





Abstract
Candida albicans STE13ca gene was identified by its homology to the Saccharomyces cerevisiae STE13 gene that encodes for the dipeptidyl aminopeptidase A (DAP A) involved in the maturation of α-factor mating pheromone. Our study revealed that C. albicans ATCC 10231 depicts dipeptidyl aminopeptidase activity. We also analyzed the expression of the STE13ca gene homologue from this pathogenic yeast. This gene of 2793 pb is homozygotic and encodes for a predicted protein of 930 amino acids with a molecular weight of 107,035 Da. The predicted protein displays significant sequence similarity to S. cerevisiae Ste13p. This C. albicans gene is located in chromosome R. STE13ca gene increases its levels of expression in conditions of nutritional stress (proline as nitrogen source) and during formation of the germinal tube, suggesting a basic biological function for the STE13ca in this yeast.
1 Introduction
The yeast Candida albicans is carried in the microflora of most healthy individuals as a commensal [1]. It is also the most common human fungal pathogen. When the defense mechanisms of an individual are compromised, this opportunistic pathogen can increase in number and penetrate tissues at one or more body locations, causing a variety of yeast-related diseases, such as mucosal and systemic infections [2,3]. The factors that contribute to the pathogenesis of C. albicans are not known completely, despite more than a decade of molecular genetic analyses. C. albicans was thought to be asexual until the sequencing of its genome revealed that it contains homologous genes to those in the MAT locus in Saccharomyces cerevisiae designated MTL (matig type-like) locus (http://www-sequence.stanford.edu/group/candida).
Sequencing of C. albicans genome and a comparative genome analysis between C. albicans and S. cerevisiae showed that, in addition to the MTL locus, this yeast contains homologues of many of the genes involved in the mating process in S. cerevisiae[4]. Recent reports have demonstrated mating between MTLa and MTLα C. albicans strains [5–8]. STE13ca gene has been identified by its homology to the S. cerevisiae STE13 gene from the Candida albicans Genome Sequencing Project. The S. cerevisiae STE13 gene encodes the dipeptidyl aminopeptidase A (Ste13p), which is a membrane-bound enzyme, involved in α-factor precursor processing by removing dipeptides from the N-terminus of the pheromone precursor [9]. Mutants lacking Ste13p activity (ste13) were sterile when their mating type was MATα because cells secreted incompletely processed forms of the α-factor pheromone [9,10]. Recently, Bennett et al. [11] described a mating pheromone encoded by the MFα gene in C. albicansα cells. This pheromone was required to mate by α cells, but not by a cells. They also identified that the STE2 gene encodes the receptor for this mating pheromone and showed that this receptor is required for the mating of a cells, but not for α cells. Cells of the a mating-type respond to the α mating pheromone by producing long polarized projections similar to those observed in mating mixtures of C. albicans a and α cells [11]. This paper reports the dipeptidyl aminopeptidase activity and the bio-informatic and molecular analysis of STE13ca gene homologue from this pathogenic yeast.
2 Materials and methods
2.1 Strains and culture media
Strains used in this study were Candida albicans ATCC 10231, Candida dubliniensis CD36 [12], and Candida tropicalis CTR10 [13]. Yeast forms were routinely maintained on YEPD plates (1% yeast extract, 2% peptone, 2% glucose, and 2% agar) at 37 °C. The DAP activity of C. albicans ATCC 10231 membrane fraction was measured growing yeasts at 37 °C in YEPD and Minimal medium broths containing 0.17% yeast nitrogen base without amino acids neither ammonium sulfate, but adding 2% glucose without or with 5% nitrogen source (proline, peptone, and ammonium sulfate) according to manufacturer's instructions. Induction of hyphae formation in yeast cells was performed according to the protocol of Lee et al. [14]. To evaluate cell growth, absorbances (A600) of culture samples were measured in a Perkin–Elmer Lambda 1A spectrophotometer.
2.2 Preparation of crude extracts and differential centrifugation
Crude extracts and membrane fractions were prepared according to Suarez-Rendueles et al. [15,16].
2.3 Enzyme assays and protein determination
The enzymatic activity of ycaDAP was determined using l-alanil-prolyl-4-nitroanilide (Ala-pro-4-NA) as substrate. One unit of X-prolyl-dipeptidyl aminopeptidase was defined as the amount of enzyme that liberates 1 µmol p-nitroaniline from the substrate in 1 min at 37 °C in our assay conditions [15].
