





Abstract
Following incubation with ATP and a cAMP-dependent protein kinase under optimal conditions of lipid acceptor, phospholipid and metal ion requirements, the transfer activity of partially purified dolichol phosphate mannose synthase (DPMS) increased about 60% and this activation correlated with a 50% increase in Vmax with no alteration in the apparent Km for GDP-Manose. Phosphorylation with [γ-32P]ATP resulted in the labeling of several polypeptides, one of which exhibited the molecular weight of the enzyme (30 kDa) and was also recognized using a specific anti-DPMS monoclonal antibody. This and the fact that the phosphate label could be removed by an alkaline phosphatase indicate that Candida DPMS may be regulated by phosphorylation-dephosphorylation, a mechanism that has been proposed for the enzyme in other organisms.
1 Introduction
Over the past few years, our laboratory has focused on enzymes that catalyze essential steps in glycoprotein biosynthesis in pathogenic organisms. One of this enzymes is dolichol phosphate mannose synthase (DPMS, EC 2.4.1.83), the glycosyltransferase responsible for the synthesis of dolichol phosphate manose (Dol-P-Man) from GDP-Man and dolichol monophosphate (Dol-P). Dol-P-Man is a mannosyl donor in protein O- and N-glycosylation and in the biosynthesis of glycosylphosphatidylinositol membrane anchors [1–3]. The enzyme has been studied in many organisms and partially purified in some cases [4–7]; however, most of our knowledge of DPMS comes from studies in Saccharomyces cerevisiae where the structural gene, DPM1, has been shown to encode for an essential 30-kDa polypeptide of the endoplasmic reticulum [8,9]. Both the native and recombinant yeast DPMS have been purified and some of their properties investigated [10,11]. Further interest on DPMS has arisen from the proposal that it can be a suitable target for inhibitors of pathogenic eukaryotes [12].
Previous studies from this laboratory demonstrated that subcellular fractions from the pathogenic yeast Candida albicans catalyzed the transfer of mannose from GDP-Man into Dol-P-Man and from this intermediate into mannoproteins in sequential coupled reactions involving DPMS and protein mannosyl transferases (PMT) [13,14]. In order to purify and characterize DPMS and PMT separately, several attempts were made to dissociate these transferases employing differential extraction with detergents. We observed that incubation of yeast membranes with Nonidet P-40 at a detergent/protein ratio as low as 0.063 (0.1%) released a soluble fraction enriched with DPMS that channeled over 80% of the total transferred radioactivity from GDP-Man into Dol-P-Man, whereas only a minor fraction of mannoproteins was labeled [3,15]. DPMS solubilized under these conditions was, however, very unstable thus difficulting enzyme purification. A slight modification of this protocol (see below) rendered more stable enzyme preparations that still catalyzed the preferential labeling of the mannolipid and could be used for the partial purification of DPMS using a single step of preparative non-denaturing electrophoresis [16]. Using this preparation, we investigated some properties of the mannosyl transfer reaction with emphasis on enzyme regulation by phosphorylation and dephosphorylation.
2 Materials and methods
2.1 Chemicals
Guanosine diphosphate mannose [mannose-1-14C] and adenosine 5′-triphosphate [γ-32P] were obtained from American Radiolabeled Chemicals (St. Louis, MO). GDP-Man, dolichol monophosphate, the catalytic subunit of cAMP-PK, the PK inhibitor, alkaline phosphatase, E-64, Nonidet P-40 and all phospholipids were from Sigma Chemical Co. (St. Louis, MO). Bio-Beads SM-2 and all electrophoresis reagents were from Bio-Rad Laboratories (Hercules, CA). Luminol was from Amersham (Buckinghamshire, England). The anti-DPMS monoclonal antibody # 5C5-A7 was obtained from Molecular Probes (Eugene, OR). All other products were from reliable commercial sources.
2.2 Organism and culture conditions
C. albicans, strain ATCC 26555, was maintained and propagated in the media and in the conditions previously described [13].
