





Abstract
While recent studies have suggested that fish mucus microbiota play an important role in homeostasis and prevention of infections, very few studies have investigated the bacterial communities of gill mucus. We characterised the gill mucus bacterial communities of four butterflyfish species and although the bacterial diversity of gill mucus varied significantly between species, Shannon diversities were high (H = 3.7–5.7) in all species. Microbiota composition differed between butterflyfishes, with Chaetodon lunulatus and C. ornatissimus having the most similar bacterial communities, which differed significantly from C. vagabundus and C. reticulatus. The core bacterial community of all species consisted of mainly Proteobacteria followed by Actinobacteria and Firmicutes. Chaetodonlunulatus and C. ornatissimus bacterial communities were mostly dominated by Gammaproteobacteria with Vibrio as the most abundant genus. Chaetodonvagabundus and C. reticulatus presented similar abundances of Gammaproteobacteria and Alphaproteobacteria, which were well represented by Acinetobacter and Paracoccus, respectively. In conclusion, our results indicate that different fish species present specific bacterial assemblages. Finally, as mucus layers are nutrient hotspots for heterotrophic bacteria living in oligotrophic environments, such as coral reef waters, the high bacterial diversity found in butterflyfish gill mucus might indicate external fish mucus surfaces act as a reservoir of coral reef bacterial diversity.
INTRODUCTION
The associations between metazoans and commensal microorganisms are among the most ancient and successful in nature (McFall-Ngai et al. 2013; Lowrey et al. 2015). Metazoa and their associated microorganisms have coevolved in response to environmental selective pressures over hundreds of millions of years (Zilber-Rosenberg and Rosenberg 2008). Host-associated microorganisms affect host physiology and health, and therefore maintaining microbiota homeostasis is a key factor to avoid pathogen proliferation and diseases (Kamada et al. 2013; Sommer and Bäckhed 2013). Recent studies on human gut microbiota show that microbiota benefit their hosts, particularly by regulating immune balance (Wu and Wu 2012). In fish, disruption of microbiota homeostasis (dysbiosis) results in a decrease of probiotic-like bacteria and an increase in pathogenic bacteria (Boutin, Audet and Derome 2013; Boutin et al. 2014). Recent studies also show that bacteria isolated from fish gut and skin display antibacterial and antifungal activities against human and fish pathogens, suggesting a protective role of fish native microbiota against pathogens (Sanchez et al. 2012; Lowrey et al. 2015).
To date, most studies on fish microbiota have investigated fish gut and skin bacterial communities (Smith, Danilowicz and Meijer 2007; Larsen et al. 2013; Larsen, Mohammed and Arias 2014; Giatsis et al. 2015), while gill bacterial diversity and their functional roles remain poorly understood. Gills are the main respiratory organ in fish, and are composed of four pairs of vascularised gill arches with hundreds of gill filaments, which increase their surface area for oxygen diffusion by folding into secondary lamellae. In addition to respiration, gills perform other functions including osmoregulation, pH balance, ammonia excretion, hormone regulation, detoxification and immune defence (Maina 2002). Gills are constantly in contact with water, and therefore they are continuously exposed to pathogens (Xu et al. 2016). The gills are covered by a mucus layer that acts as the first physical and biochemical barrier against pathogens (Roberts and Powell 2003). Gill mucus is mainly composed of glycoproteins, but also contains numerous other molecules including immune-related proteins such as immunoglobulins and antimicrobial peptides (AMPs) (Iijima et al. 2003; Xu et al. 2016). Gill mucus harbours a complex community of commensal microorganisms that play an important role in maintaining mucus homeostasis and protection against pathogens (Boutin, Audet and Derome 2013; Gomez, Sunyer and Salinas 2013; Llewellyn et al. 2014). Recent studies are starting to show the important role of commensal bacteria in the production of bioactive metabolites. For example, fish intestinal bacteria were found to synthesise an antimicrobial fatty acid, while some rainbow trout (Oncorhynchus mykiss) skin-associated bacterial strains displayed antifungal activity (Sanchez et al. 2012, Lowrey et al. 2015). Gill mucus production increases following gill infection, and a recent study reported decreased oxygen diffusion and an increase of pathogenic bacteria in fish exposed to high sediment concentrations, impairing normal gill function (Ferguson et al. 1992; Hess et al. 2015). However, despite their importance in conserving gill homeostasis and preventing gill infection, the microbiota associated with gill mucus remains poorly studied (Hess et al. 2015; Lowrey et al. 2015; Tarnecki, Patterson and Arias 2016).
In this study, we aimed to cast light on the gill mucus bacterial communities of four sympatric butterflyfish species from Moorea (French Polynesia). Butterflyfishes (family Chaetodontidae) are a diverse and conspicuous family of coral reef fishes distributed widely in all tropical seas. Butterflyfish ecology and behaviour have been extensively studied, and although they can consume a variety of prey, including algae, polychaetes, crustaceans and coral, most species feed primarily if not exclusively on scleractinian corals (Pratchett 2005). Three strict corallivore species (two generalists, Chaetodonlunulatus and C. ornatissimus, and one specialist, C. reticulatus) and one omnivore species (C. vagabundus) were selected for this study (Pratchett 2005; Berumen and Pratchett 2006; Cole, Pratchett and Jones 2008). Furthermore, the two generalist corallivore species were selected for their similarity in most ecological (e.g. diet and habitat) and phylogenetic traits except for their monogenean loads. Chaetodonornatissimus is always parasitised by gill monogeneans (Dactylogyridae), while C. lunulatus is the only butterflyfish species where these parasites have never been observed (Reverter et al. 2016). Coral reefs are among the most productive and biologically diverse ecosystems on Earth, and although coral microbiomes have attracted increasing attention in recent years, most microbial communities of reef-associated organisms remain poorly studied (Ainsworth et al. 2015). The main objective of this study was to characterise and compare the bacterial diversity and taxonomic composition of the gill mucus of four sympatric butterflyfishes.