Protein was estimated according to method of Lowry using crystalline bovine serum albumin as standard [17].
2.4 Bioinformatic analysis
C. albicans STE13ca gene was identified by similarity to the S. cerevisiae STE13 gene. Sequence data for C. albicans were obtained from the Stanford Genome Technology Center's website at http://www-sequence.stanford.edu/group/candida. Sequencing of C. albicans was accomplished with the support of the NIDR and the Burroughs Wellcome Fund. The Bioinformatic analysis was performed using several softwares. Search of ORF, reverse complementary sequence, restriction map, translation using alternative yeast nuclear genetic code, molecular weight, and codons usage frequency were performed with the DNAMAN version v. 3.0 (Lynnon BioSoft, 1994–1997). Prediction of regulatory regions was searched by means of MatInspector version 2.2 [18] (http://www.gsf.de/biodv/matinspector.html); codon adaptation index (CAI) was calculated with data from NGFN (Nationales Genomforschungsnetz) at http://ngfnblast.gbf.de/cgi-bin/emboss.pl_action=manual&_app=cai. Isoelectric point, prediction of hydrophobicity, and helical membranous regions were searched with Antheprot 2000 version 5.2. Prediction of motif was searched with Prosite [19] at http://www.expasy.org. Prediction of subcellular localization was searched with PSORT at http://www.psort.org/. STE13ca sequence was aligned with DAP's nucleotide sequences from S. cerevisiae, S. pombe, and other organisms available at NCBI (http://www.ncbi.nlm.nih.gov). Multiple alignments of deduced amino acid and nucleotide sequences were performed with CLUSTAL X version 1.8 [20]. A similarity tree was constructed with MEGA (molecular evolutionary genetics analysis) version 2.1 [21], using the neighbor-joining grouping method and P (Poisson) index. Statistical evaluation included 1000 bootstrap re-samplings. Percentage similitude was calculated using MEGA.
2.5 DNA Extraction and PCR to specific amplification of STE13ca gene
Total DNA was extracted according to Lehmann et al. [22]. Southern blot of C. albicans genomic DNA was performed with DNA digested with AluI, ClaI, DdeI, DraI, EcoRI, HincI, and HindII restriction enzymes to detect the number of copies of the STE13ca gene.
PCR was used to prepare hybridization probes for Southern blotting and chromosome assignment. A fragment of 466 bp in the ORF of STE13ca was amplified from C. albicans ATCC 10231. Pair of primer designed was: STE13sense: 5′-ATTTGGCTACTGGCG and STE13antisense: AAATAC-3′and 5′-CTGGTTCTGTCTGATTGTTTC-3′. The PCR was performed with 10 ng total DNA and conditions of amplification included a hot start at 94 °C (5 min); 40 cycles of denaturation (1 min), annealing at 57 °C (1 min) and polymerization at 72 °C (1 min); a final polymerization time was applied at 72 °C (5 min).
2.6 Pulsed-field gel electrophoresis (PFGE) and chromosome assignment
Cell preparation and separation of chromosomes were performed essentially as described by Wickes et al. [23] and run in a Bio-Rad CHEF-DRII apparatus. Each chromosome was eluted and purified as described by Sambrook et al. [24]. The localization of STE13ca gene was determined by specific PCR of STE13ca gene (see above) using each DNA chromosomes as template.
2.7 Southern hybridization
DNA was transferred to a Nylon membrane positively charged (Amersham Pharmacia Biotech, UK) as described by Sambrook et al. [24]. Hybridization was performed at 65 °C with a STE13ca-specific probe labeled with digoxigenin-dUTP (2′-deoxyuridine 5′-triphosphate) using a random-primed digoxigenin (DIG) DNA labeling detection kit (Roche, Mannheim, Germany). Hybridization and immunological detection were performed as recommended by the supplier.
2.8 Denaturing gradient gel electrophoresis (DGGE)
A fragment of 466 bp STE13ca gene amplified from C. albicans ATCC 10231 was used in search of a homozygotic/heterozygotic character for this gene by DGGE (Dcode System for DGGE, Bio-Rad Laboratories, USA) according to the manufacturer. The denaturing gradient (40–65%) was formed parallel to the direction of the electrophoresis.