2.3 Preparation of cell membranes and enzyme solubilization and purification
Yeast cells were collected, broken and the homogenate was subjected to differential centrifugation according to previously described protocols [14] except that cells were disrupted in the presence of 1 µM trans-epoxysuccinyl-l-leucylamido-(4-guanidino)butane (E-64) to minimize proteolytic activity. The high-speed pellet consisting of a mixed membrane fraction was resuspended in 8–9 ml of 50 mM Tris–HCl buffer, pH 7.5 (Tris buffer) and used for DPMS solubilization and purification in the conditions described previously [16]. Briefly, the fraction solubilized with Nonidet P-40 at a detergent/protein ratio of 0.3 (0.25%), routinely 3 ml, was freeze-dried, resuspended in 300 µl of electrophoresis loading buffer and applied onto a preparative 5% non-denaturing polyacrylamide minigel that was run at 80 V for 4 h at 4 °C according to standard protocols [17]. To locate and recover the enzyme, the gel was cut in 0.5 cm-wide fragments that were placed in assay tubes and homogenized with a Potter in 1 ml of Tris buffer containing 0.05% Nonidet P-40. After 30 min at 4 °C, the gel remnants were sedimented by low-speed centrifugation, the supernatant was aspirated with a Pasteur pipette and after elimination of detergent excess using Bio-Beads SM-2 adsorbents (1 g of beads for 5 ml of solution), it was used as the source of DPMS.
2.4 Assay of DPMS activity
Unless otherwise stated, activity of partially purified DPMS was measured in reaction mixtures containing 7.5 mM MgCl2, 35 µg Dol-P, 10 µg phosphatidylinositol (PI), 0.01 µCi (about 22,000 cpm) of GDP-[14C]Man (sp. act., 267 mCi mmol−1), the enzyme (17–32 µg protein) and Tris buffer in a total volume of 300 µl. When activity was assayed in the solubilized fraction, similar reaction mixtures were prepared except that these contained 260–300 µg of protein, 3–5 µg Dol-P and no exogenous phospholipid was added. After 30 min of incubation at 28 °C, the reaction was stopped and the mannolipid was extracted with the solvents and in the conditions described elsewhere [13]. Unless otherwise indicated, DPMS activity was expressed as cpm incorporated into Dol-P-Man in one min. Specific activity was referred to one mg of protein.
2.5 Phosphorylation of DPMS
Enzyme phosphorylation was carried out in reaction mixtures containing the purified enzyme (32–64 µg protein), 7.5 mM MgCl2, 35 µg Dol-P, 0.05% Nonidet P-40, 10 µg PI, 0.2 mM ATP, 12.5 U cAMP-PK and Tris buffer in a total volume of 200 µl. Control mixtures in which ATP and/or cAMP-PK were omitted or the cAMP-PK inhibitor was added were run in parallel. After 30 min of incubation at 30 °C (the optimum temperature for cAMP-PK), 0.01 µCi of GDP-[14C]Man were added and the volume was adjusted to 300 µl with Tris buffer. The transfer reaction was let to proceed for 30 min at 28 °C and the mannolipid was extracted and quantified as described above.
To reveal the polypeptides phosphorylated by cAMP-PK, reaction mixtures equal to those described above were prepared except that cold ATP was substituted by 5 µCi of [γ-32P]ATP (sp. act., 1000–3000 Ci mmol−1; 37–111 TBq mmol−1) and an additional reaction mixture containing 50 mU of alkaline phosphatase was run in parallel. After 1 h at 30 °C, the samples were analyzed as previously described [7], except that the analytical gel was stained with Coomassie Blue and the time of exposure of the films to the labeled gels was reduced to 23 h.
2.6 Protein determination
Protein content was determined by the modified Lowry method [19] using the Bio-Rad DC protein assay kit and bovine serum albumin as standard.