MATERIALS AND METHODS
Mucus sampling and DNA extraction
Four butterflyfish species (C. lunulatus, C. ornatissimus, C. reticulatus and C. vagabundus) were spearfished and killed immediately by brain spiking in order to minimise suffering (5 fish/species, a total of 20 fish) on the island of Moorea (French Polynesia). Sampling protocols were pre-approved by animal experimentation experts of our institute, following the European Union directive 2010/63UE. Fish were put in individual plastic bags with seawater and brought immediately to a laboratory for dissection. Gill mucus was carefully scraped with a sterile spatula into sterile tubes and placed on ice until DNA extraction (within the hour following collection). One millilitre of mucus DNA was extracted using the DNeasy Blood & Tissue kit (Qiagen, Courtaboeuf, France). DNA concentrations were quantified by measuring the absorbance at 260 nm (any sample with less than 100 ng/mL would have been discarded) and purity was checked by measuring absorbance at 260:280 nm (>1.8) and 230:260 nm (>1.8). DNA samples were sent for 454 pyrosequencing at MRDNALab (Shallower, TX, USA, http://www.mrdnalab.com) using a modified version of a previously published protocol (Croué et al2013). Briefly, 15 ng DNA was used in 20 μL PCR reactions (94°C, 3 min, followed by 28 cycles of 94°C for 30 s, 53°C for 40 s and 72°C for 1 min; and a final elongation step at 72°C for 5 min) that were performed using the HotStarTaq Plus Master Mix Kit (Qiagen) and primers 27F.1 (5΄ AGRGTTTGATCNTGGCTCAG 3΄; Kuske et al2006) and Gray519R (5΄ GTNTTACNGCGGCKGCTG 3΄; Kostka et al. 2011). The forward primers contained 8-mer tags at the 5΄ end to allow multiplexing. The reactions amplified the hypervariable V1–V3 region of the 16S rRNA gene. Products from different samples were mixed in equimolar concentrations and purified using Agencourt Ampure beads (Agencourt Bioscience Co.). Tag-encoded FLX amplicons were sequenced on a Roche 454 FLX sequencer using Titanium (Roche) reagents by MrDnaLab. Samples were identified by the fish initials and an order number (Ex. CL1–CL5 for C. lunulatus replicates).
Data treatment
Multiplex1 raw SFF (Standard Flowgram Format) files were analysed using a hybrid analysis pipeline as previously described (Croué et al. 2013) with some modifications. In brief, denoising was done by AmpliconNoise V1.25 (Quince et al. 2011) implemented in Qiime V1.5 (Caporaso et al. 2010) with a small modification to allow it to be run in an iDataplex (IBM) cluster. De novo chimera detection and removal was performed with the uchime module (Edgar 2010; Edgar et al. 2011) of usearch 5.2 (http://drive5.com/usearch/). Non-chimeric sequences were unweighted and grouped into operational taxonomic units (OTUs) with two different sequence identity percentages (97 and 98%) using the usearch v5.2 method implemented in Qiime V1.5. The longest sequence in the OTU was selected as a representative. OTUs were classified using the rdp_classifier software implemented in Qiime V1.5 (Wang et al. 2007) and a database based on the Greengenes August 2013 taxonomy (http://greengenes.secondgenome.com) modified to exclude orders, and corrected to comply with current official nomenclature [(list of prokaryotic names with standing in nomenclature (http://www.bacterio.net)]. Based on this classification, OTUs representing chloroplasts and mitochondria were removed. The resulting OTU table was further treated to remove (using custom bash and awk scripts) OTUs for which the representative sequence was shorter than 372 bp (400 bp minus the length of the primer), OTUs that were represented by a single sequence in the ensemble of samples and a ‘Root’ taxonomy status by the rdp_classifier analysis, as well as sequences failing aligning by pynast aligner implemented in Qiime. Sequences were aligned and filtered using the Lane mask (Lane 1991) using mothur v1.38.0 (Schloss et al. 2009) and a neighbour joining tree necessary to calculate unifrac distances (Lozupone et al. 2011) was constructed using phylip v3.6a3 (Felsenstein 1989). A principal component analysis was performed on the preliminary OTU table followed by the calculation of Mahalanobis distances to detect the presence of outliers. One of the samples from C. vagabundus was clearly an outlier, and therefore the sample was removed from the OTU table along with all OTUs present exclusively in this sample, as was one OTU abundant in sample CV1 corresponding to a chloroplast, but not identified as such by the RDP classifier. Alpha-diversity indices (observed richness, Chao 1, Shannon diversity index) were calculated using an OTU table rarefied to 1499 sequences (lowest number of sequences that passed all the quality control described) using the single_rarefaction.py function of Qiime v1.5 and summarised per fish species (mean and standard deviation). t-Tests were used to assess the difference in diversity indices between the two sequence identity percentages. ANOVA and Tukey posthoc tests were used to detect significant differences in diversity indices between fish species. Principal coordinates analysis (PCoA) using weighted unifrac distances, which takes into account both the OTUs’ abundance and the phylogenetic distance between the OTUs, was used to assess the differences between the microbiomes of the different fish species using a relativised (percentage of reads per sample) OTU table. Permutational multivariate analysis of variance (PERMANOVA, function adonis of the vegan package for R) and pairwise comparisons between group levels with corrections for multiple testing (function pairwise.perm.manova of the RVAideMemoire package for R) were used to evaluate statistically significant differences of PCoA groups between fish species. The number of shared OTUs among all fish species combinations was calculated and represented using a Venn diagram (using the rarefied OTU table). Microbiome composition of the four butterflyfish species was characterised and results are displayed by bacterial phyla abundances. The bacterial families and genera with abundances higher than 5% in at least one of the samples were also identified, and significant differences between fish species were assessed using the Kruskal–Wallis test and Kruskal posthoc test (non-normal data).