2.9 RT-PCR
Three expression conditions were chosen: (a) growth on YEPD medium during logarithmic, early stationary and late stationary phases; (b) growth on YNB medium without nitrogen and YNB medium supplemented with different nitrogen sources (ammonium, proline and peptone); (c) during the dimorphism from yeast-like to mycelium on Lee's medium. Total RNA was extracted and purified from C. albicans ATCC 10231 using the protocol Tripure isolation reagent (Roche, CA, USA) according to the manufacturer. To minimize the risk of contaminating DNA, RNAs (10 µg total) were digested with 10 U of DNase I (Invitrogen, CA, USA) in accordance to manufacturer's instructions. RT-PCR was performed with Thermoscript™ RT-PCR System (Invitrogen, CA, USA) from 2 µg of total RNA by using STE13sense/STE13antisense-specific primers for STE13ca gene (expected product size, 466 bp) and 18S rDNA-universal primers (LVT-1: 5′-CCTGCCAGTAGTCATATGCTTGTCT-3′ and LV-2: 5′-CACCTACGGAAACCTTGTTACGACT-3′) for 18S rDNA gene (expected product size, 1694 bp). Expression of STE13ca was normalized against levels of 18S rDNA cDNA.
3 Results
3.1 DAP activity determination of C. albicans ATCC 10231 membrane fraction
C. albicans ATCC 10231 presented ycDAP intracellular activity in the membrane fraction. The highest level of total and specific ycaDAP activity was reached in the YNB medium with proline or peptone as nitrogen source in the yeast-like phase and during early stationary growth (Table 1a). During dimorphism, the levels of ycaDAP activity were on the increase during formation of the germinal tube (1 and 2 h) and pseudomycelium formation (3 h). However, the highest levels of ycaDAP activity were reached during mycelium formation (Table 1b). No activity was detected in the extracellular fraction (supernatant of the culture medium) in any of the assayed conditions (data not shown).
Total and specific activity of DAP in C. albicans ATCC 10231 (membrane fraction)
Cellular fractions were prepared from cells growing in the corresponding medium. Biomass from the culture medium was recovered by centrifugation at 5 °C (2000g), washed twice with 0.1 M Tris buffer, pH 7.0, and disrupted with glass beads to produce a cell homogenate. This fraction was centrifuged at 23,000g, and the resulting supernatant was centrifuged at 100,000g to separate the soluble fraction (cytoplasmic) from the membrane fraction. The 100,000g fraction (membranes) was used to determine DAP enzymatic activity against Ala-pro-pNA as substrate.
Total activity (mU ml−1).
Specific activity (mU mg protein−1).
Total and specific activity of DAP in C. albicans ATCC 10231 (membrane fraction)
Cellular fractions were prepared from cells growing in the corresponding medium. Biomass from the culture medium was recovered by centrifugation at 5 °C (2000g), washed twice with 0.1 M Tris buffer, pH 7.0, and disrupted with glass beads to produce a cell homogenate. This fraction was centrifuged at 23,000g, and the resulting supernatant was centrifuged at 100,000g to separate the soluble fraction (cytoplasmic) from the membrane fraction. The 100,000g fraction (membranes) was used to determine DAP enzymatic activity against Ala-pro-pNA as substrate.
Total activity (mU ml−1).
Specific activity (mU mg protein−1).
3.2 Sequence analysis of C. albicans STE13ca gene
The nucleotide sequences from Contig19-10235 and 20235 of 28191 and 28192 pb, respectively, were analyzed. An ORF of 2793 bp in the reverse complementary sequence that showed homology with S. cerevisiae STE13 gene was found. ORFs from both contigs were homozygotic. The deduced amino acid sequence showed that this gene encodes for a protein of 931 amino acid residues, molecular weight of 107,035 Da, and pI of 5.195. Inspection of the deduced amino acid sequence of C. albicans Ste13 revealed a single hydrophobic domain, and a helical membranous region, beginning 87 residues from the NH2 terminus that could potentially span a lipid bilayer. In addition, eight probably sites for asparagine-linked (N-linked) glycosylation; seven probably sites for N-myristoylation and an active site of serine proteases belonging to subtilases family with one allowed mismatch in the Lys (k) amino acid were recognized (Fig. 1). The possible site of the subcellular localization of the predicted protein of gene STE1ca could be the Golgi apparatus with a 0.9 certainty factor. Codon usage frequency and CAI (0.239) of STE13ca indicated that this gene belongs to the low-expression genes (data not shown) [25]. A region of 2000 pb upstream of the start codon ATG was analyzed to recognize regulatory regions. Transcription factor binding sites for STRE, NIT-2, MAT1-MC, MATa1, MCM1, RAP1 were detected (Table 2). The BLAST program [26] was used to identify other homologous genes in GenBank database at NCBI (http://www.ncbi.nlm.nih.gov). The protein encoded by this ORF exhibits a high similarity with Ste13 from S. cerevisiae. The nucleotide and amino acid sequences of C. albicans and S. cerevisiae genes shared 50.70% and 31.83% of similarity, respectively. The constructed similarity tree revealed that the predicted amino acid sequence of STE13ca is more related to the Ste13 protease from S. cerevisiae than with those from other organisms (Fig. 2). Because of the similarity between C. albicans and S. cerevisiae STE13, this gene was also designated STE13ca.