2.7 Immunoblotting
Polypeptides present in the enzyme fraction were separated in two equal 10% SDS–PAGE gels that were run in parallel following standard protocols [17]. One of the gels was stained with Coomassie Blue and the other was electrotransferred to nitrocellulose as described elsewhere [18]. Electrotransfer was assessed by staining the membrane with Ponceau S diluted 1:10 in water. After removal of the dye by washing with water, the membrane was gently shaken for 2–4 h with 5% skim milk in PBS. The blocker excess was removed by washing thrice with PBS-Tween 20 (0.05%) and the membrane was incubated with a monoclonal antibody (IgG1,k mouse isotype) raised to recombinant DPMS from S. cerevisiae, diluted 1:200 in PBS-Tween 20. After 12–14 h at 4 °C, the excess antibody was removed, the membrane was washed thrice with PBS and further incubated for 2 h with a peroxidase-conjugated, rabbit anti-mouse serum diluted 1:3000 in PBS. After washing with PBS, immunoreacting polypeptides were revealed by chemiluminiscence with the Luminol substrate according to the manufacturer's instructions (Amersham Life Science booklet).
3 Results and discussion
3.1 Optimization of DPMS assaying conditions
Prior to determine the effect of phosphorylation on DPMS, which was the major scope of this study, we optimized the enzyme assaying conditions by testing the effect of phospholipids, exogenous Dol-P and metal ions on the transfer reaction.
Activity of solubilized glycosyltransferases is usually restored by phospholipids bearing distinct polar heads [5,7,20–22]. Here, although some activity was observed in the absence of added phospholipids, this was largely stimulated by PI and to a lower extent by phosphatidylethanolamine (PE) at optimal concentrations of 6.25 and 12.5 µg per assay, respectively. Other phosphoglycerides such as phosphatidylcholine and phosphatidylglycerol had no effect on DPMS. In different experiments, stimulation with PI and PE ranged from 4 to 10- and 3 to 7-fold, respectively, a result probably due to variations in the amount of endogenous phospholipids present in the different enzyme preparations. To test whether the activating effect was additive, transfer activity was measured as a function of optimal amounts of individual PE or PI or mixtures thereof at different ratios. Optimal stimulation was observed at PE/PI ratios of 2.5–5.0 and this was not substantially higher than that attained with the individual phospholipids. In terms of activation by PE, Candida DPMS is reminiscent of that from mammalian [5], yeast [22] and protozoan [7] cells, suggesting that this phosphoglyceride, and also PI in the case of Candida, may be a critical component of the enzyme hydrophobic environment in intact membranes. Activity of solubilized DPMS, on the other hand, did not depend on exogenous phospholipids suggesting that these were solubilized along with the enzyme (not shown).
Clearly, a component eliminated during purification was the lipid acceptor, as partially purified DPMS exhibited a nearly absolute dependence of exogenous Dol-P. Thus, transfer activity increased as a function of the amount of the lipid acceptor reaching saturation at about 40 µg per assay, with half-maximum activation observed at 14 µg per assay.
DPMS and other polyprenol phosphate sugar transferases from a number of sources require a metal ion for activity, preferably Mg2+ or Mn2+, optimal concentrations commonly ranging from 10 to 20 mM and 1 to 2 mM, respectively [7,20,23]. In agreement with these findings, the enzyme studied here depended on Mg2+ which stimulated activity up to 8-fold at concentrations in the range of 2.5–10.0 mM. Mn2+ and Ca2+, on the other hand, were ineffective.
3.2 Phosphorylation of DPMS
Accumulated evidence indicates that DPMS from organisms as divergent as fungi [6], protozoans [7] and mammalians [24,25] can be activated in vitro by enzyme phosphorylation to levels that range from 40–80% in rat parotid microsomes [24,25] to 40–380% in E. histolytica[7] and that these changes correlate with an increase in Vmax with no effect on the enzyme affinity for GDP-Man. We therefore considered important to investigate if this was also the case for the Candida transferase. To this aim, the partially purified enzyme was incubated with ATP, cAMP-PK and other additions as described in Section 2. Following incubation with cAMP-PK and ATP, DPMS activity increased 60% over the control and addition of the kinase inhibitor abolished this activation. As expected, kinase alone had no effect on enzyme activity (Table 1). In some experiments, incubation with ATP in the absence of cAMP-PK brought about a low activation (15–25%) suggesting the presence of some endogenous kinase activity. To investigate the effect of phosphorylation on DMPS catalytic properties, apparent Km and Vmax values for GDP-Man were determined under conditions of phosphorylation as well as in conditions where the kinase was omitted. Results indicated that phosphorylated and unphosphorylated enzymes transferred mannose from GDP-Man into Dol-P-Man with hyperbolic kinetics but the former was 40–45% more active (Fig. 1(a)). Analysis of double reciprocal plots of data revealed Km and Vmax values of 0.2 mM GDP-Man and 2000 pmol min−1 mg−1 protein, respectively, for the phosphorylated enzyme. Corresponding values for the unphosphorylated DPMS were 0.2 mM GDP-Man and 3000 pmol min−1 mg−1 protein (Fig. 1(b)).