RESULTS
Diversity of bacterial communities
A total of 1940 OTUs were obtained from the gill mucus of four butterflyfish species (C. lunulatus, C. ornatissimus, C. reticulatus and C. vagabundus). Alpha-rarefaction indices showed that one of the C. vagabundus samples possessed a remarkably different microbiome. This sample (CV5) presented 476 unique OTUs (including five unique phyla), which accounted for over 50% of all C. vagabundus OTUs (931). CV5 presented 7.5 times more unique OTUs than the average number of OTUs per C. vagabundus sample (64 OTUs). Furthermore, when this sample was compared with all gill mucus samples, it still had 384 unique OTUs, which represented 20% of the total OTUs found in butterflyfish gill mucus. A principal component analysis followed by the calculation of the Mahalanobis distances clearly identified this sample as an outlier (Supplementary Fig. S1). The high number of OTUs in CV5 was most likely due to contamination from an external source with higher OTU diversity, and after removal of this outlier sample, 1450 OTUs were obtained in the full OTU table and 1041 in the rarefied table.
Alpha diversity indices were highly similar between the two identity cut-offs used (97 and 98%, Table 1), and none of the studied diversity indices showed a significant difference between them (P < 0.05, Supplementary Table 1). Therefore, the results presented hereafter refer to the highest sequence identity cut-off used (98%). Shannon diversity varied significantly (P < 0.05; ANOVA, Tukey post-hoc test) between fish species, with the highest diversity found in C. vagabundus (H = 5.7 ± 0.5) and the lowest in C. reticulatus (H = 3.7 ± 0.7) (Table 1). Chaetodonlunulatus and C. ornatissimus presented similar diversities (H = 4.7 ± 1.2 and H = 4.8 ± 0.5, respectively. Observed and estimated richness (Chao1) were both highest in C. lunulatus followed by C. ornatissimus, C. vagabundus and C. reticulatus (Table 1).
Alpha-diversity estimates for the four butterflyfish species (mean ± standard deviation). Same letters indicate values that are significantly different between them (P < 0.05, ANOVA, Tukey posthoc test).
| OTU sequence cut-off | Fish species | Chao 1 | Shannon (H) | Observed richness |
|---|---|---|---|---|
| 98% | C. lunulatus | 254.0 ± 137.7a | 4.7 ± 1.2 | 140.6 ± 61.5a |
| C. ornatissimus | 184.8 ± 95.9 | 4.8 ± 0.5 | 113.2 ± 40.9 | |
| C. reticulatus | 49.1 ± 23.2a | 3.7 ± 0.7a | 46.0 ± 22.5a,b | |
| C. vagabundus | 154.6 ± 79.4 | 5.8 ± 0.4a | 137.0 ± 55.6b | |
| 97% | C. lunulatus | 211.4 ± 87.5a | 4.8 ± 1.2 | 140.8 ± 60.9a |
| C. ornatissimus | 170.4 ± 67.9b | 4.7 ± 0.7 | 111.2 ± 37.7 | |
| C. reticulatus | 44.9 ± 19.6a,b | 3.7 ± 0.7a | 43.2 ± 17.3a,b | |
| C. vagabundus | 147.0 ± 67.9 | 5.6 ± 0.3a | 131.3 ± 51.1b |
| OTU sequence cut-off | Fish species | Chao 1 | Shannon (H) | Observed richness |
|---|---|---|---|---|
| 98% | C. lunulatus | 254.0 ± 137.7a | 4.7 ± 1.2 | 140.6 ± 61.5a |
| C. ornatissimus | 184.8 ± 95.9 | 4.8 ± 0.5 | 113.2 ± 40.9 | |
| C. reticulatus | 49.1 ± 23.2a | 3.7 ± 0.7a | 46.0 ± 22.5a,b | |
| C. vagabundus | 154.6 ± 79.4 | 5.8 ± 0.4a | 137.0 ± 55.6b | |
| 97% | C. lunulatus | 211.4 ± 87.5a | 4.8 ± 1.2 | 140.8 ± 60.9a |
| C. ornatissimus | 170.4 ± 67.9b | 4.7 ± 0.7 | 111.2 ± 37.7 | |
| C. reticulatus | 44.9 ± 19.6a,b | 3.7 ± 0.7a | 43.2 ± 17.3a,b | |
| C. vagabundus | 147.0 ± 67.9 | 5.6 ± 0.3a | 131.3 ± 51.1b |
Alpha-diversity estimates for the four butterflyfish species (mean ± standard deviation). Same letters indicate values that are significantly different between them (P < 0.05, ANOVA, Tukey posthoc test).