Nucleotide and deduced amino acid sequences of the C. albicans STE13ca gene. The deduced amino acid sequence is given beneath the nucleotide sequence of the STE13ca coding region. Nucleotide residues are numbered in the 5′-to-3′ direction, with the A of the predicted ATG; initiating methionine given the number 1. The amino acid sequence comprises the putative membrane-spanning domain (marked with ), the potential sites of N-linked glycosylation (), N-myristoylation () and serine proteases active site ().
Possible binding sites to transcription factors of gene STE13ca from C. albicans
2000 pb upstream of the starting codon (ATG) were analyzed.
S: Positive/negative strand.
Basepairs marked in bold show high information content, i.e., the matrix exhibits a high conservation at this position.
The “core sequence” of a matrix is defined as the (usually 4) consecutive highest conserved positions of the matrix.
The analysis was performed with a “core similarity” of 0.9. The maximum core similarity of 1.0 is only reached when the highest conserved bases of a matrix match exactly in the sequence. More important than the core similarity is the matrix similarity, which takes into account all bases over the whole matrix length. A perfect match to the matrix gets a score of 1.00 (each sequence position corresponds to the highest conserved nucleotide at that position in the matriz), a good match to the matriz usually has a similarity of >0.80.
Basepairs in capital letters denote the core sequence used by MatInspector.
Possible binding sites to transcription factors of gene STE13ca from C. albicans
2000 pb upstream of the starting codon (ATG) were analyzed.
S: Positive/negative strand.
Basepairs marked in bold show high information content, i.e., the matrix exhibits a high conservation at this position.
The “core sequence” of a matrix is defined as the (usually 4) consecutive highest conserved positions of the matrix.
The analysis was performed with a “core similarity” of 0.9. The maximum core similarity of 1.0 is only reached when the highest conserved bases of a matrix match exactly in the sequence. More important than the core similarity is the matrix similarity, which takes into account all bases over the whole matrix length. A perfect match to the matrix gets a score of 1.00 (each sequence position corresponds to the highest conserved nucleotide at that position in the matriz), a good match to the matriz usually has a similarity of >0.80.
Basepairs in capital letters denote the core sequence used by MatInspector.
![Similarity tree of deduced amino acid sequence of STE13ca gene of C. albicans, and other DAPs decribed in A. fumigatus, A. oryzae, A. niger, B. taurus, C. elegans, D. melanogaster, F. catus, H. sapiens, M. musculus, M. grisea, N. crassa, R. norvegicus, S. cerevisiae, Schizosaccharomyces pombe, S. scrofa, and X. leavis. DAP2 from C. albicans is another gene identified to encode the putative DAP B. The tree was defined by MEGA version 2.2 software [21]. Analysis was based on multiple alignment (data no shown) using neighbor-joining grouping method and P (Poisson) index. Statistical evaluation included 1000 bootstrap resamplings. The bar indicates number of nucleotide changes per 100 nucleotides.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femspd/45/3/10.1016_j.femsim.2005.05.020/1/m_FIM_459_f2.jpeg?Expires=1750704402&Signature=ksqgxDyH7sOxK9nqdZVxWHldSNnqtPtTn3NtwuH1rXziNVjKNYzLDyuBoss3t2Mjpn6eCYgwdal-RXWi9GQMrNRS2MozK9pg3fwNXN0HOVOnErUtf5rVvkihAGtnmgBQtajbBtigKTTX9UOHFZN22NW1EZdS8gyho-Hw5ThSU0umpY8npFm8VmjbKH7SluLOkqjVWYQQ63IQJVQFdCqUqdiKCDuS1kKyv--jokQqQ8-jZ-y5XJl7bNCJDpYDRIofzGvePstqiXjxg8lSaeq1kdb6nnRGsDQ0cpfQCGRNdmLkUhboa0e0NyyWfvFJERF6UG1I5mEqfmQJ3jTzV2o43Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Similarity tree of deduced amino acid sequence of STE13ca gene of C. albicans, and other DAPs decribed in A. fumigatus, A. oryzae, A. niger, B. taurus, C. elegans, D. melanogaster, F. catus, H. sapiens, M. musculus, M. grisea, N. crassa, R. norvegicus, S. cerevisiae, Schizosaccharomyces pombe, S. scrofa, and X. leavis. DAP2 from C. albicans is another gene identified to encode the putative DAP B. The tree was defined by MEGA version 2.2 software [21]. Analysis was based on multiple alignment (data no shown) using neighbor-joining grouping method and P (Poisson) index. Statistical evaluation included 1000 bootstrap resamplings. The bar indicates number of nucleotide changes per 100 nucleotides.
