





Abstract
Arsenic concentrations (450–600 μmol L−1) at the El Tatio Geyser Field in northern Chile are an order of magnitude greater than at other natural geothermal sites, making El Tatio an ideal location to investigate unique microbial diversity and metabolisms associated with the arsenic cycle in low sulfide, > 50 °C, and circumneutral pH waters. 16S rRNA gene and arsenite oxidase gene (aioA) diversities were evaluated from biofilms and microbial mats from two geyser-discharge stream transects. Chloroflexi was the most prevalent bacterial phylum at flow distances where arsenite was converted to arsenate, corresponding to roughly 60 °C. Among aioA-like gene sequences retrieved, most had homology to whole genomes of Chloroflexus aurantiacus, but others were homologous to alphaproteobacterial and undifferentiated beta- and gammaproteobacterial groups. No Deinococci, Thermus, Aquificales, or Chlorobi aioA-like genes were retrieved. The functional importance of amino acid sites was evaluated from evolutionary trace analyses of all retrieved aioA genes. Fifteen conserved residue sites identified across all phylogenetic groups highlight a conserved functional core, while six divergent sites demonstrate potential differences in electron transfer modes. This research expands the known distribution and diversity of arsenite oxidation in natural geothermal settings, and provides information about the evolutionary history of microbe–arsenic interactions.
Introduction
Microbes contribute to arsenic metalloid speciation and mobility through several energy conservation reactions and detoxification mechanisms (Bentley & Chasteen, 2002; Oremland et al., 2004; Silver & Phung, 2005; Fisher et al., 2008; Ledbetter & Magnuson, 2010; Slyemi & Bonnefoy, 2011). There have been a number of studies of microbe–arsenic geochemistry and geomicrobiology from diverse environments, including soils, mine waters, geothermal fluids, wastewater, and animal feed lots (Oremland & Stolz, 2003). Most of what is known about microbe–arsenic geomicrobiology has resulted from studies of continental geothermal, sulfidic fluids, or groundwater at temperatures < 50 °C and at extreme pH conditions (Ballantyne & Moore, 1988; Langner et al., 2001; Macur et al., 2004; D'Imperio et al., 2007; Inskeep et al., 2007; Cai et al., 2009; Sultana et al., 2012). One study of Alvord Hot Spring in Oregon (USA), discharging waters with low total arsenic concentrations averaging 60 μmol L−1, low sulfide, > 70 °C, and circumneutral pH waters, still described a microbial community associated with cycling sulfur compounds and diversity similar to sulfidic, high arsenic geothermal sites in Yellowstone National Park (USA; Connon et al., 2008). Microbial arsenic cycling from nonsulfidic, circumneutral pH fluids at temperatures above 50 °C is not well understood (Langner et al., 2001; Webster & Nordstrom, 2003). Such investigations are important because sulfide and sulfite, both substrates used for reduced sulfur oxidation, have been found to inhibit enzymatic arsenite oxidation (Lieutaud et al., 2010).
An order of magnitude greater arsenic concentrations (450–600 μmol L−1) than Alvord Hot Spring has been reported from El Tatio Geyser Field (ETGF), located at an elevation of c. 4300 m in the Altiplano-Puna Volcanic Complex in the Andean Cordillera Region II, Chile (Cusicanqui et al., 1975; Lahsen & Trujillo, 1975; Glennon & Pfaff, 2003; Fernandez-Turiel et al., 2005; Dunckel et al., 2009; Landrum et al., 2009). Arsenic contamination in the region has been known about for decades (Romero et al., 2003). Most of the > 100 nonsulfidic hot springs, pools, mud pots, and geysers have pH values from 6.8 to 7.2 at or near local boiling (86 °C at 460 mm Hg atmospheric pressure), and low dissolved sulfate (Landrum et al., 2009). Detailed physicochemistry and site descriptions have been reported by Landrum et al. (2009). Colorful microbial mats and biofilms occupy nearly all of the geyser features and stream channels at ETGF (Fig. 1). The geothermal fluids discharge arsenite [As(III)] that progressively oxidizes to arsenate [As(V)] downstream. The speciation of As in solution is affected by pH, temperature, evaporation, and a variety of removal and transformation mechanisms, including sorption, photooxidation of As(III) to As(V), and/or microbial metabolism. Compared to other geothermal sites, such as those from Yellowstone that are characterized as having acid-chloride-sulfate waters (Inskeep et al., 2007), the distinctive geochemistry of ETGF was expected to yield unique information regarding the diversity of microbes associated with microbial arsenic metabolism, and specifically As(III) oxidation. Because As(V) is less toxic, less mobile, and more strongly partitions (i.e. sorbs) to organic matter and ferric oxyhydroxides that can remove As from fluids, it is important to understand the nature and behavior of arsenic in as many sites as possible.

(a) Tower geyser-discharge stream at ETGF, Chile. (b) Close-up of orange, filamentous microbial mats at the c. 30 m sampling location. Tips of tweezers approximately 1 cm apart for scale.