| OTU sequence cut-off | Fish species | Chao 1 | Shannon (H) | Observed richness |
|---|---|---|---|---|
| 98% | C. lunulatus | 254.0 ± 137.7a | 4.7 ± 1.2 | 140.6 ± 61.5a |
| C. ornatissimus | 184.8 ± 95.9 | 4.8 ± 0.5 | 113.2 ± 40.9 | |
| C. reticulatus | 49.1 ± 23.2a | 3.7 ± 0.7a | 46.0 ± 22.5a,b | |
| C. vagabundus | 154.6 ± 79.4 | 5.8 ± 0.4a | 137.0 ± 55.6b | |
| 97% | C. lunulatus | 211.4 ± 87.5a | 4.8 ± 1.2 | 140.8 ± 60.9a |
| C. ornatissimus | 170.4 ± 67.9b | 4.7 ± 0.7 | 111.2 ± 37.7 | |
| C. reticulatus | 44.9 ± 19.6a,b | 3.7 ± 0.7a | 43.2 ± 17.3a,b | |
| C. vagabundus | 147.0 ± 67.9 | 5.6 ± 0.3a | 131.3 ± 51.1b |
| OTU sequence cut-off | Fish species | Chao 1 | Shannon (H) | Observed richness |
|---|---|---|---|---|
| 98% | C. lunulatus | 254.0 ± 137.7a | 4.7 ± 1.2 | 140.6 ± 61.5a |
| C. ornatissimus | 184.8 ± 95.9 | 4.8 ± 0.5 | 113.2 ± 40.9 | |
| C. reticulatus | 49.1 ± 23.2a | 3.7 ± 0.7a | 46.0 ± 22.5a,b | |
| C. vagabundus | 154.6 ± 79.4 | 5.8 ± 0.4a | 137.0 ± 55.6b | |
| 97% | C. lunulatus | 211.4 ± 87.5a | 4.8 ± 1.2 | 140.8 ± 60.9a |
| C. ornatissimus | 170.4 ± 67.9b | 4.7 ± 0.7 | 111.2 ± 37.7 | |
| C. reticulatus | 44.9 ± 19.6a,b | 3.7 ± 0.7a | 43.2 ± 17.3a,b | |
| C. vagabundus | 147.0 ± 67.9 | 5.6 ± 0.3a | 131.3 ± 51.1b |
Microbiota differences between butterflyfishes
Among the 1041 OTUs, only 26 were found in all four butterflyfishes. This ‘core microbiota’ (26 OTUs) represented between 18 and 35% of all bacterial sequences in each fish species. The core microbiota (at the OTU level) consisted of mainly Proteobacteria, Actinobacteria and Firmicutes (class Clostridia) and one OTU identified as a Cyanobacteria (Table 2). Chaetodonlunulatus was the species with the highest number of unique OTUs (273 OTUs), which represented between 5 and 19% of its bacterial community (Fig. 1). Chaetodonvagabundus had the second highest number of unique OTUs (249 OTUs, 26–66% of all OTUs), followed by C. ornatissimus (143 OTUs, 6–15% of all OTUs), and finally C. reticulatus, with only 80 unique OTUs that represented between 16 and 62% of its bacterial community (Fig. 1). Chaetodonlunulatus and C. ornatissimus shared the highest number of OTUs (a total of 167 shared OTUs), while C. lunulatus and C. reticulatus shared the lowest (40 OTUs) (Fig. 1).
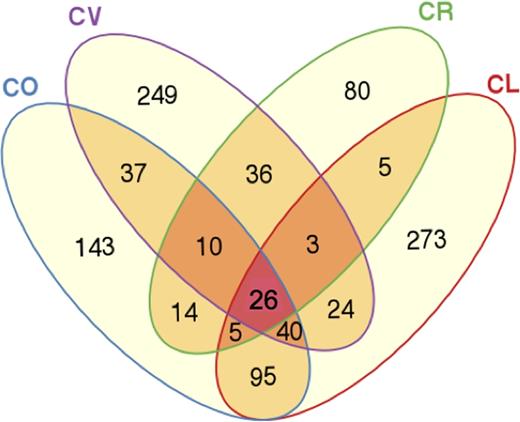
Venn diagram representing shared operational taxonomic units (OTUs) between fish species. CL, Chaetodonlunulatus (n = 5); CO, C. ornatissimus (n = 5); CR, C. reticulatus (n = 5); CV, C. vagabundus (n = 4).
Core microbiota (OTUs and their classification) of the four butterflyfish species (C. lunulatus, C. ornatissimus, C. reticulatus and C. vagabundus).
| OTUs | Phylum | Class | Family | Genus |
|---|---|---|---|---|
| 44, 68 | Corynebacteriaceae | Corynebacterium | ||
| 131 | Actinobacteria | Actinobacteria | Micrococcaceae | Micrococcus |
| 3 | Propionibacteriaceae | Propionibacterium | ||
| 114 | Cyanobacteria | Synechococcophycideae | Synechococcaceae | Synechococcus |
| 1, 25, 36, 38, | ||||
| 47, 219, 4467 | Firmicutes | Clostridia | ||
| 58, 208 | [Mogibacteriaceae] | |||
| 280 | Nautella | |||
| Alphaproteobacteria | Rhodobacteraceae | |||
| 71, 269 | Ruegeria | |||
| 854 | Betaproteobacteria | Burkholderiaceae | Ralstonia | |
| 696 | ||||
| 79 | Acinetobacter | |||
| 42 | Proteobacteria | Moraxellaceae | Acinetobacter | |
| 142 | Enhydrobacter | |||
| Gammaproteobacteria | ||||
| 67 | Salinisphaeraceae | |||
| 5 | Photobacterium | |||
| 0 | Vibrionaceae | Vibrio | ||
| 426 | Vibrio |
| OTUs | Phylum | Class | Family | Genus |
|---|---|---|---|---|
| 44, 68 | Corynebacteriaceae | Corynebacterium | ||
| 131 | Actinobacteria | Actinobacteria | Micrococcaceae | Micrococcus |
| 3 | Propionibacteriaceae | Propionibacterium | ||
| 114 | Cyanobacteria | Synechococcophycideae | Synechococcaceae | Synechococcus |
| 1, 25, 36, 38, | ||||
| 47, 219, 4467 | Firmicutes | Clostridia | ||
| 58, 208 | [Mogibacteriaceae] | |||
| 280 | Nautella | |||
| Alphaproteobacteria | Rhodobacteraceae | |||
| 71, 269 | Ruegeria | |||
| 854 | Betaproteobacteria | Burkholderiaceae | Ralstonia | |
| 696 | ||||
| 79 | Acinetobacter | |||
| 42 | Proteobacteria | Moraxellaceae | Acinetobacter | |
| 142 | Enhydrobacter | |||
| Gammaproteobacteria | ||||
| 67 | Salinisphaeraceae | |||
| 5 | Photobacterium | |||
| 0 | Vibrionaceae | Vibrio | ||
| 426 | Vibrio |
Square brackets indicate that nomenclature requires to be updated.