3.3 Determination of the number of copies of gene STE13ca
Southern analysis revealed that gene STE13ca fromC. albicans ATCC 10231 presents as a single copy, since the number and size of the hybridization bands of the genome digested with enzymes: AluI, DdeI, DraI, Hinc II, EcoRI corresponded to that expected according to the restriction map (Fig. 3(a)). Digestion of the genome with enzyme ClaI presented a larger size hybridization band, and the digestion with enzyme HindIII depicted, besides the expected band, and extra band of larger size (Fig. 3(b)). C. dubliniensis CD36 and C. tropicalis (CTR10) presented Southern hybridization bands with the probe of the gene STE13ca (data not shown).
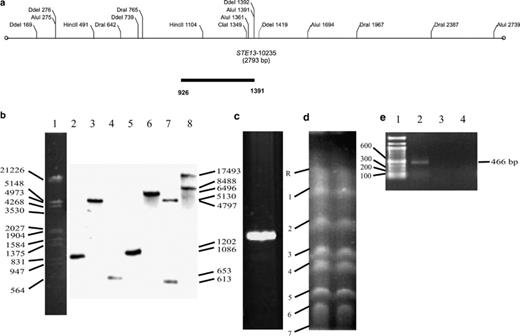
Southern blot of total DNA, DGGE analysis, and electrophoretic karyotype of STE13ca gene from C. albicans ATCC 10231. (a) Restriction map of STE13ca gene. (b) Southern blot of total DNA digested with several restriction enzymes and probed with STE13ca-specific probe: (1) DNA marker, (2) AluI, (3) ClaI, (4) DdeI, (5) DraI, (6) EcoRI, (7) HincII, (8) HindII. (c) DGGE analysis of the fragment of 466 bp STE13 probe amplified by PCR. A single band indicates absence of nucleotide changes and homozygotic character of STE13ca. (d) Electrophoretic karyotype. Electrophoretic conditions were 120, 240, and 360 s of initial and final pulsed time per 24 h each, all at 150 V, 11 °C, and TBE buffer 0.5×. Numbers refer to chromosomes. (e) PCR of the chromosomes: (1) DNA marker, (2) Chromosome R, (3) chromosome 1, and (4) chromosome 2. STE13ca amplified on chromosome R.
3.4 Determination of the homo/heterozygotic character and chromosome location of gene STE13ca
The DNA fragment of the PCR amplified gene STE13ca of 466 bp and subjected to DGGE depicted one band only, confirming the homozygosity of this gene in C. albicans (Fig. 3(c)). The eight chromosome pairs of C. albicans ATCC 10231 were separated by PFGE (Fig. 3(d)). The gene STE13ca was located in chromosome R by STE13ca-specific PCR. No PCR product was obtained when other chromosomes DNA were used as template (Fig. 3(e)).