Therefore, to enhance our understanding of As metabolism, the current study aimed to compare the bacterial diversity of 16S rRNA gene sequences with sequences of the large-subunit of the arsenite oxidase structural gene (aioA, based on nomenclature recently proposed by Lett et al., 2012) from microbial mats at ETGF. To assess the potential functional importance of significant residue variations among amino acid sites within the AioA protein, we performed evolutionary trace (ET) analyses to identify phylogenetically significant divergent residues from the retrieved aioA gene diversity at ETGF; we then mapped these residues onto the protein crystal structure from Alcaligenes faecalis (Ellis et al., 2001) to evaluate potential functional diversity. Previous research has focused on the phylogenetic diversity of aioA genes, or the functional variations in amino acid residues from cultured strains, but ET analyses of aioA gene functional diversity from a natural setting, from microbial groups that occupy the same general habitat, have not been done. As such, this research expands the known distribution and diversity of arsenite-oxidizing bacteria in natural geothermal settings, as well as provides insight into microbe–arsenic interactions.
Materials and methods
Sample acquisition and DNA isolation
Aqueous geochemistry methods for field and laboratory (e.g. for arsenic concentration determination) measurements were described in Landrum et al. (2009). Two transects are used in this study, a geyser feature and stream in the Lower Basin and another geyser feature and stream at the Tower Geyser in the Middle Basin (Fig. 1). Microbial mats and geyser sediment samples were collected separately, and each sample was placed in sterile lysis buffer and kept as close to 4 °C as possible after collection and during transport.
Total environmental DNA was extracted from microbial mat and sediment samples using the Qiagen DNeasy extraction kit, following manufacturer's instructions (Qiagen Inc., Valencia, CA) within 1 week of collection. Each sample was aseptically homogenized in tissue lysis buffer with a tissue homogenizer, supplemented with proteinase K, and then agitated on a shaker table at 225 r.p.m. at 55 °C overnight. Nucleic acids were precipitated in ethanol and eluted into TE buffer. Samples were stored at −20 °C until analysis. The quality and quantity of DNA were determined using a ThermoScientific NanoDrop spectrophotometer. At least three DNA extractions per sample location were combined for each sample.
16S rRNA gene amplification
Bacterial 16S rRNA gene sequences were PCR-amplified multiple times from each composite sample with the 16S rRNA gene primers 8F (5′-AGAGTTTG ATCCTGGCTCAG-3′) and 1510R (5′-GGTTACCTTGTTACGACTT-3′; Ausubel et al., 1990). PCR was performed on a MJ Research Dyad Disciple thermal cycler with 1 U Taq DNA polymerase (5 PRIME, Inc., Gaithersburg, MD), 1× PCR buffer, 10 mg mL−1 bovine serum albumin (BSA), 10 mM dNTPs, 1 mM MgCl2, and c. 50 ng environmental DNA in a 50 μL final volume. PCR conditions for 16S rRNA genes included an initial hot start at 94 °C for 4 min, denaturation at 94 °C for 1 min, primer annealing at 47 °C for 1 min, and chain extension at 72 °C for 3 min; these steps were repeated 29 times with a final extension step at 72 °C for 20 min.
aioA gene amplification
There had been considerable confusion in the literature regarding the naming of the arsenite oxidase proteins and genes. Recently, Lett et al. (2012) propose a new nomenclature to circumvent the confusion based on the fact that the structures of all currently known arsenite oxidases are similar and have two subunits (Ellis et al., 2001; Silver & Phung, 2005). The smaller Rieske [2Fe–2S] type subunit is encoded by the gene aioB (previously referred to in the literature as aoxA, aroB, or asoB genes), and the larger subunit gene aioA (previously referred to as aoxB, aroA, or asoA) contains a molybdenum-bis (pyranopterin guanine dinucleotide) (Mo-bisPGD) cofactor and [3Fe–4S] cluster. AioA is associated with the dimethylsulfoxide reductase enzyme family (Ellis et al., 2001; Lebrun et al., 2003; Santini & vanden Hoven, 2004; Silver & Phung, 2005; Inskeep et al., 2007; Duval et al., 2010; van Lis et al., 2012a). These Aio genes are distinct from the respiratory arsenate reductase or molybdenum oxidoreductases, like the arsenite reductase (ArxA), genes found from the nitrate-reducing arsenite-oxidizing Alkalilimnicola ehrlichii (Hoeft et al., 2007; Zargar et al., 2010).
In this study, microbial groups that could contribute to As(III) oxidation, based on the presence of aioA genes, from the microbial mats at Tower Geyser in the Middle Basin were examined using ‘aroA’ primers from Inskeep et al. (2007), aroA#1F (5′-GTSGGBTGYGGMTAYCABGYCTA-3′) and aroA#1R (5′-TTGTASGCBGGNCGRTTRTGRAT-3′). PCR amplifications were performed using a MJ Research Dyad Disciple thermal cycler (Biorad) with 5 U μL−1 Taq DNA polymerase (5 PRIME, ThermoFisher Scientific), 10 mg mL−1 BSA (Fisher Scientific), 10 mM dNTPs, 1 mM MgCl2, and c. 45 ng μL−1 environmental DNA. The concentration of each primer was 1 μM. PCR conditions included a hot start at 95 °C for 4 min, nine cycles of 95 °C for 45 s, 50 °C (decreased by 0.5 °C after each cycle) for 45 s, 46 °C for 45 s, 72 °C for 50 s, and a final extension step at 72 °C for 5 min (Inskeep et al., 2007).
Clone library construction and Sanger sequencing
PCR products of the correct size (c. 1500 base pairs for 16S rRNA genes and c. 600 base pairs for aioA genes) were gel-purified using the Wizard PCR prep DNA purification kit (Promega Corp.), following manufacturer's instructions.