Core microbiota (OTUs and their classification) of the four butterflyfish species (C. lunulatus, C. ornatissimus, C. reticulatus and C. vagabundus).
| OTUs | Phylum | Class | Family | Genus |
|---|---|---|---|---|
| 44, 68 | Corynebacteriaceae | Corynebacterium | ||
| 131 | Actinobacteria | Actinobacteria | Micrococcaceae | Micrococcus |
| 3 | Propionibacteriaceae | Propionibacterium | ||
| 114 | Cyanobacteria | Synechococcophycideae | Synechococcaceae | Synechococcus |
| 1, 25, 36, 38, | ||||
| 47, 219, 4467 | Firmicutes | Clostridia | ||
| 58, 208 | [Mogibacteriaceae] | |||
| 280 | Nautella | |||
| Alphaproteobacteria | Rhodobacteraceae | |||
| 71, 269 | Ruegeria | |||
| 854 | Betaproteobacteria | Burkholderiaceae | Ralstonia | |
| 696 | ||||
| 79 | Acinetobacter | |||
| 42 | Proteobacteria | Moraxellaceae | Acinetobacter | |
| 142 | Enhydrobacter | |||
| Gammaproteobacteria | ||||
| 67 | Salinisphaeraceae | |||
| 5 | Photobacterium | |||
| 0 | Vibrionaceae | Vibrio | ||
| 426 | Vibrio |
| OTUs | Phylum | Class | Family | Genus |
|---|---|---|---|---|
| 44, 68 | Corynebacteriaceae | Corynebacterium | ||
| 131 | Actinobacteria | Actinobacteria | Micrococcaceae | Micrococcus |
| 3 | Propionibacteriaceae | Propionibacterium | ||
| 114 | Cyanobacteria | Synechococcophycideae | Synechococcaceae | Synechococcus |
| 1, 25, 36, 38, | ||||
| 47, 219, 4467 | Firmicutes | Clostridia | ||
| 58, 208 | [Mogibacteriaceae] | |||
| 280 | Nautella | |||
| Alphaproteobacteria | Rhodobacteraceae | |||
| 71, 269 | Ruegeria | |||
| 854 | Betaproteobacteria | Burkholderiaceae | Ralstonia | |
| 696 | ||||
| 79 | Acinetobacter | |||
| 42 | Proteobacteria | Moraxellaceae | Acinetobacter | |
| 142 | Enhydrobacter | |||
| Gammaproteobacteria | ||||
| 67 | Salinisphaeraceae | |||
| 5 | Photobacterium | |||
| 0 | Vibrionaceae | Vibrio | ||
| 426 | Vibrio |
Square brackets indicate that nomenclature requires to be updated.
PCoA based on weighted Unifrac distances (both at 97 and 98% OTU sequence identity cut-off) showed significant microbiota differences between the pairs C. lunulatus–C. ornatissimus and C. reticulatus–C. vagabundus (P < 0.05, Adonis test and pairwise comparisons; Fig. 2; Table 3).
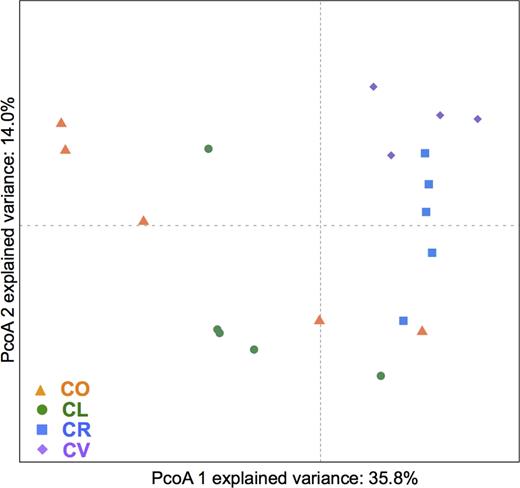
PCoA analyses of the gill mucus microbiota of four species of butterflyfish. CL, Chaetodonlunulatus (n = 5); CO, C. ornatissimus (n = 5); CR, C. reticulatus (n = 5); CV, C. vagabundus (n = 4); based on weighted Unifrac distances.
Pairwise P-values between the different PCoA groups (butterflyfish species) estimated using Adonis test and pairwise comparisons. *P < 0.05. CL, Chaetodon lunulatus; CO, C. ornatissimus; CR, C. reticulatus; CV, C. vagabundus.
| OUT sequence cut-off | Species | CL | CO | CR |
|---|---|---|---|---|
| 98% | CO | 0.406 | ||
| CR | 0.039* | 0.049* | ||
| CV | 0.038* | 0.039* | 0.067 | |
| 97% | CO | 0.245 | ||
| CR | 0.012* | 0.049* | ||
| CV | 0.052* | 0.060* | 0.324 |
| OUT sequence cut-off | Species | CL | CO | CR |
|---|---|---|---|---|
| 98% | CO | 0.406 | ||
| CR | 0.039* | 0.049* | ||
| CV | 0.038* | 0.039* | 0.067 | |
| 97% | CO | 0.245 | ||
| CR | 0.012* | 0.049* | ||
| CV | 0.052* | 0.060* | 0.324 |
Pairwise P-values between the different PCoA groups (butterflyfish species) estimated using Adonis test and pairwise comparisons. *P < 0.05. CL, Chaetodon lunulatus; CO, C. ornatissimus; CR, C. reticulatus; CV, C. vagabundus.
| OUT sequence cut-off | Species | CL | CO | CR |
|---|---|---|---|---|
| 98% | CO | 0.406 | ||
| CR | 0.039* | 0.049* | ||
| CV | 0.038* | 0.039* | 0.067 | |
| 97% | CO | 0.245 | ||
| CR | 0.012* | 0.049* | ||
| CV | 0.052* | 0.060* | 0.324 |
| OUT sequence cut-off | Species | CL | CO | CR |
|---|---|---|---|---|
| 98% | CO | 0.406 | ||
| CR | 0.039* | 0.049* | ||
| CV | 0.038* | 0.039* | 0.067 | |
| 97% | CO | 0.245 | ||
| CR | 0.012* | 0.049* | ||
| CV | 0.052* | 0.060* | 0.324 |
Composition of bacterial communities
We are fully aware that the percentage of OTUs does not exactly represent the percentage of cellular abundances, and thus hereafter, abundances will refer to the percentage of total sequence reads in the final OTU table. The number of sequences in unidentified OTUs (unclassified bacterial OTUs) was higher at 97% OTU cut-off (12.2 ± 9.0%) than at 98% cut-off (3.2 ± 4.1%). Upon further examination, this was the case since in this type of analysis a single sequence (i.e. the OTU) represents an entire cluster of sequences. Thus, some sequences representing OTUs constructed at 97% (selected since they were the longest sequence in these OTUs) were not classifiable by the rdp_classifier (and therefore all sequences in the OTU were labelled as unclassified). At 98% cut-off these longer unclassifiable sequences were placed in separate OTUs with fewer sequences, and shorter, more abundant classifiable sequences were placed in other OTUs, increasing the percentage of identifiable sequences.
