallmark of A. phagocytophilum in humans, horses, and in mice is the early appearance of IFN-γ and IL-10 in serum or plasma (Dumler et al., 2000; Martin et al., 2000, 2001; Scorpio et al., 2008). While the proinflammatory cytokines TNF-α and IL-1β can be induced from peripheral blood leukocytes after infection ex vivo, these cytokines are generally not detected at high quantities in serum or plasma with active infection (Dumler et al., 2000, 2007a, b; Kim & Rikihisa, 2000). In the murine model of A. phagocytophilum infection, both IFN-γ and IL-10 are present in plasma during active infection and appear as early as several hours postinfection, peaking between days 2 and 7, typically considered an interval prior to adaptive immune response (Akkoyunlu & Fikrig, 2000; Martin et al., 2001; Choi et al., 2007a, b; Pedra et al., 2008). As anticipated, IFN-γ peaks shortly after maximum A. phagocytophilum bacteremia in mice; however, histopathologic injury, as reflected in hepatic inflammatory lesions, tracks more closely with IFN-γ levels than with bacteremia (Martin et al., 2001). Among IFN-γ knockout mice, the level of bacteremia is 7–8 times higher than in wild-type mice, yet IFN-γ knockout mice lack hepatic histopathology that is present in wild-type mice. Interestingly, IL-10 knockout mice do not have altered bacteremia compared with wild-type mice, yet develop significantly worse hepatic inflammatory lesions, providing key evidence for an immunopathologic role in the balance between Th1 and Th2 responses, or with relative roles of both IFN-γ and IL-10.
Given the clear importance of IFN-γ and IL-10 in histopathologic inflammatory lesions in the murine model of HGA, and their early appearance postinfection, potentially important cellular sources include CD8 T lymphocytes and the innate immune effector cells, NK and NKT cells (Choi et al., 2007a, b; Pedra et al., 2008). Splenic CD8 T lymphocytes from infected mice become more activated than from mock-infected mice by day 4 postinfection, when IFN-γ is peaking in plasma. However, the NK1.1-labeled splenic lymphocyte population, which includes both NK and NKT cells, is even more significantly activated with infection vs. mock infection, peaking in near-precise concordance with plasma IFN-γ levels. Infection in Jα18−/− (Choi et al., 2007a, b) and CD1d−/− (unpublished data) mice that lack NKT cells leads to complete loss of hepatic inflammatory lesions on days 4–7 postinfection, and antibody depletion of NK cells also suppresses hepatic inflammation at day 7 compared with infection in wild-type or control antibody-treated mice. These results clearly indicate that innate immune induction of inflammatory lesions, which correspond to disease manifestations, is a key component of disease pathogenesis with HGA.
A role for cytotoxic lymphocytes in disease pathogenesis with A. phagocytophilum?
How do cytotoxic cells, like NK, NKT, and CD8 cells, contribute to inflammatory disease manifestations with activation? The underpinning for IFN-γ-driven effector response is twofold: (1) induction of proinflammatory cytokines and chemokines that activate local effector cells for phagocytosis and enhanced cellular killing and for recruitment of more effector cells; and (2) production of effector molecules for microbial killing, including the oxygen-dependent production of superoxide radicals and hypochlorite and of nitric oxide and peroxynitrates (Ismail et al., 2002; Mosser, 2003; Mantovani et al., 2004; Lang, 2005; Browning et al., 2006). When sequestered, these effector molecules and pathways are potent in their antimicrobial capacity; however, when excessive or exposed to the external host milieu, they can lead to ‘collateral’ host inflammatory injury (Grom, 2004; Larroche & Mouthon, 2004; Janka & zur Stadt, 2005; Menasche et al., 2005; Ravelli et al., 2005).
The balance between microbial killing and host injury is a major determinant of disease outcome in many infections. One class of effector cells recruited and activated for many infectious agents, particularly intracellular infectious agents like viruses and intracellular bacteria, is the cytotoxic lymphocyte. Cytotoxic lymphocytes have a potent capacity to both amplify the response by further release of IFN-γ and to directly kill cellular targets identified by specific cytotoxic cell–target cell interactions. This occurs as with CD8 T lymphocyte recognition of foreign peptides presented in the context of MHC class I molecules or with NK lymphocyte recognition of altered HLA expression or TLR2 signaling or with NKT cell recognition of specific glycolipids in the context of MHC class I CD1d (Scharton & Scott, 1993; Crowe et al., 2003; Davis & Dustin, 2004; Godfrey & Kronenberg, 2004; Kronenberg, 2005; Mattner et al., 2005; Voskoboinik et al., 2006).