Purified PCR products were cloned using the TOPO Cloning Kit with the PCR2.1-TOPO® vector, following manufacturer's instructions (Invitrogen Corp.). Resulting clones were PCR-amplified using manufacturer primers. The correct sized inserts were sequenced in both directions by capillary Sanger sequencing at the High-throughput Genomics Unit, University of Washington (USA). To obtain complete coverage of 16S rRNA gene sequences, sequencing was done with the internal primers 704F (5′-GTAGCGGTGAAATGCGTAGA-3′) and 907R (5′-CCGTCAATTCCTTTRAGTTT-3′).
Sequence analyses and phylogenies
Full sequences were assembled using Contig Express, a component of Vector NTI Advance 10.3.0 (Invitrogen Corp.). For the 16S rRNA gene sequences, sequences were screened for chimera using greengenes (DeSantis et al., 2006) and aligned using MAFFT (Katoh et al., 2002). A Phylip distance matrix was constructed in mobyle 0.96 (Néron et al., 2009) for the evaluation of distance-based operational taxonomic units (OTUs) defined by dotur (Schloss & Handelsman, 2005) at the 97% sequence identity level.
The aioA-like genes were translated to amino acid sequences and aligned with homologues obtained from GenBank using clustalw (Thompson et al., 1994) in mobyle. A phylogenetic tree was reconstructed using phyml (Guindon & Gascuel, 2003) under the LG + I + G amino acid substitution model (G = 1.276, I = 0.007) determined from ProtTest (Abascal et al., 2005).
Evolutionary trace analyses and protein modeling
Both conserved and functionally divergent amino acid residues within known arsenite oxidase proteins were investigated using ET analysis (Lichtarge et al., 1996), one of the most validated approaches currently available for identifying functional sites within a protein (Wilkens et al., 2012). The ET method identifies functionally important amino acid residues by analyzing evolutionary patterns of sequence variations and then ranking every residue by relative importance. We used ET to evaluate the evolution of functional diversification of arsenite oxidase in two steps. First, we used ET report_maker (Mihalek et al., 2006) to investigate the function of important residues using existing arsenite oxidase sequences. Second, we combined previously characterized arsenite oxidase genes with our new sequences isolated from the ETGF mats and used the ET Viewer (Morgan et al., 2006) to run predictions of functional sites in arsenite oxidase based on our alignments. Using these two methods, we identified amino acid sites that are functionally important across all arsenite oxidase genes, as well as within major phylogenetic groups. The structural and functional impacts of residues identified by ET analyses were evaluated by mapping them onto the crystal structure of arsenite oxidase from A. faecalis (structure 1g8k, 1.64 Å; Ellis et al., 2001) using swiss-pdb viewer (http://spdbv.vital-it.ch) (Guex & Peitsch, 1997). Herein, all amino acid numbers were referred to as the site number in the A. faecalis protein.
Nucleotide sequence accession numbers
The nucleotide sequences reported have been deposited in the GenBank database under accession numbers GU437233–GU437738 (cloned 16S rRNA genes) and GU437739–GU437822 (cloned aioA-like sequences).
Results
16S rRNA gene sequence diversity
On the basis of field methods described by Landrum et al. (2009), waters from the Lower Basin were dominated by Na and Cl ions (i.e. Na–Cl type) and had specific conductance values between 16.9 and c. 23 mS cm−1, depending on geyser eruption cycles. pH values ranged from 7.03 to 7.47 depending on temperature, which decreased from the spring orifice downstream, and location along the stream channel. Microbial mat samples were collected 11 m apart from one another. Water from the Tower Geyser was also Na–Cl type, with specific conductance values of 22–23 mS cm−1. pH values were between 6.9 and 7.2, depending on temperature and distance along the outflow channel. Microbial mat samples were taken from 7, 30, and 60 m downstream along the channel, generally corresponding to the temperature profile from the Lower Basin geyser feature. At 60 m, two samples were taken, one of a hydrated mat and another of a dry mat. Total arsenic concentrations for the Lower Basin geyser feature ranged from 354 to 391 μM, whereas concentrations at the Tower Geyser were c. 400 μM, with slightly higher total concentrations downstream owing to evaporation. Along the streams, the concentration of As(III) decreased while the concentration of As(V) increased (Landrum et al., 2009).
Microbial diversity was determined from surveys of 16S rRNA gene sequences. A total of 556 clones were retrieved from six samples (Fig. 2). The majority of retrieved sequences from all of the samples were affiliated with the Chloroflexi (34.7%; Fig. 2), with additional representation from the phyla Proteobacteria (23.6%) and Deinococcus-Thermus (18.8%). Within the Proteobacteria,Alphaproteobacteria was the dominant class (13.9%), followed by Gammaproteobacteria (5.4%), Betaproteobacteria (3.3%), and the Deltaproteobacteria (1.0%). Other phyla included the Bacteroidetes/Chlorobi (10.3%), Firmicutes (3.8%), Cyanobacteria (1.5%), Candidate Division groups (3.8%), Planctomycetes (1.5%), Acidobacteria (1.3%), Verrucomicrobia (0.4%), and Deferribacteres (0.2%). Despite previous isolation of Hydrogenobacter sp. 153II-6 and Sulfuryhydrogenibium str. 153IV-9 (Aquificales) from ETGF (Ferrera et al., 2007), no clones affiliated with this phylum were retrieved in the current study. The TAT-08-009-43 library was of dried microbial mats out of the active stream flow and was dominated by Alphaproteobacteria and Bacteroidetes, which was distinct from the rest of the hydrated microbial mat samples (Fig. 2; Supporting information, Table S1).