Proteobacteria was the most abundant phylum in the four butterflyfish species (Fig. 3). Gammaproteobacteria dominated the microbial assemblage of C. lunulatus and C. ornatissimus (90% and 80% of the total Proteobacteria sequences), while Alphaproteobacteria was more abundant in C. vagabundus (59% of total Proteobacteria sequences). Chaetodonreticulatus, whose community presented the highest abundance of Proteobacteria (67% of total sequences), had 41% of Alphaproteobacteria, 19% of Betaproteobacteria and 39% of Gammaproteobacteria. In addition to Proteobacteria, only Actinobacteria and Firmicutes were present in all samples of butterflyfish gill mucus, being particularly well represented in C. vagabundus, C. reticulatus and C. ornatissimus. Chaetodonlunulatus and C. ornatissimus presented a higher abundance of Bacteroidetes (11% and 17% respectively). Chaetodonlunulatus also presented a notably higher abundance of Fusobacteria (4%), which was nearly absent in the other fish species (Fig. 3).
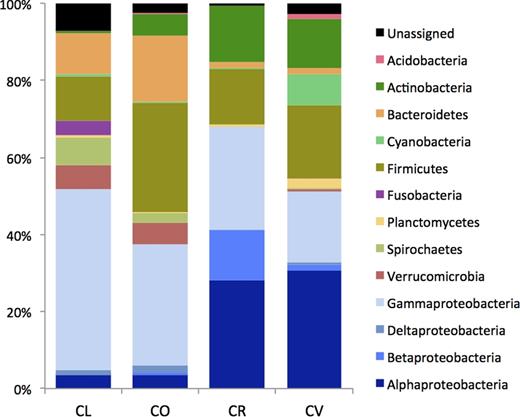
Bacterial diversity at the phylum level and class level (only Proteobacteria) determined by 454 pyrosequencing. CL, Chaetodonlunulatus; CO, C. ornatissimus; CR, C. reticulatus; CV, C. vagabundus.
At the family level, we saw consistent associations between some bacterial families and Chaetodon species, but also high inter-individual variation (Fig. 4). As expected from the PCoA analysis, dominant families were most similar between C. lunulatus and C. ornatissimus, and C. reticulatus and C. vagabundus.

Abundant taxaof the gill mucus microbiome: (a) family and (b) genus. Only taxa with abundance ≥5% at least in two of the samples are plotted. CL, Chaetodonlunulatus; CO, C. ornatissimus; CR, C. reticulatus; CV, C. vagabundus.
Associations of the most abundant taxa with C. lunulatus and C. ornatissimus includethe following: (i) Vibrionaceae (mostly Vibrio spp.), generally the most abundant microbial taxon in most C. lunulatus samples (between 25 and 90% of total sequences in four samples, and 3% in the remaining sample) and C. ornatissimus (between 1 and 41%); (ii) Verrucomicrobiaceae (mostly Akkermansia spp.) with abundances up to 15%; (iii) Hahellaceae (mostly Endozoicomonas spp.), which reached an abundance of 25% in one individual, while absent in C. reticulatus samples; (iv) Spirochaetaceae (Spirochaeta spp.) with abundances ranging from 1 to 10% in these species, and rarely present in C. vagabundus and totally absent in C.reticulatus; and (v) Ruminococcaceae (genus undetermined), which represented around 5% of the total bacterial assemblage of C. lunulatus and C. ornatissimus, but were absent in C. reticulatus (Fig. 4).
Associations of the most abundant taxa with C. reticulatus and C. vagabundus includethe following: (i) Moraxellaceae (mainly Acinetobacter spp.); (ii) Propionibacteriaceae (mostly Propionibacterium spp.) with abundances between 2 and 8% in C. reticulatus and between 0.5 and 16% in C. vagabundus, and 13% in one C. ornatissimus sample; and (iii) Corynebacteriaceae (genus Corynebacterium), which represented around 10% of the microbiota in these species while found to be nearly absent in most C. lunulatus and C. ornatissimus, but for one sample of C. ornatissimus (11%) (Fig. 4).
Many bacterial families appeared to have been particularly associated with a single Chaetodon species. Anaplasmataceae were abundant (33 and 70%) in two samples of C. reticulatus, and were mainly constituted of OTUs belonging to the Neorickettsia genus (Fig. 4). Burkholderiaceae (genus Ralstonia) were also significantly more frequent in C. reticulatus (up to 25%, P < 0.01) than in the other fish species. Rhodobacteraceae were present in all fish species, but were particularly abundant in C. vagabundus, where they reached abundances up to 32%. Similarly, ‘Exiguobacteraceae’, mainly the Exiguobacterium genus, were significantly more abundant in C. vagabundus (P < 0.05), displaying abundances lower than 1% in the other fish species. Finally, Fusobacteriaceae (genus undetermined) were significantly more abundant in in C. lunulatus (0.4–7.1%, P < 0.05), while they were nearly absent in the other three fish species.