When the typical homeostatic mechanism of inflammatory dampening occurs after the inflammatory stimulus has been controlled and reduced in quantity, inflammatory signaling ceases and the underlying pathologic processes is altered to that of repair and reconstitution of function. However, significant and unremitting inflammatory injury occurs in macrophage activation and hemophagocytic syndromes (Francois et al., 1997; Fisman, 2000; Grom, 2004; Larroche & Mouthon, 2004; Janka & zur Stadt, 2005; Menasche et al., 2005; Behrens et al., 2007; Arceci, 2008), genetic and acquired conditions that share the phenotype of (1) active cytotoxic lymphocyte participation, (2) leukopenia, (3) thrombocytopenia, (4) anemia, and in hemophagocytic syndromes, (5) the presence of hemophagocytic cells. Macrophage activation and hemophagocytic syndromes are believed to result from an underlying genetic or acquired defect in the ability of the cytotoxic lymphocyte, whether a CD8 T lymphocyte, an NK cell, or an NKT lymphocyte, to mobilize or deliver functional secretory granules that contain both perforin (which creates a membrane pore in the target cell) and granzyme (which enters via the perforin pore and causes DNA degradation, inducing apoptosis and death of the target cell) (Fisman, 2000; Grom, 2004; Larroche & Mouthon, 2004; Janka & zur Stadt, 2005; Menasche et al., 2005; Arceci, 2008). Such genetic conditions include PRF1 (perforin) mutations, RAB27A mutations (secretory vesicle trafficking; Griscelli syndrome), LYST mutations (lysosomal trafficking regulator; Chediak–Higashi syndrome), and SH2D1A mutations (development of NKT cells; X-linked lymphoproliferative syndrome). When the target is an infected cell, the pathogen is killed. However, these cytotoxic cells do not directly recognize intact pathogen; rather pathogen molecular patterns are the ligands in these immunologic synapses (Davis & Dustin, 2004; Voskoboinik et al., 2006). Thus, antigen-presenting cells, whether MHCI- or CD1d-restricted, are also targets. This process is believed to be a normal homeostatic mechanism to contain unrestrained inflammatory activation. Thus, when cytotoxic cells are activated, they release the macrophage activating, proinflammatory IFN-γ that drives further inflammatory injury, but killing of antigen-presenting cell targets initiates homeostatic control; with defective vesicular trafficking or inactive perforin mutants, the inability to deliver perforin and granzyme and to kill antigen-presenting cells permits an unrestrained, amplifying proinflammatory response without benefit of cytolytic homeostatic resolution (Fisman, 2000; Grom, 2004; Larroche & Mouthon, 2004; Menasche et al., 2005; Arceci, 2008). The result is a poorly controlled, in cases, relentless downward spiral of inflammatory injury and severe or fatal disease.
In fact, patients with HGA have many features of macrophage activation, as evidenced by high levels of IFN-γ, IL-10, IL-12, and serum ferritin, as well as the key clinical features leukopenia, thrombocytopenia, anemia, and hepatic injury, and periodically including the detection of hemophagocytic cells in bone marrow, spleen, lymph node and liver, where examined (Lepidi et al., 2000; Dumler et al., 2007a, b) (Fig. 1). Moreover, HGA severity is correlated with the level of serum ferritin, serum IL-12, and the ratio of IL-10 to IFN-γ (Dumler et al., 2007a, b). The latter feature is highly similar to observations in the murine model (Martin et al., 2001; Choi & Dumler, 2007). As mice do not develop disease, the horse model, where clinical disease is strikingly similar to that in humans, is studied. Here, infected horses are studied after the IFN-γ:IL-10 axis is pharmacologically manipulated by dexamethasone treatment that alters STAT3 and STAT1 signaling to reduce propagation of effector signaling — functionally polarizing the response to favor anti-inflammatory IL-10 domination. As anticipated, horses treated with dexamethasone had increased IL-10:IFN-γ and IL-10:IL-12 ratios, higher overall bacteremias than untreated animals, but significantly less clinical signs and disease (Davies et al., 2011).
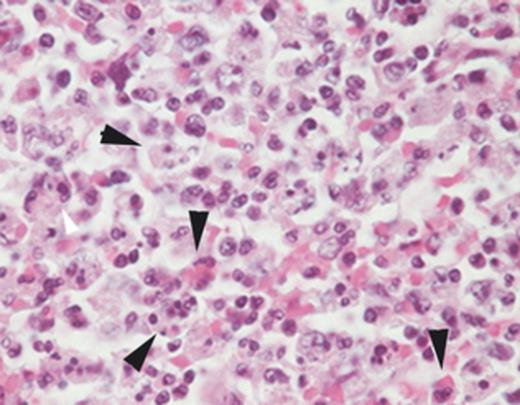
Splenic histopathology from a patient who died after HGA. Note the hemo- and leukophagocytic cells denoted by the arrows. Hematoxylin and eosin stain; original magnification ×400.