3.5 Differential expression of gene STE13ca
Three expression conditions were chosen: growth phases, different nitrogen sources and during the dimorphism from yeast-like to mycelium (Fig. 4). RT-PCR results indicated that this gene expresses in both the yeast-like stage and the mycelial stage in the diploid C. albicans ATCC 10231 strain. During the different growth phases, it expresses preferentially at 6 and 12 h, which correspond, respectively, to the logarithmic and early stationary phases of the cell growth cycle, but is not expressed during the late stationary phase (Fig. 4(d)-I). The gene STE13ca did not express in either YNB medium without nitrogen source or in YNB with peptone; maximal expression levels were obtained in the YNB medium with proline. Although a moderate expression was obtained in the YNB medium with ammonium, STE13ca is a gene mainly induced by proline (Fig. 4(d)-II). In stage G1, expression levels of gene STE13ca were nil. During germinal tube formation induction (1 h), levels of expression increased until the germinal tube had been formed (2–3 h); thereafter, levels decreased slightly during formation of the pseudomycelium (5 h), and increased anew to maximal levels during formation of the true mycelium (24 h) (Fig. 4(d)-III).
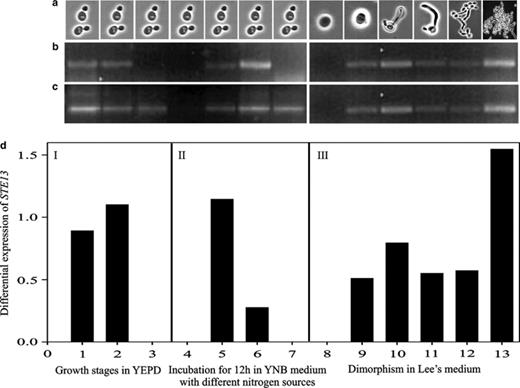
Differential expression of STE13ca gene in C. albicans ATCC 10231 during different growth phases, nitrogen sources and during dimorphism using RT-PCR. (a) Digital imagery of cellular morphology, (b) cDNA STE13ca gene, (c) 18S rDNA cDNA, and (d) STE13ca gene expression. (d)-I Growth phases in YEPD: (1) logarithmic, 6 h; (2) early stationary, 12 h; (3) late stationary, 24 h. (d)-II Incubation for 12 h in YNB medium with different nitrogen sources: (4) YNB, (5) YNB–proline, (6) YNB–NH2, (7) YNB–peptone. (d)-III Dimorphism in Lee's medium: (8) phase G1, (9–10) germinal tube formation, (11–12) pseudomycelium formation, (13) mycelium formation. The intensity of bands STE13ca cDNA was normalized to the intensity of 18S rDNA cDNA.
4 Discussion
In S. cerevisiae, a membrane-bound X-prolyl dipeptidyl aminopeptidase activity has been described [15,27], which is associated to two different enzymes: yscDAP A, located in the membrane of the Golgi apparatus and coded by gene STE13, [28,29]; and yscDAP B, detected on the vacuolar membrane and coded by gene DAP2[30]. The present results show that C. albicans ATCC 10231 has ycaDAP membrane activity in both the yeast-like and the mycelial stages. Similarly to S. cerevisiae, this activity was intracellular in all the assayed conditions, since the activity could not be detected in the supernatants of liquid cultures. During dimorphism, the highest levels of specific ycaDAP activity were encountered during formation of the germinal tube. These results could suggest the participation of this enzyme during dimorphism. During the yeast-like stage, the highest ycaDAP activity levels were found with proline as sole nitrogen source, an amino acid considered as a non-preferential nitrogen source [31].
Although it has been proven that C. albicans possesses a diploid genome (http://www-sequence.stanford.edu/group/candida), the gene STE13ca resulted homozygotic. Our DGGE results with the amplified fragment of this gene coincided with the homozygosity of the sequence. This result supports the hypothesis of Hull and Johnson about heterozygosity of the MTL locus of C. albicans, and the homozygosity of the rest of the genome [5]. The ORF of the gene STE13ca (2793 pb) encodes a predicted 930 amino acids protein homologous to the Ste13p of S. cerevisiae (2796 pb and a 931 amino acids protein) [32,33].