DISCUSSION
Gills are organs essential to fish well-being and their associated microbiota play an essential role in gill functioning, and thus information on the bacterial communities of gills is of great importance to understanding gill homeostasis and fish health (Lowrey et al. 2015; Tarnecki, Patterson and Arias 2016; Xu et al. 2016). Fish gill mucus is in constant contact with the environment, providing a habitat for heterotrophic bacteria that is rich in nutrients when compared with the surrounding oligotrophic coral reef waters. In this study we characterised and compared the gill mucus bacterial communities of four sympatric coral reef fish species of Moorea (French Polynesia) using tag 454-pyrosequencing. We compared two different sequence cut-offs (97 and 98%) for OTU building, which resulted in very similar alpha diversity values. We chose 98% identity for OTU building since the method (AmpliconNoise) and parameters for denoising allowed the use of this sequence cut-off, and since we also consider that it better represents a sequence cut-off for an ecologically coherent group of organisms, in particular for genera where 16S rRNA gene similarity is high among its members (ex. Vibrio sp.).
Although the bacterial diversity of gill mucus of butterflyfishes varied significantly between species, Shannon diversities were consistently high (H = 4.7–5.6, 26 phyla, with the exception of C. reticulatus, which displayed a significantly lower diversity, H = 3.7) when compared with gill microbiome of cultured rainbow trout (Oncorhynchus mykiss) (H < 4.5, 11 phyla; Lowrey et al. 2015), gut microbiota of surgeonfishes (H = 0.5–3.9; Miyake, Ngugi and Stingl 2015), tropical sponges (H = 1.6–4.3; Moitinho‐Silva et al. 2014), coral reef waters (H = 0.6–3.2) and coral reef sediments (H = 0.8; Glasl, Herndl and Frade 2016). Nonetheless regarding the difference from rainbow trout, it is expected that the microbiome of cultured fish would be less diverse than wild fish, which have higher genetic variability as well as higher dietary plasticity (Givens et al. 2015). Furthermore, higher bacterial diversity in fish external surfaces (skin, gills) was already observed by Lowrey et al. 2015, who suggested that external fish surfaces could reflect the environmental diversity, while the gut may offer more stable habitats for specialised microbial communities. Bacterial diversity of coral species (H = 0.8–6.1) is highly variable depending on the studied species and the geographic location, but some values are similar to those found in butterflyfish gill mucus (e.g. Kimes et al. 2010; McKew et al. 2012; Morrow et al. 2012; Bayer et al. 2013; Glasl, Herndl and Frade 2016).
Chaetodonvagabundus presented the most diverse bacterial community (Shannon's diversity, which takes into account how evenly the OTUs are distributed). This species has also the most varied diet of the four butterflyfish species studied, consuming a wide variety of prey items (Harmelin-Vivien 1988). Different dietary patterns are associated with distinct combinations of bacteria in the digestive tracts of humans and fish (Wu et al. 2011; Sun et al. 2013; Liu et al. 2016), and it seems that more diverse diets can lead to a more diverse microbiome (Heiman and Greenway 2016). Although the effect of diet on external surfaces (e.g. skin, gills) of organisms is much less understood, some studies indicate that the metabolic reactions induced by different dietary intakes might influence bacterial communities of fish external surfaces (Landeira-Dabarca, Sieiro and Alvarez 2013; Schommer and Gallo 2013). Chaetodonlunulatus presented the highest number of unique OTUs, and the highest Chao number, indicating the presence of a large number of rare OTUs. It is an obligate generalist corallivore, like C. ornatissimus, being able to feed on a wide diversity (up to 51 species) of scleractinian corals (Pratchett 2005, 2007). Furthermore, C. lunulatus is the only butterflyfish species that is not parasitised by monogenean gill parasites (Reverter et al. 2016), a particularity that might also be related to the diversity and composition of C. lunulatus gill bacterial communities. The lowest bacterial diversity was observed in C. reticulatus, which is a strict corallivore fish that feeds mostly on Acropora corals (Harmelin-Vivien 1988; Berumen and Pratchett 2006).
Although all fish species were collected at the same sampling site, they presented significant microbiome differences. Butterflyfishes are mostly territorial species (Roberts and Ormond 1992), and therefore a difference in composition of their bacterial communities must be related to the host-specific characteristics of each species. While we saw high inter-individual variation, by comparing at least four individual microbiomes per species, we were able to highlight some general trends regarding the Chaetodon bacterial communities. Chaetodonlunulatus and C. ornatissimus were clearly the two species with the most similar bacterial communities. These two species are the closest phylogenetic relatives and they are both obligate corallivores (Littlewood et al. 2004; Pratchett 2005; Fessler and Westneat 2007). In contrast, C. vagabundus is an omnivorous species that belongs to a different phylogenetic clade (Pratchett 2005; Fessler and Westneat 2007). Therefore, a similarity between C. lunulatus and C. ornatissimus gill bacterial communities is not unexpected and these results suggest that gill mucus microbiome of fishes might be influenced by phylogeny and/or ecological characteristics of the host species. On the other hand, we cannot explain why C. reticulatus, which is ecologically and phylogenetically closer to C. lunulatus and C. ornatissimus, presents a bacterial community that is more similar to that of C. vagabundus. Overall, our results show that gill mucus microbiota is somewhat host-specific, but more studies should assess temporal and spatial variability of gill mucus microbiome to confirm the associations presented here.
The bacterial communities associated to the gill mucus of the four butterflyfish species were dominated by Proteobacteria, which is in accordance with previous studies on the gill, skin and gut of several fish species (Sullam et al. 2012; Larsen et al. 2015; Lowrey et al. 2015; Tarnecki, Patterson and Arias 2016). Studies on the coral microbiome have also revealed a high relative abundance of Protebacteria, but it has been observed that mucus of different coral species enriches for different bacterial communities (McKew et al. 2012). Gammaprotebacteria represented over 80% of the total Proteobacteria in C. lunulatus and C. ornatissimus gill mucus, which was mainly represented by families Vibrionaceae, Pseudomonadaceae and Hahellaceae. In a recent study, Vibrio was identified as the most common bacterial genus in the gills of red snapper (Lutjanus campechanus) (Tarnecki, Patterson and Arias 2016). Vibrio species are widespread in the marine environment and are also known to be associated with coral mucus and tissues and in some cases they seem responsible for coral disease (reviewed in Rosenberg et al2007). Unfortunately, it is very difficult if not impossible to discriminate between Vibrio species based on 16S rRNA sequences alone, so we prefer not to speculate on the origin or the role of the OTUs associated with C. lunulatus and C. ornatissimus gill mucus (Sawabe et al. 2013).