Effect of in vitro phosphorylation on DPMS activity
| Addition | Activity | |
| Specific | Relative | |
| None (control) | 2895 | 1.0 |
| cAMP-PK | 2815 | 1.0 |
| ATP | 3665 | 1.3 |
| cAMP-PK + ATP | 4724 | 1.6 |
| cAMP-PK + ATP + Inh cAMP-PK | 3204 | 1.1 |
| Addition | Activity | |
| Specific | Relative | |
| None (control) | 2895 | 1.0 |
| cAMP-PK | 2815 | 1.0 |
| ATP | 3665 | 1.3 |
| cAMP-PK + ATP | 4724 | 1.6 |
| cAMP-PK + ATP + Inh cAMP-PK | 3204 | 1.1 |
Effect of in vitro phosphorylation on DPMS activity
| Addition | Activity | |
| Specific | Relative | |
| None (control) | 2895 | 1.0 |
| cAMP-PK | 2815 | 1.0 |
| ATP | 3665 | 1.3 |
| cAMP-PK + ATP | 4724 | 1.6 |
| cAMP-PK + ATP + Inh cAMP-PK | 3204 | 1.1 |
| Addition | Activity | |
| Specific | Relative | |
| None (control) | 2895 | 1.0 |
| cAMP-PK | 2815 | 1.0 |
| ATP | 3665 | 1.3 |
| cAMP-PK + ATP | 4724 | 1.6 |
| cAMP-PK + ATP + Inh cAMP-PK | 3204 | 1.1 |
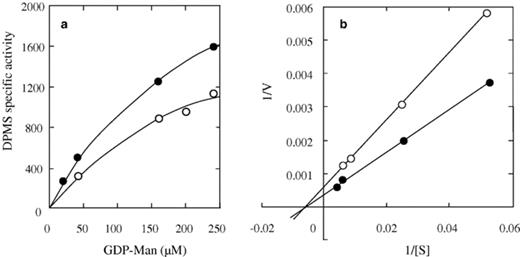
Effect of substrate concentration on catalytic activity of phosphorylated and unphosphorylated DPMS. The electrophoresis-purified enzyme was incubated at 30 °C with (closed circles) or without (open circles) cAMP-PK and other components of the phosphorylation mixture as described in the text. After 30 min, the labeled sugar donor was added at the indicated concentrations and incubation continued for another 30 min at 28 °C. After this time, Dol-P-Man was extracted and quantified. (a) Enzyme velocity versus GDP-Man concentration; (b) Reciprocal plots of data from (a). Lines drawn resulted from a least-squares analysis of data. Here, enzyme activity is expressed as Pmol of mannose incorporated into Dol-P-Man min−1 mg−1 of protein. Results are representative of three independent experiments run in duplicate.