The search for binding sites to the transcription factors of gene STE13ca suggested a possible regulatory action promoted by stress, nitrogen source, and processes related with mating. Thus, a binding site to Stre (stress response element) encoded by STRE, which regulates stress response genes in S. cerevisiae was located. STREs regulate the induction of transcription due to thermal shock, nitrogen inanition, oxidative and osmotic stress, low external pH, sorbate, benzoate, or ethanol induced stress [34,35]. As in S. cerevisiae, this could indicate that expression of STE13ca in C. albicans is induced when the cell is subjected to diverse stress conditions. Also, four binding sites to Nit-2, which activates the nitrogen-regulated genes, were found. NIT-2 is the major positive-acting regulator of nitrogen in Neurospora crassa. This regulator turns on genes when primary sources of nitrogen (ammonium or glutamine) are not available [36]. The present study on STE13ca gene expression revealed higher expression levels (at mRNA level) in the presence of proline as nitrogen source, suggesting that gene STE13ca could be under the transcriptional control of Nit-2. Other binding sites to the transcription factors MAT1-MC and MATa1 were located. These genes act in mating determination in S. pombe and S. cerevisiae, respectively [37–39]. These binding sites suggest that the gene STE13ca could be involved in C. albicans mating. As well a binding site to MCM1, which together with MATα2 (α2/Mcm1p) represses aspecific genes in the a/α diploid of S. cerevisiae, was detected [40]. Upstream sequence of STE13ca gene possesses a binding site to Rap1 (Tuf1), which displays a transcriptional activation and silencing of MAT genes, among other functions. The rap1 strains are also deficient in α-pheromone production. Elimination of the binding site for RAP1 in the HMR locus (silent mating-type) has an opposed effect on this locus, which becomes partially depressed [41]. Rap1 activates and represses MAT genes according to the situation, allowing us to infer that gene STE13ca from C. albicans might be activated by the action of Rap1.
Codons usage has been characterized in S. cerevisae, where highly expressed genes depict a use of synonym codons strongly directed to those codons efficiently translated by the most abundant tRNA species [42]. The CAI value for STE13ca was 0.239, which when was compared with values of already reported genes [25], was placing among the low-expression genes.
Analysis of the hydrophobicity profile and the prediction of transmembrane regions of the predicted Ste13 protein from C. albicans indicate that this protein could be associated to membranes, a prediction that was proven by detecting yscDAP activity in the membrane fraction of C. albicans ATCC 10231. Similarly, in S. cerevisiae, Ste13p contains a transmembrane segment close to the amino acid terminus [33]. Analysis of the possible subcellular site indicated that this predicted protein from C. albicans, just as in S. cerevisiae, might be located in the Golgi apparatus membrane. Prediction of “motif” sequences revealed that similarly to the Ste13p from S. cerevisiae, the Ste13p from C. albicans possesses an active site of serine proteases. This prediction was also confirmed since the ycaDAP membrane activity was inhibited by the presence of PMSF, an inhibitor of serine proteases (data not shown).
As can be observed in the tree showing the similarity relations between the amino acid sequences of proteins with DAP activity of diverse organisms, the ORF deduced from the gene STE13ca showed a high similarity with the amino acid sequence of DAP A, from S. cerevisiae. As mentioned before, in S. cerevisiae, the function of the gene STE13 is to process the precursor of the α sexual factor by removing the dipeptide from the end of the amino terminal of the pheromone precursor. Since, the mating process has been described in C. albicans, the STE13ca gene could perform the same function in this yeast, where a possible homologous of α sexual factor has been described [11].
We also investigated the presence of homologous genes to STE13 in other yeasts of the Candida genus. In this way, gene STE13 was PCR-amplified in C. dubliniensis. The Southern analysis confirmed the presence of a homologous gene to STE13 in C. dubliniensis and C. tropicalis (data not shown). This is not surprising, since C. dubliniensis is a species phylogenetically related to C. albicans[12,43,44].
Southern analysis indicated that there is probably only one copy of the STE13ca gene in each C. albicans chromosome R, discarding the possibility of genes originated by duplication events (paralogous genes) as in the secreted aspartyl proteinases genes family (SAP1–SAP10) [45].
The STE13ca gene of C. albicans is located in chromosome R. This chromosome varies in its electrophoretic motility among C. albicans strains; in general, it runs equally, slower or faster than chromosome 1 [23]. Therefore, chromosome R was PCR identified based on the location of the SAP2 gene [13], located in chromosome R [45,46].
We found that expression of the gene STE13ca is regulated by the growth phase, the nitrogen source, and the dimorphism from yeast to mycelium. The highest mRNA levels of gene STE13ca were encountered during the logarithmic and early stationary growth phases, in the presence of proline as sole nitrogen source, and during formation of the germinal tube. It must be mentioned that proline has been classified as a hard to assimilate nitrogen source, and has been used as an inductor of nutritional stress [31,47]. The increase in gene STE13ca expression could be related with several stress conditions.