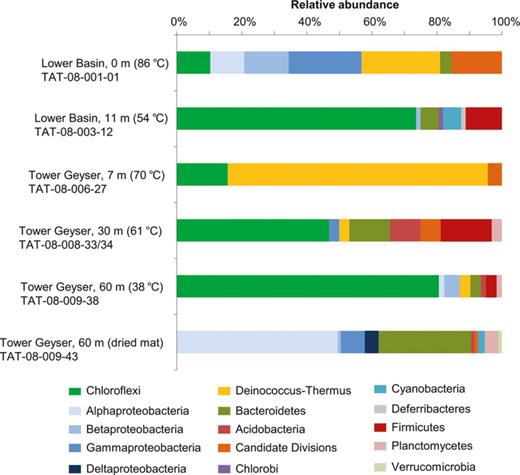
Distribution of major taxonomic groups based on 16S rRNA gene sequence clones, originating from microbial mats in two geyser and discharge stream transects at ETGF, Chile. For each transect, the distances from the geyser feature and water temperatures are noted.
From all of the sequences, there were 137 OTUs defined at 97% sequence identity (corresponding to genus-level affinities; Table S1). OTU groups were comprised of clones retrieved from different stream transects that had similar temperatures; for example, one OTU consisted of clones from the Tower Geyser 61 °C clone library and the 67 °C Lower Basin library (Table S1). Four OTUs formed from three libraries. Three were affiliated with Chloroflexi and one with the Candidate Division ‘Toll’ identified from the Coffee Pots Hot Spring in Yellowstone National Park (USA), clone pCOF_65.7 Toll (accession no. EU156156; Cron et al., 2008).
Most of the ETGF sequences closely related to Chloroflexus spp. were from samples < 70 °C (Table S1; Fig. 2) and were closely related (97–100% sequence identity) to Chloroflexus sp. Y-400-fl (accession no. CP001364; syn. Chloroflexus aurantiacus ATCC 29366), a facultative aerobe that is also capable of anoxygenic photosynthesis. In all, sequences affiliated with only two of the five classes within the Chloroflexi phylum, Chloroflexi and Caldilineae, were retrieved, but sequences related to Caldilinea aerophila (97–98% sequence identity) were generally rare (Table S1). Also among the Chloroflexi class, clones related to Roseiflexus sp. RS-1 (accession no. CP000686; 94–97% sequence identity) were retrieved. This organism does not have chlorosomes, which distinguishes it from C. aurantiacus (Hanada & Pierson, 2006).
A temperature-related distribution, similar to Chloroflexi OTUs, was also evident for OTUs affiliated with Deinococcus-Thermus. Almost all of the retrieved sequences affiliated with this phylum were from the most upstream, hottest samples (≥ 80 °C) in both transects (Table S1; Fig. 2). The two largest OTUs within the Deinococcus-Thermus phylum from the Lower Basin and the Tower Geyser transects were moderately related to clone pCOF_65.7_N (accession no. EU156154) from Coffee Pots Hot Spring, Yellowstone National Park (93–94% sequence similarity; Cron et al., 2008) and clone A3UB-E8 (accession no. DQ645240) from Alvord Hot Spring in Oregon (94–95% sequence similarity; Connon et al., 2008), respectively. Less than a quarter of the sequences affiliated with this phylum, most being represented by single sequences, were related to Thermus spp., ranging 87–95% sequence identity to Thermus isolate W28 (accession no. L10068) and 94–95% sequence similarity to Thermus isolate Tok20A.1 (accession no. L09665) (Table S1).
Some OTUs were represented by singleton clone sequences, being affiliated with the Proteobacteria, Bacteroidetes,Firmicutes, and other major phyla (e.g. Deferribacteres, Plantomycetes, and Verrucomicrobia; Table S1). Excluding the dry mat sample from the Tower Geyser transect where Alphaproteobacteria comprised almost 50% of the clone library and were related (92–99% sequence identity) to iodide-oxidizing bacteria and the halotolerant anoxygenic phototrophs, Roseovarius spp. (Table S1), there were several OTUs related to Alphaproteobacteria from the Lower Basin transect, but rarely for the Tower Geyser transect. Of the Bacteroidetes, two OTUs, comprised of clones from two libraries each, were related to Rhodothermus spp. (96% sequence identity). One OTU within the Firmicutes consisted of sequences from two libraries, one from each transect. This OTU was related to clone M2D03 (accession no. AF445720) from Angel Terrance hot spring in Yellowstone National Park.