Finally, to determine whether DPMS stimulation was due to enzyme phosphorylation, the polypeptides phosphorylated following incubation with cAMP-PK and labeled ATP were analyzed as described in Section 2. Inspection of autoradiograms revealed the presence of several 32P-labeled polypeptides showing molecular masses in the range of 12–15 (E) to 45.5 (A) kDa (Fig. 2(a), lane 3) including one of 30 kDa (D), a molecular weight closely comparable to that estimated for DPMS from other organisms [6,7,9,25]. Phosphorylation in the presence of alkaline phosphatase (Fig. 2(a), lane 1) or the cAMP-PK inhibitor (Fig. 2(a), lane 2) markedly reduced labeling of all polypeptides and, as expected, no labeled bands were observed in the absence of the kinase (Fig. 2(a), lane 4). A polypeptide of 30 kDa was also recognized by a mouse monoclonal antibody raised to recombinant yeast DPMS in both the solubilized (Fig. 2(b), lane 2) and the partially purified (Fig. 2(b), lane 3) enzyme samples strongly suggesting that this species corresponds to the enzyme protein. Failure to detect this polypeptide in mock incubations lacking cAMP-PK or containing alkaline phosphatase or a specific inhibitor of the kinase argues against unspecific labeling. The nature of the immunoreacting 26.7-kDa polypeptide, which was also observed in the yeast preparation used as a positive control (Fig. 2(b), lane 1), remained undetermined. However, it may very well be a degradation product of DPMS retaining the recognizable epitope. No bands were detected in mock incubations lacking the primary antibody.
![Analysis of polypeptides phosphorylated by cAMP-PK and [γ-32P]ATP and immunodetection of the DPMS polypeptide. (a) Samples of the electrophoresis-purified DPMS were incubated at 30 °C with [γ-32P]ATP and other additions as indicated. After 30 min, proteins were precipitated with TCA and processed to reveal the labeled polypeptides by autoradiography as described in the text. Results are representative of two independent experiments. (b) Proteins in a membrane fraction from S. cerevisiae (positive control, lane 1, 45.4 µg protein), the solubilized fraction (lane 2, 39 µg protein) and the partially purified DPMS (lane 3, 39 µg protein) were separated by SDS–PAGE, electrotransferred to nitrocellulose membranes and incubated with an anti-DPMS monoclonal serum in the conditions described in Section 2. Immunoreacting bands were visualized with a peroxidase-conjugated, rabbit anti-mouse secondary antibody.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femspd/45/3/10.1016_j.femsim.2005.05.016/1/m_FIM_429_f2.jpeg?Expires=1750780200&Signature=F0j7t54B3AlvfnE7olxSBrZngspvfMknK9D4F-hE2YZkID4wy~ZcztgHv3G~ya4BUNazWT2RiOxHcVhxNb6jDsbT6ua2UFCFRPpFIi79Kf2YaH9yUTcLaevUnZutJDa1xoyEvT35LIpnEw4HwoArDXjGsiXC-MDk04t5n~os2NQBYvMsecvbZufKNSdsW5DBelTH1dRIju2HY8kVV9olmvjQ4zeGj1vuNY-SdkFvqCpdC85spgLBbYgmI-ykC9WXhLXOSK-cCer2OeszMpJASuQetqV3~HS7Kdm8JkvsCI6ANN6Uktu1xA4tEmvSSqsa~GaTo~befpWKTCwzBmuV5A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Analysis of polypeptides phosphorylated by cAMP-PK and [γ-32P]ATP and immunodetection of the DPMS polypeptide. (a) Samples of the electrophoresis-purified DPMS were incubated at 30 °C with [γ-32P]ATP and other additions as indicated. After 30 min, proteins were precipitated with TCA and processed to reveal the labeled polypeptides by autoradiography as described in the text. Results are representative of two independent experiments. (b) Proteins in a membrane fraction from S. cerevisiae (positive control, lane 1, 45.4 µg protein), the solubilized fraction (lane 2, 39 µg protein) and the partially purified DPMS (lane 3, 39 µg protein) were separated by SDS–PAGE, electrotransferred to nitrocellulose membranes and incubated with an anti-DPMS monoclonal serum in the conditions described in Section 2. Immunoreacting bands were visualized with a peroxidase-conjugated, rabbit anti-mouse secondary antibody.
Taken together, our results indicate that DPMS from C. albicans, like that from other organisms, can also be regulated in vitro by phosphorylation–dephosphorylation. The physiological implication of this mechanism in vivo will be the subject of future research.
Acknowledgments
We thank Dr. Julio C. Villagómez-Castro for expert technical assistance. This work was partially supported by Grant No. 458100-5-27818N from the Consejo Nacional de Ciencia y Tecnología (CONACyT), México. All authors except BLAF are National Investigators, México.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.femsim.2005.05.016.
References

{kind=link}
{kind=link}