In early stationary phase, it was detected high ycaDAP activity in YNB + peptone medium, although no mRNA was produced for the same conditions, perhaps the measured ycaDAP activity corresponds to the protein encoded by DAP2ca gene that is able to attack Ala-pro-4-NA substrate. C. albicans DAP2ca gene was identified by similarity to the S. cerevisiae DAP2 gene and it is expressed in early stationary phase in YNB + peptone medium, it is not expressed in YNB + proline, and DAP2 mRNA does not increase during dimorphism (data not shown).
Aimed at assigning a possible function to the product of gene STE13ca from C. albicans, we decided to analyze the amino acids sequence of the peptide proposed as α sexual factor, encoded by the MFα1 gene. The α-sexual factor of S. cerevisiae contains four equally spaced repeats of the peptidic sequence of the mature α-sexual factor, this protein is processed and the four peptides are released [48]. The first three are flanked in their carboxyl terminus by spacing peptides, each one starting with the processing site by protease Kex2 KR (Lys-Arg)1, followed by the processing sites for protease Ste13p EA (Glu-Ala)2, EA (Glu-Ala)3 or -E-A-D-E-A-sequence or (−Glu-Ala-Asp-Ala-Glu-Ala-sequence). The last copy ends in an amino acid before the terminus of the deduced protein [49]. In C. albicans, MFα1 encodes a precursor protein containing three equally spaced repeats of the α-pheromone sexual factor [50]. Although the number and sequence of amino acids differ between the pheromones (α-sexual factors) of C. albicans and S. cerevisiae, the general arrangement of the amino acid sequences is similar in both peptides. Fig. 5 depicts the possible cutting sites proposed here of the DAP activity of C. albicans for the maturation of this factor. Future studies will allow testing this hypothesis. On the other hand, disruption in KEX2 (another gene involved in processing the S. cerevisiae pheromone Mfα) prevented mating in MTLα but not in MTLa cells [51] and diminished C. albicans virulence [52]. Therefore, the functional complementation of a ste13Δ mutation in S. cerevisiae with STE13ca and/or disruption in STE13ca is very important to demonstrate the function of this gene in C. albicans: its possible role in α-sexual factor maturation, maturation of another proteins, as well as its role in nitrogen metabolism under nutritional stress conditions.
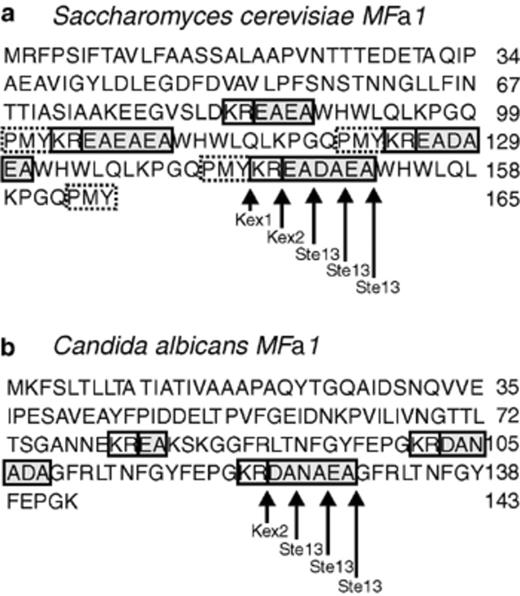
Potential maturation sites of α-pheromone by protease Ste13 from C. albicans. Deduced amino acid sequence of MFα1 gene from (a) S. cerevisiae, and (b) C. albicans. The C. albicansα-pheromone contains three equally spaced sequences encoding α-pheromone peptides: two encoding 13-mers and one encoding a 14-mer. The arrows indicate sites of α-pheromone maturation by Kex1, Kex2 and Ste13 in S. cerevisiae, and potential maturation sites of α-pheromone by Ste13 in C. albicans.
Acknowledgments
This work was supported by grant from CGPI-IPN 20040560 and 20050005, COFAA, EDI, and EDD from IPN. Bautista-Muñoz was fellow from CONACYT and PIFI-IPN. The authors acknowledge Dr. Javier Zavala Díaz de la Serna for performing computer analysis and Dr. Laura Ongay from IFC-UNAM for providing CHEF-DRII, Bio-Rad equipment.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}