Arsenite oxidase gene diversity
From the 84 new aioA-like amino acid sequences retrieved in this study, there were six distinct AioA-like clades (Fig. 3). Approximately 49% of the sequences had strong homology to aioA amino acid sequences from the whole genomes of C. aurantiacus (99–100% sequence identity) and to previously identified environmental aioA-like sequences affiliated with the Chloroflexi, including clone W1–8 retrieved from arsenical pesticide contaminated soil in Washington (USA) (accession no. DQ380638) and the clone VD-7 retrieved from the Madison River Valley soil in Montana (USA; accession no. DQ380639; Peryea, 2002; Macur et al., 2004) (Fig. 3). Among the Chloroflexi-like aioA sequences, those from ETGF formed two clades, demonstrating that there was potentially additional diversity (data not shown). The remaining sequences were homologous to previously retrieved isolates and environmental clones belonging to the undifferentiated group of Betaproteobacteria and Gammaproteobacteria, as well as to the Alphaproteobacteria (Macur et al., 2004; Cai et al., 2009; Hamamura et al., 2009). Numerous proteobacterial strains oxidize arsenite (e.g. see Fig. 3 for Accession numbers; Heinrich-Salmeron et al., 2011; Sultana et al., 2012). Some of these aioA-like sequences from ETGF were consistent with 16S rRNA gene taxonomic identities. For instance, high aioA amino acid similarities and 16S rRNA gene sequence similarities were noted for Roseovarius spp. (Alphaproteobacteria) and Marinobacter spp. (Gammaproteobacteria; Fig. 3; Table S1). But two additional aioA clades could not be taxonomically assigned to any previously identified proteobacterial genera, and it was unclear from 16S rRNA genes which groups they might be.
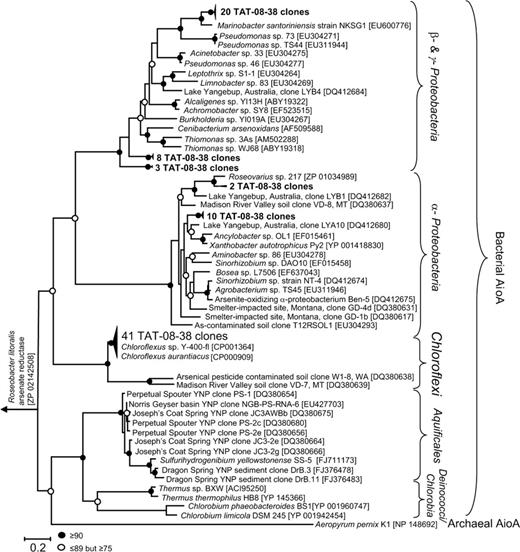
Phylogenetic tree of Aio-like sequences from amplified isolates and clones. Topology inferred using phyml with the LG+I+G model for amino acid substitution (G = 1.276, I = 0.007). Bootstrap values (per 100 replicates) of major branch points are shown. The tree was rooted with Roseobacter litoralis arsenate reductase gene. Accession numbers for isolates and clones are shown in brackets.
Protein ET analyses
To understand how the differences in the amino acid sequences translated to differences in the potential function of the AioA protein, ET analyses were performed to identify significant amino acid residues across the known arsenite oxidase protein diversity and within major phylogenetic groups identified from ETGF. Herein, all amino acid numbers were referred to as the site number in the A. faecalis protein. We sequenced residues 25–195 of the large subunit of arsenite oxidase (Fig. 4), comprising portions of domain I that binds the [3Fe–4S] cluster and the Rieske subunit with domain II involved in hydrogen bonding (Ellis et al., 2001). Identified residues were mapped onto the protein crystal structure from A. faecalis (Ellis et al., 2001) to determine the position of residues relative to known functional centers in the protein involved in electron transfer, following the scheme: Mo → [3Fe–4S] → [2Fe–2S] (van Lis et al., 2012b). Within the sequenced segment of the protein, ET analyses identified 15 sites with conserved residues across all of the aioA genes from our alignment, mainly associated with the [3Fe–4S] cluster-binding motif and Mo-bisPGD cofactor that highlight the functionally constrained core common to all genes (Fig. 4b). For instance, one conserved residue was Ser99, which has also been found to be highly conserved among all bacterial groups because it is where electrons are passed to His62 of AioB through a hydrogen bridge with one of the cluster-bridging S atoms from the [3Fe–4S] center to the [2Fe–2S] center (Duval et al., 2010; van Lis et al., 2012b). Other conserved residues have been noted previously from cultured microbial groups by other researchers (Santini & vanden Hoven, 2004; van Lis et al., 2012b), but have not been confirmed from amino acids coexisting from a single natural habitat.