Chaetodonreticulatus and C. vagabundus had a much higher proportion of Alphaproteobacteria, that corresponded mostly to higher abundances in Rhodobacteraceae (mainly Paracoccus) and remarkable abundances of Neorickettsia in two samples of C. reticulatus. Some Rhodobacteraceae are known coral pathogens that have been found in higher abundances in diseased corals (Roder et al. 2014). Paracoccus strains have been previously identified in fish skin, fish gut and corals (Sheu et al. 2011; Liu et al. 2013; Larsen et al. 2015). Neorickettsia species are normally intracellular pathogens that cause severe illnesses in mammals and are transmitted by flukes (Platyhelminthes: Digenea) that infect fish (Vaughan, Tkach and Greiman 2012). A potential new species of Neorickettsia has been identified as species specific from skin microbiota of striped mullet (Mugil cephalus), but their role and importance in fish microbiota and coral reefs remains unknown (Larsen et al. 2015).
Actinobacteria and Firmicutes, which seem to constitute the fish core microbiota along with Proteobacteria, were abundant in gill mucus of all fish species, especially in C. reticulatus and C. vagabundus. Interestingly, 10% of the microbial communities of C. vagabundus and C. reticulatus were composed of Corynebacterium, a genus containing pathogenic species that was found to increase in gill mucus of clownfish exposed to high concentrations of suspended sediment (Hess et al. 2015) and was highlighted as a potential pathogen involved in coral disease (Sweet et al. 2013).
Verrucomicrobia species, which were abundant in C. lunulatus and C. ornatissimus (1–15%, mainly an Akkermansia unidentified species) have been found in low abundances in different fish tissues (gills, gut and skin, <1%) and coral tissues (1–3%) (Kooperman et al. 2007; Chiarello et al. 2015; Miyake, Ngugi and Stingl 2015; Lawler et al. 2016; Tarnecki, Patterson and Arias 2016). Verrucomicrobia are ubiquitous (although rarely in very high proportions) in the marine environment, both in the water column and in sediment, but little is known of the functional role they might play in association with organisms such as fish (Freitas et al. 2012). In a recent study, Glasl, Herndl and Frade (2016) showed that Verrucomicrobia abundances increased in corals where the microbiome had been disrupted, along with other opportunistic pathogens of the family Vibrionaceae, indicating the potential pathogenicity of Verrucomicrobia to corals.
Gill mucus of butterflyfishes was also found to host several coral-associated bacteria. Endozoicomonas species are often found as part of the coral holobiont or in close association to other marine invertebrates (e.g. bivalves, sea slugs; Kurahashi and Yokota 2007; Bayer et al. 2013; Hyun et al. 2014). We also found remarkably high abundances of Propionibacterium and Ralstonia mostly in C. reticulatus and C. vagabundus, which have been described as being closely associated to coral dinoflagellate symbionts but presenting very low abundances, and being undetectable on water surrounding corals (Ainsworth et al. 2015).
The nearly specific presence of Fusobacteria in C. lunulatus was also remarkable. Fusobacteria are anaerobic bacteria found in large quantities in fish gut (Clements et al. 2014), and to the best of our knowledge this is the first record of Fusobacteria in external surfaces of fish. Fusobacteria are known to produce a short-chain fatty acid, butyrate, which is the end-product of fermentation of carbohydrates including those found in mucins (Bennett and Eley 1993). In mammals, butyrate provides many benefits to the host, enhancing mucus production and acting as an anti-carcinogen and anti-inflammatory (von Engelhardt et al. 1998; Andoh, Bamba and Sasaki 1999). Some Fusobacteria species close to those found in C. lunulatus, such as Cetobacterium somerae, are also known to produce abundant amounts of vitamin B12 (Merrifield and Rodiles 2015). Although the role of Fusobacteria and butyrate are poorly understood in fish external surfaces such as gills, the previous studies might indicate that they could display protective roles in fish gills.
In conclusion, we found a high bacterial diversity in gill mucus of butterflyfishes, many species of which are also found in the coral holobiont, either as pathogens or coral-associated bacteria. Mucus layers are nutrient hotspots for marine heterotrophic bacteria living in oligotrophic environments such as coral reefs. Therefore, we hypothesise that external fish mucus surfaces could act as a reservoir for coral reef bacterial diversity. Our study shows that different butterflyfish species possess different microbiomes, indicating the presence of species specificities in some bacterial OTUs that might arise from bacterial–host coevolution and ecological parameters. However, there is a part of the bacterial community (18–35%) that is common in the four fish species studied. Bacterial chemotaxis has been shown in coral-associated bacteria (e.g. Endozoicomonaceae, Rhodobacteraceae and Vibrionaceae) in response to coral mucus amino acids (Tout et al. 2015). Fish mucus is known to contain a wide array of proteins, including the same amino acids found in coral mucus (Valdenegro-Vega et al. 2014). Pathogenic bacterial chemotaxis towards fish mucus has been observed before (e.g. Bordas et al. 1998; Larsen, Larsen and Olsen 2001), and therefore it would be interesting to investigate whether naturally occurring coral reef bacteria might also display chemotaxis towards fish mucus, and whether there might be a bacterial transfer between sympatric organisms. Although this study is focused on a specific coral reef fish family, the Chaetodontidae, these results cast light on the bacterial communities of fish gills, bringing new insights on the possible relationship between fish microbiota, fish ecology and the surrounding natural environment.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSEC online.
Acknowledgments
The authors would like to acknowledge Christine Sidobre for her assistance in DNA extraction and Sonja Fagervold for her suggestions for the manuscript.
FUNDING
This work was supported by the labex Corail project (MECANO, to Pierre Sasal). This research is part of an EPHE thesis supported by a labex ‘Corail‘ doctoral grant awarded to Miriam Reverter.
Conflict of interest. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}