It is reasonably understood why genetically linked syndromes that result from specific mutations in key pathways important for secretory vacuole delivery or effector function result in a macrophage activation or hemophagocytic syndrome. However, why this occurs with acquired infections, especially those associated with bacteria, is not well understood (Francois et al., 1997; Fisman, 2000; Larroche & Mouthon, 2004; Janka & zur Stadt, 2005). To determine whether such a principle might apply to A. phagocytophilum, the murine model was studied using ex vivo splenic lymphocytes from infected and mock-infected animals. These cells were examined for their ability to discharge secretory vacuole contents (CD107a) when activated in vitro, a well-recognized correlate of cellular cytotoxicity. Ex vivo splenic NKT cells from infected animals stimulated for degranulation were significantly less cytotoxic than mock-infected controls at days 7 and 10, as were CD8 T lymphocytes at day 4 (Fig. 2; Dumler, unpublished data). While neither NKT nor CD8 T lymphocytes are targets of A. phagocytophilum infection, these preliminary results suggest the basis for an acquired cytotoxicity defect with A. phagocytophilum infection that plausibly provides a tool to examine the periodic occurrence of severe inflammatory disease complications in HGA and to investigate mechanisms of acquired cytotoxic lymphocyte function with nonviral infections.
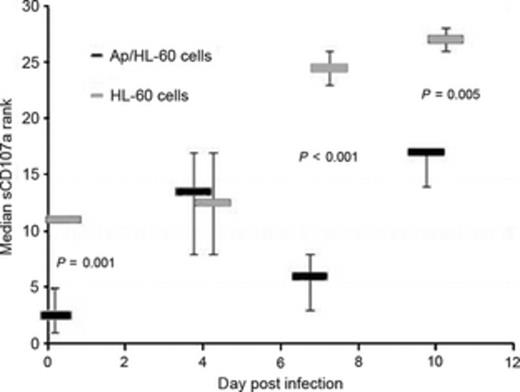
Splenic NKT cells from Anaplasma phagocytophilum-infected mice (Ap/HL-60 cells) have abrogated cytotoxic responses, based on reduced delivery of vesicles (CD107a) to membranes after degranulation stimulated by ionomycin C, as compared with mock-infected (HL-60 cells) mice at days 7 and 10 postinfection. P-values were calculated using the Wilcoxon test for nonparametric analysis, and a value < 0.05 was considered significant.
Conclusions
In summary, HGA is an important emerging tickborne infection that occurs worldwide. While most infections seem only moderate in severity and are easily treated with doxycycline, a minority will have very significant and debilitating disease, despite otherwise adequate antimicrobial treatment. The inflammatory basis for disease is most likely a result of immunopathologic injury after IFN-γ activation of effector cells not adequately tempered by the dampening effects of IL-10 (Martin et al., 2001; Dumler et al., 2007a, b). A subset of patients that does poorly owing to serious inflammatory injury could occur because of a higher degree of polarization in this IL-10:IFN-γ axis, resulting in a macrophage activation-like syndrome, that if accompanied by defects in cytotoxic effector molecule delivery, whether genetically predisposed or acquired via infection, could lead to the most polar of severe infection complications (Dumler et al., 2007a, b; Davies et al., 2011). How this happens, what ligands drive the inflammatory process, the relative role of innate and adaptive immunity, and how they can be controlled are now important questions to address (Choi & Dumler, 2007). The ability to toggle disease progression by targeting anti-inflammatory treatment during infection in horses could provide additional guidance to help for those severely affected (Scorpio et al., 2006; Davies et al., 2011). Moreover, the dissection of the fundamental biological processes that drive this syndrome with HGA at the level of cytotoxic lymphocyte dysfunction offers a new possibility to study the pathogenesis of acquired nonviral hemophagocytic syndromes. Once again, obligate intracellular bacteria have lessons to teach, and as good students, we should pay careful attention.
Acknowledgements
This work was supported in part by grant R01 AI44102 from the National Institutes of Allergy and Infectious Diseases. Thanks to many colleagues including Johan S. Bakken, M.D., Diana G. Scorpio, DVM MPH, Kyoung Seong Choi, PhD DVM, and John E. Madigan, DVM for long help and support.
References
Author notes
Editor: Gilbert Greub

{kind=link}
{kind=link}