![Three-dimensional crystal structure of Acaligenes faecalis (Ellis et al., 2001) with residues identified by ET analyses mapped onto the protein. (a) Arsenite oxidase cluster showing the larger subunit I (blue), the smaller Rieske subunit (green), the [3Fe–4S] cluster (yellow), and the molybdenum-bis (pyranopterin guanine dinucleotide; Mo-bisPGD) cofactor (purple). (b–d) Arsenite oxidase large subunit illustrating the portion of the protein represented by our sequences (black) relative to the rest of the protein (gray), as well as the Mo-bisPGD (purple) cofactor and the [3Fe–4S] cluster (yellow). (b) Residues (light blue) conserved across all of the AroA sequences in our alignment. (c) Residues (green) divergent between the Aquificales group and the remaining sequences. (d) Residues unique to the Alphaproteobacteria (orange near the [3Fe–4S] cluster), divergent between the Proteobacteria and the Chloroflexi+ Aquificales group (orange near the Mo-bisPGD), or divergent among all groups (red).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsec/83/3/10.1111_1574-6941.12030/2/m_fem12030-fig-0004.jpeg?Expires=1750366867&Signature=c~u4B21QPC6eysWZIm~J-IvyMAqGlVt-RpHEiScphM6L9bMG9cEmejzxhgNiO4-v92HcqAc4-jccui9StzR14xrFk~JDbsBQcbUlkAqNQbHz2xzcAfLEas7R38QNsyEOCJzX3DyK5X5fSoYf13nZy9ToQ3BJa7311uIVV5hb5hqG2kBAKD2Rv5XG3ZRPr7tsWXAlppAenHYQPT9fNWObie368SxrLIPfU6BT2tQcZaFFVZhgkwxXy9SidEM7CllJ3xn0R9PJstvjL5~DMf2I74hSZuiR2b8q6Qo80w4kdzDkrUkqz~jj9-ef74BbVJhilUxAsxNN6XXaDAuh11HkSg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Three-dimensional crystal structure of Acaligenes faecalis (Ellis et al., 2001) with residues identified by ET analyses mapped onto the protein. (a) Arsenite oxidase cluster showing the larger subunit I (blue), the smaller Rieske subunit (green), the [3Fe–4S] cluster (yellow), and the molybdenum-bis (pyranopterin guanine dinucleotide; Mo-bisPGD) cofactor (purple). (b–d) Arsenite oxidase large subunit illustrating the portion of the protein represented by our sequences (black) relative to the rest of the protein (gray), as well as the Mo-bisPGD (purple) cofactor and the [3Fe–4S] cluster (yellow). (b) Residues (light blue) conserved across all of the AroA sequences in our alignment. (c) Residues (green) divergent between the Aquificales group and the remaining sequences. (d) Residues unique to the Alphaproteobacteria (orange near the [3Fe–4S] cluster), divergent between the Proteobacteria and the Chloroflexi+ Aquificales group (orange near the Mo-bisPGD), or divergent among all groups (red).
ET analyses also revealed six residues that exhibited variation concordant with functional diversification among the major phylogenetic groups represented in our tree, which included gene sequences from ETGF and from previously published strains and environmental clones (Fig. 3). One site (residue 176) was near the Mo-bisPGD cofactor and differentiates the Proteobacteria from the Chloroflexi and Aquificales groups (Fig. 4d). The other five residues were near the [3Fe–4S] cluster (Fig. 4c and d). Three sites (88, 91, and 93) differentiated the Aquificales group from the rest of the sequences; one site (103) contained a unique residue in the Alphaproteobacteria. Lastly, one site (98) was unique in every group, with each taxonomic group containing a different amino acid residue (Table 1). Because of its proximity to Ser99, residue 98 is also central to electron transfer from the large subunit [3Fe–4S] cluster to the small subunit Rieske [2Fe–2S] center (Ellis et al., 2001). Among the four phylogenetic groups of aioA genes identified from ETGF, Proteobacteria had small, slightly polar to neutral residues at the 98 position, but Aquificales and Chloroflexi had large, aromatic, slightly hydrophobic/polar residues. The presence of a His at this site in the Chloroflexi was considered unique, as histidines are the most common amino acids in protein active, particularly metal binding, sites (Holm & Sander, 1997). Overall, the significant property differences among the residues were large and demonstrate potential divergence in modes of electron transfer among the different phylogenetic groups (Ellis et al., 2001; Duval et al., 2010).
Arsenite oxidase subunit 1 sites exhibiting functional divergence, as identified by ET analyses, and shown in Fig. 4
| Amino acid position (rank) | 88 (1.77) | 91 (1.77) | 93 (2.66) | 98 (3.01) | 103 (2.16) | 176 (2.03) |
| Beta-/Gammaproteobacteria | D | C | V | S | G | T |
| Alphaproteobacteria | D | C | V | G | A | T |
| Chloroflexi | D | C | V | H | G | N |
| Aquificales, Deinococci, Chlorobi | A | S | I | Y | G | N |
| Significant property changes | + | + | − | + | + | + |
| Amino acid position (rank) | 88 (1.77) | 91 (1.77) | 93 (2.66) | 98 (3.01) | 103 (2.16) | 176 (2.03) |
| Beta-/Gammaproteobacteria | D | C | V | S | G | T |
| Alphaproteobacteria | D | C | V | G | A | T |
| Chloroflexi | D | C | V | H | G | N |
| Aquificales, Deinococci, Chlorobi | A | S | I | Y | G | N |
| Significant property changes | + | + | − | + | + | + |
Indicated are the amino acid position based on the Alcaligenes faecalis crystal structure numbering (Ellis et al., 2001), the residue identity in each phylogenetic group, the ET ranks by relative evolutionary importance (i.e. the smaller the number, the higher the potential evolutionary importance), and amino acid differences that represent significant property changes (e.g. size, hydrophobicity, polarity, charge, side group composition).
Arsenite oxidase subunit 1 sites exhibiting functional divergence, as identified by ET analyses, and shown in Fig. 4
| Amino acid position (rank) | 88 (1.77) | 91 (1.77) | 93 (2.66) | 98 (3.01) | 103 (2.16) | 176 (2.03) |
| Beta-/Gammaproteobacteria | D | C | V | S | G | T |
| Alphaproteobacteria | D | C | V | G | A | T |
| Chloroflexi | D | C | V | H | G | N |
| Aquificales, Deinococci, Chlorobi | A | S | I | Y | G | N |
| Significant property changes | + | + | − | + | + | + |
| Amino acid position (rank) | 88 (1.77) | 91 (1.77) | 93 (2.66) | 98 (3.01) | 103 (2.16) | 176 (2.03) |
| Beta-/Gammaproteobacteria | D | C | V | S | G | T |
| Alphaproteobacteria | D | C | V | G | A | T |
| Chloroflexi | D | C | V | H | G | N |
| Aquificales, Deinococci, Chlorobi | A | S | I | Y | G | N |
| Significant property changes | + | + | − | + | + | + |
Indicated are the amino acid position based on the Alcaligenes faecalis crystal structure numbering (Ellis et al., 2001), the residue identity in each phylogenetic group, the ET ranks by relative evolutionary importance (i.e. the smaller the number, the higher the potential evolutionary importance), and amino acid differences that represent significant property changes (e.g. size, hydrophobicity, polarity, charge, side group composition).
Discussion
The goal of this study was to enhance our understanding of potential arsenic-based metabolism from ETGF, a unique, natural geothermal site with exceptional arsenic concentrations compared to other natural and contaminated environments globally (Webster & Nordstrom, 2003; Landrum et al., 2009). The distinctive geochemistry of ETGF was hypothesized to support novel groups of microbes capable of cycling arsenic. Although there has been recent effort to combine 16S rRNA gene surveys with functional arsenic gene analyses from other habitats (Hamamura et al., 2009; Sultana et al., 2012; Meyer-Dombard et al., in press), and there is considerable interest in isolating potential arsenite-oxidizing bacteria from different environments, the results from our investigation expand the current knowledge of arsenite oxidase diversity in geothermal environments and reveal how significant differences in amino acid residues across the retrieved diversity correspond to potential differences in arsenite oxidase protein function for major phylogenetic groups at ETGF. Collectively, the results provide us a direction for future attempts to isolate arsenite-oxidizing bacteria from ETGF, particularly Chloroflexi.
On the basis of the relative abundances of microbial groups from the 16S rRNA gene sequence surveys, representation of major taxonomic groups changed downstream from the geyser orifice pools. These changes not only corresponded to temperature decreases, but also to where the concentrations of As(V) exceeded As(III) in the discharge waters (Landrum et al., 2009). As Landrum et al. (2009) demonstrate, microbial metabolic processes are likely responsible for changes in arsenic speciation. On the basis of the previous work (Silver & Phung, 2005; Inskeep et al., 2007; Ledbetter & Magnuson, 2010; van Lis et al., 2012b), we considered likely arsenite-oxidizing bacterial groups at ETGF to include members of the phyla Proteobacteria, Deinococcus-Thermus, Aquificales, Chlorobi, Chloroflexi, and even Archaea. Proteobacteria capable of arsenite oxidation are probably the most frequently detected from natural and contaminated settings (Macur et al., 2004; Cai et al., 2009; Hamamura et al., 2009; Heinrich-Salmeron et al., 2011; Sultana et al., 2012). Detection of various proteobacterial clades at ETGF was expected, and the occurrence of potentially novel groups was also unsurprising (Fig. 3). However, because populations of any of the Proteobacteria were not significantly abundant in the microbial mats (Fig. 2), it is highly unlikely that these groups could be capable of the observed changes in arsenic speciation downstream at the Tower Geyser, but could be more important at the Lower Basin site > 50 °C. Of the other potential arsenite-oxidizing lineages, we did not retrieve aioA-like genes homologous to Aquificales, Chlorobi, or Archaea (e.g. Pyrobaculum). This starkly contrasts previous work done with geothermal fluids in Yellowstone National Park and other geothermal settings (Macur et al., 2004; Inskeep et al., 2007; Hamamura et al., 2009), as well as from marine hydrothermal vents (Meyer-Dombard et al., in press). No archaeal strains have been experimentally shown to oxidize arsenite, although some are associated with arsenic cycling (van Lis et al., 2012b). On the basis of the archaeal taxonomic groups from ETGF from 16S rRNA gene sequence clone libraries and 454 tag pyrosequences (Franks, 2012), archaeal taxa associated with arsenic or that possess aioA genes are not prevalent in the microbial mats from Tower Geyser, so it understandable that we did not retrieve aioA homologs to Archaea.
We also did not detect aioA genes affiliated with Deinococcus-Thermus, despite 16S rRNA gene sequence clones to this phylum being relatively more abundant upstream (Fig. 2), which is similar to their distribution in other hot spring and arsenic-laden systems (Connon et al., 2008; Meyer-Dombard et al., in press). Our inability to detect aioA genes from Thermus spp. could be because, to our knowledge, retrieved arsenite oxidase gene diversity among Thermus spp. is limited overall, although some are considered to be capable of rapid arsenite oxidation (Chung et al., 2000; Gihring et al., 2001). Only one unpublished investigation (Accession numbers FJ231195–FJ231198) and two published studies using similar primers (Inskeep et al., 2007; Hamamura et al., 2009) have resulted in a total of six aioA-like gene sequences available in GenBank that are closely related to cultured representatives from Thermus, including T. thermophilus,T. aquatics, and T. scotoductus.
There was an unexpected convergence among aioA gene sequences affiliated with the Chloroflexi (predominately C. aurantiacus) from the Tower Geyser microbial mats. Chloroflexus aurantiacus is considered to be an anaerobic photoheterotroph that assimilates CO2 with the 3-hydroxypropionate autotrophic carbon fixation pathway (Zarzycki et al., 2009), but can also be a chemoheterotroph in the dark by consuming organic carbon (e.g. acetate, lactate, propionate, and butyrate; Pierson & Castenholz, 1974; Hanada & Pierson, 2006; Tang et al., 2011). The relative abundance of Chloroflexi-like groups was higher downstream where the concentration of As(V) exceeded As(III) in the discharge streams at temperatures < 70 °C. Although Chloroflexus spp. have been found in temperatures higher than what other anoxygenic phototrophs can survive, optimal growth is considered to be between 50 and 60 °C, which matches with the 16S rRNA gene survey distribution results. The link between the distribution of Chloroflexi along the geyser streams and arsenic speciation suggests that Chloroflexi may play a more important role in arsenic cycling at ETGF than previously considered. Moreover, because only two other environmental aioA clone sequences closely related to Chloroflexus spp. were available from the public databases (Fig. 3), our study expands the known distribution of arsenite oxidase genes from Chloroflexus spp. These data should allow for future hypothesis-testing with respect to the potential metabolic implications of Chloroflexi and arsenic cycling.
Knowledge of C. aurantiacus arsenite oxidase enzyme structure and activity from natural settings is limited, despite C. aurantiacus being previously considered an ideal organism to study arsenite oxidase enzyme function and arsenic-based metabolism (Lebrun et al., 2003). Among the arsenite oxidase homologs, where other known, particularly proteobacterial, arsenite-oxidizers possess functional gene clusters comprised of several Aio genes (van Lis et al., 2012b), C. aurantiacus only possess the two aioA and aioB genes, and these genes are preceded by the transcriptional regulator gene luxR (van Lis et al., 2012b). One of the genes that C. aurantiacus is not known to possess is aioX. This gene is essential for aioBA upregulation and arsenite oxidation in Agrobacterium tumefaciens strain 5A, but also other bacterial groups (Sinorhizobium, Xanthobacter,Alcaligenes, Thiomonas, Achromobacter,Burkholderia, and T. thermophilus; Liu et al., 2012). Consequently, in the absence of aioX as the arsenite sensing mechanism (sensu stricto Liu et al., 2012), electrons from arsenite oxidation could potentially transfer reducing equivalents toward another pathway, such as photosynthesis (Lebrun et al., 2003; van Lis et al., 2012b).
Previous research dating back to experiments by Zannoni & Ingledew (1985) demonstrate the Aio-Rieske protein in Chloroflexus spp., which has a slightly higher but comparable reduction potential to the Rieske/cytb motif (Duval et al., 2010), can be re-oxidized by short illumination and can be linked to electron transfer at the photosynthetic reaction center. Incidentally, this reduction potential also closely matches what C. aurantiacus requires for auracyanin, a small blue copper protein, to transfer electrons to cytochrome c554, the electron donor of the photosynthetic reaction center (McManus et al., 1992). Because laboratory experiments with C. aurantiacus have demonstrated expression of Aio during photosynthetic growth and oxidation of auracyanin and Aio-Rieske (Zannoni & Ingledew, 1985; Lebrun et al., 2003), van Lis et al. (2012b) propose that auracyanin would accept electrons from arsenite oxidation as the reductant of the photosynthetic reaction center. Our observed changes in the amino acid residue at position 98 to a His for the Chloroflexi-like sequences may contribute to electron transfer. Use of arsenite as the photosynthetic electron donor has been demonstrated for an Ectothiorhodospira strain, although this species uses the Arx system (Kulp et al., 2008; Hoeft et al., 2010; van Lis et al., 2012b).
In summary, the current study from ETGF expands the known distribution and diversity of arsenite-oxidizing bacteria in natural, arsenic-laden geothermal settings that have nonsulfidic, circumneutral fluids at temperatures more than 50 °C. Determining whether the Chloroflexi at ETGF use As(III) during photosynthesis, or function as anoxygenic photoheterotrophs without any relevance to arsenic cycling, is a matter of future research. van Lis et al. (2012b) propose that arsenite oxidation via Aio may be an ancient metabolism, supported by the fact that Chloroflexi are among some of the deepest-branching bacterial lineages. If Chloroflexi at ETGF oxidize arsenite to reduce auracyanin as part of the anoxygenic photosynthetic pathway, then ETGF may be yet another site to explore novel arsenic oxidation pathways and could provide insight into the diversity of arsenic cycling among phylogenetically distinct groups that arose from divergent evolutionary pressures (Gupta et al., 2002; Lebrun et al., 2003; Silver & Phung, 2005; Kulp et al., 2008; Ledbetter & Magnuson, 2010).
Acknowledgements
This project was supported in part by the US National Science Foundation, with a collaborative award to A.S.E. (EAR-0544960) and Philip C. Bennett at The University of Texas at Austin (EAR-0545336). The Jones Endowment for Aqueous Geochemistry at the University of Tennessee-Knoxville to A.S.E. has recently supported this research. L.R.J. was also partially funded by a John Teagle Memorial Grant of the American Association for Petroleum Geologists Grants in Aid program. C. Omelon, M. Franks, and J. Landrum assisted in the field work. B. Donnelley and S. White were undergraduate students who assisted in laboratory analyses at LSU. Earlier versions of this manuscript benefited from input by G. Byerly and J. Hanor at LSU.
References
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Author notes
Department of Biology, University of South Dakota, Vermillion, SD USA
Editor: Cindy Nakatsu

{kind=link}
{kind=link}
{kind=link}
{kind=link}